

Abstract
The mammalian hippocampus harbours neural circuitry that is crucial for associative learning and memory. The mechanisms that underlie the development and regulation of this complex circuitry are not fully understood. Our previous study established an essential role for the zinc finger protein Zbtb20 in the specification of CA1 field identity in the developing hippocampus. Here, we show that conditionally deleting Zbtb20 specifically in mature CA1 pyramidal neurons impaired hippocampus-dependent memory formation, without affecting hippocampal architecture or the survival, identity and basal excitatory synaptic activity of CA1 pyramidal neurons. We demonstrate that mature CA1-specific Zbtb20 knockout mice exhibited reductions in long-term potentiation (LTP) and NMDA receptor (NMDAR)-mediated excitatory post-synaptic currents. Furthermore, we show that activity-induced phosphorylation of ERK and CREB is impaired in the hippocampal CA1 of Zbtb20 mutant mice. Collectively, these results indicate that Zbtb20 in mature CA1 plays an important role in LTP and memory by regulating NMDAR activity, and activation of ERK and CREB.
Key points
Zinc finger and BTB domain-containing protein 20 (Zbtb20) plays a critical role in hippocampal development.
In the present study, we generated mutant mice in which Zbtb20 knockout was restricted to mature CA1 pyramidal cells of the hippocampus.
Conditionally deleting Zbtb20 specifically in mature CA1 pyramidal neurons impaired LTP and memory.
We found that Zbtb20 controls memory formation and synaptic plasticity by regulating NMDAR activity, and activation of ERK and CREB.
Introduction
The hippocampus plays a critical role in learning and memory. Plasticity within dentate gyrus (DG)–CA3–CA1 circuit synapses is well known to underlie learning and memory via the process of long-term potentiation (LTP) (Lynch, 2004). Strong repetitive stimulation of the input to a hippocampal neuron can activate the N-methyl-d-aspartate receptor (NMDAR) and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR). The opening of NMDA channels allows Ca2+ entry, which is a critical step in the induction of LTP, as it activates numerous downstream kinases such as Ca2+/calmodulin-dependent kinases (CaMK) II (Fukunaga et al. 1993), protein kinase A (PKA) (Roberson & Sweatt, 1996) and protein kinase C (PKC) (Vaccarino et al. 1987; Roberson et al. 1999). Extracellular signal-regulated kinase (ERK) appears to act as a point of convergence for the above-mentioned signalling cascades (Lynch, 2004). Of the many downstream effectors of ERK, cAMP response element binding protein (CREB) in particular has received a great deal of attention due to its critical role in memory formation (Benito & Barco, 2010).
Zinc finger and BTB domain-containing protein 20 (Zbtb20, also known as DPZF, HOF and Zfp288) belongs to a subfamily of zinc finger proteins containing C2H2 Krüppel-type zinc fingers and BTB/POZ domains (Zhang et al. 2001; Mitchelmore et al. 2002), and plays a critical role in hippocampal development (Nielsen et al. 2007; Xie et al. 2010). Zbtb20 protein expression initiates in mouse hippocampal primordia as early as embryonic day 13.5 (E13.5), and is maintained at high levels in hippocampal neurons throughout late embryonic stages and early postnatal development. Genetic deletion of Zbtb20 leads to severe defects in hippocampal development (Xie et al. 2010), while ectopic overexpression of Zbtb20 in subiculum pyramidal neurons induces hippocampus-like corticoneurogenesis (Nielsen et al. 2007). Importantly, Zbtb20 is essential for the specification of CA1 field identity in the developing hippocampus through its repression of adjacent transitional neocortex-specific fate determination (Nielsen et al. 2010; Xie et al. 2010). In adulthood, Zbtb20 expression is specifically restricted to hippocampal neurons and glia (Mitchelmore et al. 2002), but its potential role in mature CA1 neurons and its relevance to learning and memory has not been determined.
In the present study, we generated mutant mice in which Zbtb20 knockout was restricted to mature CA1 pyramidal cells of the hippocampus (hereafter referred to as CA1-ZB20KO). These mutant mice exhibited no obvious morphological abnormalities; however hippocampus-dependent memory was impaired. We used the CA1-ZB20KO mice to determine the effect of ablating Zbtb20 activity in the CA1 on synaptic plasticity, and to analyse the activity of genes involved in memory. We found that Zbtb20 controls memory formation and synaptic plasticity by regulating NMDAR activity, and activation of ERK and CREB.
Methods
Generation of Zbtb20 mutant mice
Zbtb20flox mice were prepared as previously described (Xie et al. 2008). We crossed Zbtb20flox mice with heterozygous T29-1 transgenic mice, which have the capacity to mediate Cre/loxP recombination in the forebrain or exclusively in CA1 pyramidal cells (Tsien et al. 1996a). After repeated crossings, we obtained Zbtb20 mutant mice (Zbtb20flox/flox;CaMKII-Cre), as well as various types of siblings. From the latter, floxed/Cre-negative, non-floxed Cre-positive, and wild-type mice were used as littermate controls. Throughout the behavioural and electrophysiological experiments, observers were blind to the genotype of each individual animal. Animal experiments were conducted in accordance with the guidelines of the Second Military Medical University Animal Ethics Committee, and comply with The Journal of Physiology and UK regulations on animal experimentation (Drummond, 2009). In the experiments, except for the behavioural test, mice were anaesthetized with sodium pentobarbital (50 mg kg−1) and decapitated.
Immunohistochemistry
Immunohistochemistry was carried out as described previously (Xie et al. 2010). Briefly, forebrain cryosections were incubated overnight with the appropriate primary antibody. The signal was amplified using a tyramine amplification kit (Perkin-Elmer, TSA biotin system) according to the manufacturer's instructions and visualized with Alexa Fluor 594 (Molecular Probes). DAPI was used for nuclear counterstaining. The following primary antibodies were used: mouse monoclonal anti-Zbtb20 antibody 9A10 (1:1000), mouse monoclonal anti-NeuN (1:1000, Chemicon, Temecula, CA, USA), mouse monoclonal anti-Calbindin-D28k (1:1000, Swant, Bellinzona, Switerland), rabbit polyclonal anti-active caspase 3 (1:1000, Chemicon), rabbit monoclonal anti-CREB (1:1000, Cell Signaling Technology, Inc., Danvers, MA, USA) and rabbit monoclonal anti-pCREB (1:100, Cell Signaling Technology). Secondary antibodies were either horseradish peroxidase- conjugated anti-mouse or anti-rabbit IgG (1:8000, Vector Laboratories, Inc., Burlingame, CA, USA).
In situ hybridization
In situ hybridization was carried out as described previously (Xie et al. 2010). Antisense riboprobes labelled with digoxygenin-UTP were transcribed from cDNA clones. Sequence information for the in situ hybridization RNA probes of Man1α is as following: GenBank accession no., NM_008548; probe position, 1806–2184; probe length (bp), 379; clone ID, 214–3. After overnight hybrization with riboprobes, coronal forebrain cryosections were detected with anti-digoxygenin (Roche) antibody conjugated to alkaline phosphatase, and developed with nitroblue tetrazolium.
TUNEL assays
Serial coronal forebrain cryosections were subjected to TUNEL staining following the manufacturer's protocol (Promega Corp., Madison, WI, USA).
Behavioural methods
Behavioural experiments were conducted with age- and sex-matched CA1-ZB20KO mice and control littermates.
Water maze task
The Morris water maze apparatus consisted of a circular pool (1.2 m in diameter) containing water maintained at 24–26°C. The training protocol consisted of 8 days (2 trials per day). In order to escape from the water, the mice had to find a hidden escape platform (diameter: 11 cm) that was submerged in a fixed location, approximately 1 cm below the water surface. For each trial, the animal was placed into the maze near one of four possible points – north, south, southeast, or northwest. The location was determined randomly for each trial. Animals were allowed up to 60 s to locate the escape platform in the southwest quadrant, and their escape latency was recorded. After landing on the hidden platform, each mouse was allowed to remain for 15 s before being returned to its cage. Mice that failed to land on the platform by themselves within the time limit were manually guided to it. After the 8 day training, the mice were then subjected to probe trials on post-training day 1 and day 10, respectively. During the probe trials, the platform was removed and the mice were allowed to swim in the pool for 60 s. The time spent in each quadrant was recorded.
Novel object recognition task
The experimental protocol used was almost the same as that described previously (Tang et al. 1999; Jeon et al. 2003). Forty-four animals were individually habituated to an open-field box (40 × 40 × 40 cm) for 3 days. During training sessions, two novel objects were placed into the open field and the animal was allowed to explore for 5 min. A mouse was considered to be exploring the object when its head was facing the object within 1 inch. The time spent exploring each object was recorded. Following retention intervals (1 h, 1 day or 3 days), the animals were placed back into the same box, in which one of the familiar objects used during training was replaced by a novel object, and allowed to explore freely for 5 min. The proportion of time exploring the novel object was expressed as a proportion of the total time exploring all the objects.
Contextual fear conditioning task
The procedures used for contextual fear conditioning and cued fear conditioning were similar to those described in previous studies (Tang et al. 1999; Jeon et al. 2003). The conditioned stimulus (CS) used was an 85 dB sound at 2800 Hz, and the unconditioned stimulus (US) was a continuous scrambled foot shock at 1 mA. For conditioning, mice were placed in the fear-conditioning apparatus chamber for 2 min, and then a 20 s acoustic CS was delivered. During the last 2 s of the tone, a 1 mA shock of US was applied to the floor grid. After the CS–US pairing, the mice were allowed to stay in the chamber for another 30 s to measure immediate freezing. Retention tests were conducted 1 h, 1 day and 10 days after training. During the retention test, each mouse was placed back into the shock chamber and the freezing response was recorded for 3 min (contextual conditioning). Subsequently, the mice were put into a novel chamber and monitored for 3 min before the onset of the tone (pre-CS). Immediately after that, a tone identical to the CS was delivered for 3 min and freezing responses were recorded (cued conditioning).
In another contextual fear conditioning protocol (Dai et al. 2008), mice were placed in the box and allowed to freely explore for 2 min before receiving five foot shocks (1 mA, 2 s) with intershock intervals of 2 min. Mice were then placed back in their home cages 2 min after the final foot shock. Freezing behaviour was measured as the amount of time exhibiting freezing behaviour during each intershock interval. To study contextual fear memory, mice were placed in the conditioned fear context 30 min, 1 day, and 10 days after fear conditioning and their contextual freezing behaviour was measured for 11 min without any foot shocks. In the experiments for detecting ERK and CREB phosphorylation, we used another batch male mice because the activation of ERK and CREB of male mice is more sensitive to fear conditioning than female mice (Gresack et al. 2009; Mizuno & Giese, 2010). Sham shocked mice were killed without shock and shocked mice were killed at 1 min, 1 h and 24 h after this contextual fear conditioning protocol task. Samples from one side of hippocampal CA1 was used for Western blot and the other side hippocampus was used for immunohistochemistry.
Golgi staining and analysis of dendritic spine density
Golgi-stained neurons were obtained using the FD Rapid Golgi Stain kit (FD Neurotechnologies, Columbia, MD, USA). Brains were prepared according to the user manual, and 100 μm serial coronal sections were cut with a cryostat. The Neurolucida program was used to reconstruct spines in three dimensions, and dendritic spines on hippocampal CA1 pyramidal neurons were counted by an experimenter blind to genotype. For each experimental group, dendritic spine density of three animals was analysed.
Hippocampal slice recording
The brain was rapidly dissected and transferred into ice-cold oxygenated artificial cerebrospinal fluid (ACSF) (in mm: 124 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 11 d-glucose, pH 7.35, ∼303 mosmol l−1) for 2 min. The hippocampus was dissected and trimmed. Transverse slices (350 μm thick) were cut using a Vibratome 3000 sectioning system (St Louis, MO, USA) at 0–2°C, and then maintained in a submerged chamber with oxygenated ACSF at 30°C for at least 2 h before recording. During the experiments, individual slices were transferred to a submersion recording chamber and were continuously perfused with oxygenated ACSF at 28–30°C. The rate of perfusion control was 2–3 ml min−1. For recording field excitatory postsynaptic potentials (fEPSPs) in the CA1 region of the hippocampus, both the stimulating and recording electrodes were placed in the stratum radiatum of the CA1. The distance between the two electrodes was kept at 80–120 μm. Recording pipettes were made from borosilicate glass capillaries (B-120–69-15, Sutter Instrument Co., Novato, CA, USA) and filled with ACSF solution. The stimulating electrode was a bipolar tungsten electrode (MCE-100; Rhodes Medical Instruments, Woodland Hills, CA, USA). fEPSPs were evoked by electrical stimuli to the Schaffer collateral/commissural afferents pathway with a pulse generator (Master-8; A.M.P.I., Jerusalem, Israel) coupled through an isolator (Iso-flex; A.M.P.I.).
The stimulation intensity (0.2 ms duration) was adjusted to give fEPSP slopes approximately 30–40% of the maximum, and baseline responses were elicited three times per minute (0.05 Hz) at this intensity. Paired-pulse facilitation (PPF) was measured at various interpulse intervals (20, 50, 100, 200 and 500 ms) and the paired-pulse ratio (PPR) was calculated as the ratio of the slope of the second fEPSP (fEPSP2) in relation to the slope of the first fEPSP (fEPSP1). The stable baseline of fEPSPs was obtained at least 30 min before LTP induction. Theta burst stimulation (TBS) to the Schaffer collateral fibres was used to induce synaptic potentiation. TBS consists of five bursts of spikes (5 pulses at 100 Hz) at 5 Hz, repeated 5 times at 0.1 Hz. All data were normalized to the last group of responses recorded prior to induction. In all electrophysiological experiments, n indicates the number of slices. Each data set is from at least three animals.
Whole-cell recordings were performed using pipettes pulled from borosilicate glass capillaries with a resistance of 3–5 MΩ when filled with the following solution (in mm): 130 potassium d-gluconate, 20 KCl, 10 Hepes, 10 disodium phosphocreatine, 4 MgATP, 0.3 Na2GTP, 0.2 EGTA (pH 7.32, adjusted with KOH, ∼299 mosmol l−1). Slices were continuously perfused with oxygenated ACSF at 28–30°C (in mm: 124 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 11 d-glucose, 0.03 PTX, pH 7.35, ∼303 mosmol l−1). Excitatory postsynaptic currents (EPSCs) were evoked at a rate of 0.05 Hz by a stimulating electrode placed in the stratum radiatum. Access resistance was monitored throughout experiments and ranged from 10 to 25 MΩ. Data were discarded when access resistance varied by >20% during an experiment. AMPAR-mediated EPSCs were recorded at a holding potential of −70 mV. NMDAR-mediated EPSCs were recorded at a holding potential of +40 mV in the presence of 10 μm CNQX to block AMPAR-mediated EPSCs. Amplitude was measured as the difference between measurements with cursors placed in the prestimulus baseline period and the EPSC peak. The decay time constant was estimated from single-exponential fits to the decay phase of the current starting just after the current peak.
All electrophysiological data were analysed using Clampfit 9.2 software.
Western blot analysis
Total proteins of CA1 subregions were electrophoretically separated on 4–15% gradient SDS-PAGE gels and transferred onto nitrocellulose membranes. Membranes were probed with the following primary antibodies: mouse monoclonal anti-Zbtb20 antibody 9A10 (1:1000); rabbit monoclonal anti-ERK antibody (1:1000, Cell Signaling Technology); rabbit monoclonal anti-pERK antibody (1:1000, Cell Signaling Technology); rabbit monoclonal anti-CREB antibody (1:1000, Cell Signaling Technology); rabbit monoclonal anti-pCREB (1:1000, Cell Signaling Technology); mouse monoclonal anti-α-tubulin antibody (1:8000, Sigma-Aldrich). Secondary antibodies were either horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (1:8000, Vector Laboratories). Signals were then generated using a Chemiluminescent Detection Kit (ECL Plus; Amersham Pharmacia Biotech, Piscataway, NJ, USA) and visualized with a scanner. Expression levels of each protein of interest were normalized to that of α-tubulin.
Statistical analyses
Statistical analyses unless otherwise indicated were carried out using Student's t test or ANOVA followed by post hoc comparisons. In the second contextual fear conditioning task, two-way ANOVA with repeated measures was used to analyse freezing with five-trial conditioning. Averaged fEPSP slope values per time point (with 1 min bin, expressed as the percentage change from baseline values within 30 min prior to the induction) were analysed using the non-parametric Wilcoxon's signed-rank test when compared within one group, or the Mann–Whitney U test when data were compared between groups. In the latter analysis, LTP magnitude was measured as the mean amplitude of fEPSP slopes within the last 30 min of recording. Statistical methods for other experiments are indicated in the corresponding text or figure legend. Differences were considered significant when P < 0.05. Throughout the paper, n indicates the number of animals, and data are given as means ± SEM. In all figures, symbols with error bars indicate the mean ± SEM; *P < 0.05; **P < 0.01.
Results
Generation of CA1-specific Zbtb20 knockout mice
To evaluate the potential role of Zbtb20 in mature CA1 neurons, we generated conditional knockout mice by crossing Zbtb20flox mice to CaMKII-Cre transgenic mouse line T29-1, which has the capacity to mediate Cre/LoxP recombination and gene deletion in highly differentiated, postmitotic CA1 pyramidal cells of the hippocampus, starting from the third postnatal week as demonstrated by LacZ reporter and X-Gal staining (Tsien et al. 1996a). The CA1-ZB20KO mice grew and mated normally, and their overall behaviour was indistinguishable from that of wild-type, homozygous Zbtb20flox, or Cre transgenic mice. These characteristics of CA1-ZB20KO mice are in contrast to the premature lethality of Zbtb20 global knockout mice (Sutherland et al. 2009).
Considering that Cre recombination mediated by the transgenic Cre line T29-1 tends to expand in forebrain neurons in an age-dependent manner (Fukaya et al. 2003), we first examined Zbtb20 expression in the mutant forebrain by immunohistochemistry. Before the end of the third postnatal week, spatial distribution of the Zbtb20 protein in the mutant hippocampus was indistinguishable from that of control (Fig. 1A and B). From postnatal day 24 (P24), Zbtb20 protein expression in CA1 pyramidal cells of the mutant mice was substantially reduced relative to control, and almost undetectable at 2 months of age, while its expression in the mutant CA3 and DG regions was not significantly altered compared with control counterparts (Fig. 1C–E). At 4 months, Zbtb20 knockout in the mutant forebrain was still restricted to CA1 pyramidal cells of the hippocampus, with an apparently normal distribution of Zbtb20 protein in the mutant CA3, DG, and neocortex (Fig. 1F and Supplemental Fig. S1). Beyond the hippocampus, Zbtb20 expression was restricted to glia in the adult forebrains of control and mutant mice. Efficient deletion of the Zbtb20 in the CA1 region of adult CA1-ZB20KO hippocampi was confirmed at the protein level by immunoblot analysis with anti-Zbtb20 antibody, and the residual Zbtb20 was most likely from glia (Supplemental Fig. S2). Therefore, CA1-ZB20KO mice lack Zbtb20 expression selectively in mature CA1 pyramidal neurons, and we used 2- to 4 month-old adult mice in the further study.
Figure 1. Lack of Zbtb20 protein expression in CA1 pyramidal cells of the adult CA1-ZB20KO mouse hippocampus.
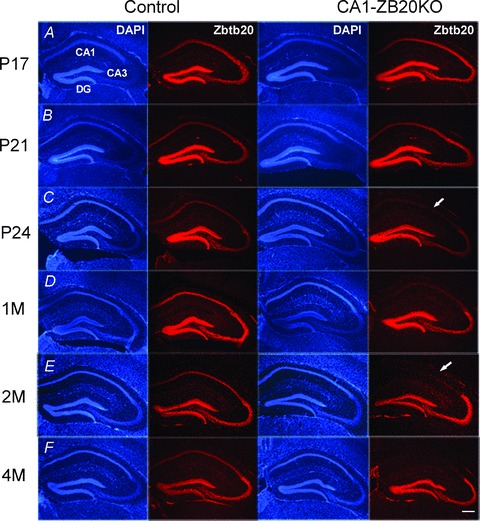
Zbtb20 expression was detected by immunohistochemistry with anti-Zbtb20 antibody 9A10 and visualized with Alexa 594 (red) on coronal forebrain sections from mice at P17 (A), P21 (B), P24 (C), 1 month (D), 2 months (E) and 4 months (F) of age. Zbtb20 expression in CA1 pyramidal cells of mutant mice began to reduce markedly at P24 and became almost undetectable by 2 months of age, but remained unaltered in the CA3 and DG regions at all ages examined. (Scale bars: 200 μm).
Normal gross hippocampal morphology in CA1-ZB20KO mice
Next, we examined whether genetic deletion of Zbtb20 in highly differentiated CA1 pyramidal cells could affect hippocampal development. Nissl staining did not reveal any obvious abnormalities in the laminar architecture, cellular density, or cellular arrangement of the hippocampus in 2- to 4 month-old adult CA1-ZB20KO mice (Supplemental Fig. S3). Immunohistochemical staining and in situ hybridization showed that the expression of NeuN protein, a general neuronal marker, and of Calbindin-D28k protein and mannosidase 1α (Man1α) mRNA, markers of mature CA1 pyramidal neurons (Lein et al. 2005; Nielsen et al. 2010), were indistinguishable between CA1-ZB20KO and control hippocampi (Supplemental Fig. S4). Moreover, TUNEL labelling and immunohistochemical staining for activated caspase-3 revealed that cell death is rare in the hippocampus of both CA1-ZB20KO and control adult mice (Supplemental Fig. S5). Taken together, these findings suggest that Zbtb20 is dispensable for CA1 pyramidal neurons to maintain their differentiation status and to survive, although it is essential for the specification of CA1 field identity during early development (Xie et al. 2010).
Impaired spatial memory of CA1-ZB20KO mice in the Morris water maze
To assess the effect of ablating Zbtb20 from the hippocampus of adult mice (2–4 months old), we first measured the performance of CA1-ZB20KO mice in the Morris water maze task, a spatial learning test of declarative memory (Kolb et al. 1994). During the acquisition phase of the test, both CA1-ZB20KO and control mice performed the task equally well, as illustrated by similar escape latencies for finding the platform throughout the 8-day training period (Fig. 2A). To test memory retention, each trained mouse was tested in probe trials on post-training days 1 and 10. During the probe tests, CA1-ZB20KO mice spent significantly less time in the target quadrant in which the platform was previously located (P < 0.05) (Fig. 2B and C). No difference in swimming speed was observed between CA1-ZB20KO and control mice during the training and probe trials (Fig. 2D). These results indicate that CA1-ZB20KO mice exhibit normal acquisition but impaired retention of spatial memory.
Figure 2. Impaired spatial memory of CA1-ZB20KO mice in the Morris water maze and novel object recognition tests.
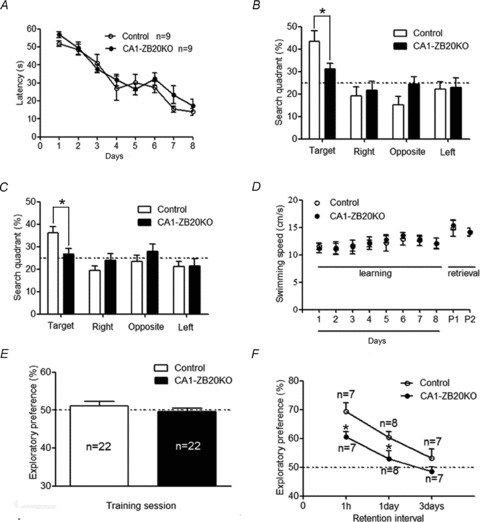
A, mean escape latency during 8 training days. CA1-ZB20KO mice showed normal acquisition of spatial reference memory in the Morris water maze task. B and C, in probe tests conducted 1 day (B) or 10 days (C) after the acquisition phase, CA1-ZB20KO mice spent significantly less time in the target quadrant than control mice (P < 0.05). D, there was no difference in swim speed between control and CA1-ZB20KO mice during the 8 training days and 2 probe tests (P1 and P2). E, exploratory preference in the training session of novel object recognition task. The dotted line represents performance at the chance level (50%). The amount of time spent exploring the two objects was the same for control and CA1-ZB20KO mice. F, memory in control and CA1-ZB20KO mice was measured after training at different time intervals as indicated. CA1-ZB20KO mice exhibited much weaker preference for the novel object than did control mice in retention tests conducted 1 h and 1 day after training. *P < 0.05.
Impaired novel object recognition memory in CA1-ZB20KO mice
We used the novel object recognition task to measure visual recognition memory, which requires the involvement of the hippocampus (Tang et al. 1999). During training, there was no significant difference between the two groups in the amount of time spent exploring the two novel objects, indicating that both mouse strains exhibit similar levels of curiosity and motivation to explore new objects (Fig. 2E). For the retention tests, which were conducted 1 h, 1 day and 3 days post-training, one of the two objects used during training was replaced by a novel object. In retention tests conducted either 1 h or 1 day post-training, CA1-ZB20KO mice exhibited a much weaker preference for the novel object than the control mice, indicating that memory is impaired in CA1-ZB20KO mice (Fig. 2F). By 3 days after training, the preference shown by control and CA1-ZB20KO mice had returned to basal levels (Fig. 2F).
Impaired contextual but not cued fear conditioning in CA1-ZB20KO mice
Contextual fear memory is hippocampus dependent, whereas cued fear memory is hippocampus independent (Phillips & LeDoux, 1992). Therefore, we next assessed whether contextual and cued fear memories were altered in CA1-ZB20KO mice, using previously described protocols (Tang et al. 1999; Jeon et al. 2003). Both contextual and cued conditioning were measured 1 h, 1 day and 10 days after training using separate groups of animals (Fig. 3). No significant difference was observed in immediate freezing between control and CA1-ZB20KO mice (Fig. 3A, C and E), but a significant difference was found in contextual freezing on post-training days 1 (P < 0.05) and 10 (P < 0.01), indicating that long-term memory of contextual fear conditioning is impaired in CA1-ZB20KO mice. On the other hand, no difference was observed between control and CA1-ZB20KO mice in cued fear conditioning assayed 1 h, 1 day and 10 days after training (Fig. 3B, D and F), indicating that impaired memory in CA1-ZB20KO mice is limited to hippocampus-dependent fear conditioning.
Figure 3. Impaired contextual fear memory but not cued fear memory in CA1-ZB20KO mice.
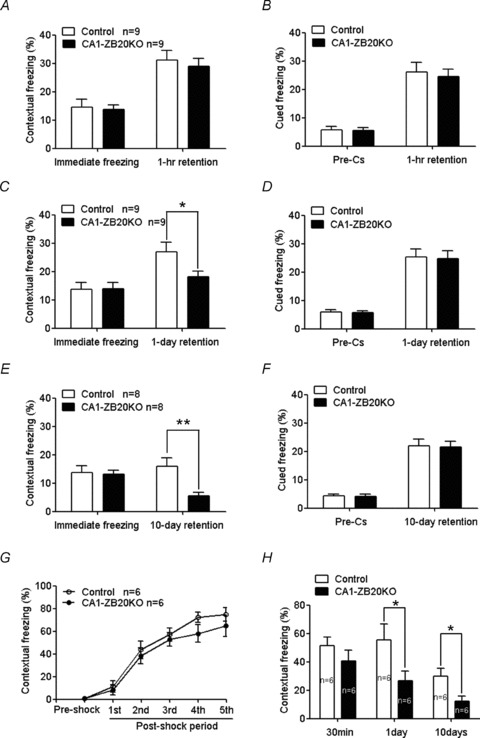
A, C and E, contextual fear conditioning 1 h (A), 1 day (C), and 10 days (E) after training. B, D and F, cued fear conditioning 1 h (B), 1 day (D) and 10 days (F) after training. G, freezing behaviour evoked by foot shocking did not differ significantly between CA1-ZB20KO and control mice during the five post-shock intervals. H, 1 day and 10 days after fear conditioning, freezing behaviour evoked by exposure to the conditioned environment was significantly impaired in CA1-ZB20KO mice compared to control mice. *P < 0.05; **P < 0.01.
To further validate our fear conditioning results, we assessed hippocampus- dependent contextual fear memory using a second protocol (Dai et al. 2008). Contextual fear memories were provoked in control and CA1-ZB20KO mice by associating a novel environment with five foot shocks at 2 min intervals, and freezing behaviour was measured as the percentage of time spent freezing during the 2 min interval between shocks. There was no significant difference in freezing behaviour between control and CA1-ZB20KO mice during the five postshock intervals (Fig. 3G). When placed back into the conditioned environment 30 min, 1 day or 10 days after conditioning, CA1-ZB20KO mice were frozen for a much shorter duration than control mice at 1 day or 10 days (P < 0.05) (Fig. 3H). Furthermore, no abnormal nociceptive responses were observed in CA1-ZB20KO mice, as their minimal amount of flinching/running, jumping or vocalizing was the same as in control mice (Supplemental Fig. S6). These results indicate that CA1-ZB20KO mice exhibit normal acquisition but impaired retention of contextual fear memory.
Synaptogenesis in CA1-ZB20KO mice
Since the synapse is widely assumed to be the cellular basis for learning and memory (Martin et al. 2000), we used Golgi staining to assess whether Zbtb20 regulates the density of dendritic spines and therefore the number of synapses. No significant difference was observed between CA1-ZB20KO mice and control mice in the density of dendritic spines along individual dendrites of hippocampal CA1 pyramidal neurons (Fig. 4), suggesting that Zbtb20 is probably not required for the formation or maintenance of dendritic spines in mature CA1 neurons, and that the deficit in hippocampus-dependent memory observed in CA1-ZB20KO mice is unlikely to result from altered spine-associated synaptogenesis.
Figure 4. CA1-ZB20KO mice show normal density of dendritic spines in CA1 pyramidal neurons.
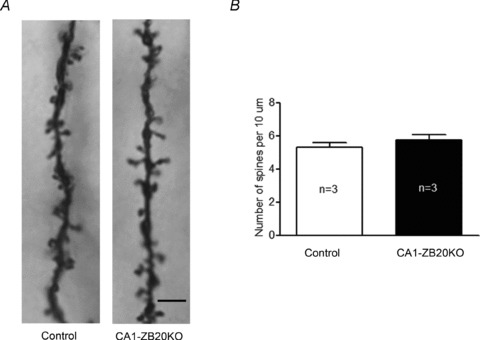
A, representative images of Golgi stained dendrites from the CA1 region of the hippocampus (Scale bar: 5 μm). B, there was no significant difference in the density of dendritic spines in CA1 pyramidal neurons between control and CA1-ZB20KO mice (Control, 27 neurons, 3 mice; CA1-ZB20KO, 27 neurons, 3 mice, P > 0.05).
Normal basal synaptic transmission and short term plasticity in CA1-ZB20KO mice
As our CA1-ZB20KO mice exhibited impairments in hippocampus-dependent memory, we next examined whether basal synaptic neurotransmission in the hippocampus of these mice was also affected. We recorded the slope of fEPSPs in the CA1 by stimulating the Schaffer collaterals of 2- to 4-month-old CA1-ZB20KO mice and their control counterparts, and computed the input–output relations of the fEPSPs to measure synaptic transmission efficiency. Input–output curves were constructed by plotting fEPSP slope against stimulation strength (Fig. 5A) or presynaptic fibre volley amplitude (Fig. 5B), or by plotting stimulation strength against presynaptic fibre volley amplitude (Fig. 5C). The curves were not statistically different between CA1-ZB20KO and control mice, suggesting that conditional ablation of Zbtb20 in mature CA1 neurons does not markedly affect basal synaptic neurotransmission and axonal excitability in the CA1 regions.
Figure 5. Normal basal synaptic transmission in CA1-ZB20KO mice.
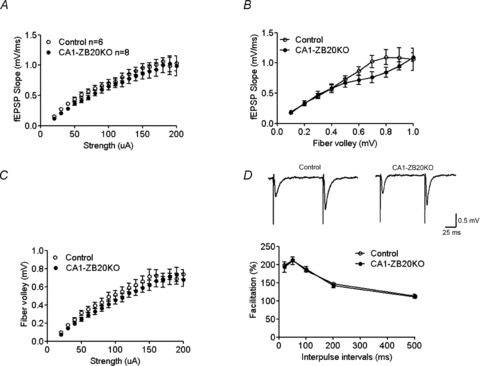
A, input–output plot of synaptic transmission between stimulation strength and corresponding fEPSP slope. B, input–output plot of synaptic transmission between fibre volley and corresponding fEPSP slope. C, plot of synaptic transmission between stimulation strength and presynaptic fibre volley amplitude. D, CA1-ZB20KO and control slices showed no significant difference in PPF of the EPSP at various interpulse intervals. Control, 13 slices, 6 mice; CA1-ZB20KO, 15 slices, 8 mice.
Next, we examined paired-pulse facilitation (PPF), a transient enhancement in neurotransmitter release induced by two closely spaced stimulations. This increase in neurotransmitter release is usually attributed to intracellular Ca2+ concentrations in the presynaptic terminal following the first stimulus (Regehr et al. 1994). Over five interstimulus intervals (20, 50, 100, 200 and 500 ms), we did not observe any statistical differences in PPF between the control and CA1-ZB20KO mice (198.1 ± 1.9%vs. 193.0 ± 14.3%, 212.1 ± 4.3%vs. 210.4 ± 10.9%, 183.8 ± 2.5%vs. 186.9 ± 7.4%, 146.6 ± 4.3%vs. 141.7 ± 5.9% and 112.9 ± 5.5%vs. 111.0 ± 4.4%, respectively; Control, n= 6; CA1-ZB20KO, n= 8; Fig. 5D). These results suggested that specifically ablating Zbtb20 expression in developed CA1 pyramidal neurons does not compromise presynaptic release probability or short-term plasticity at CA3–CA1 synapses.
Impaired LTP in CA1-ZB20KO mice
Since LTP has received much attention as a leading candidate mechanism involved in memory formation, we studied the LTP of CA3–CA1 synapses in hippocampal slices from adult control and CA1-ZB20KO mice. Theta-burst stimulation (TBS) of Schaffer collaterals was used to induce LTP (Nguyen & Kandel, 1997). In control hippocampal slices, TBS of the afferent fibres within the stratum radiatum produced a lasting potentiation of fEPSP slopes, which became steady within 2 h after TBS withdrawal (Fig. 6A). Although LTP was successfully induced by TBS in CA1-ZB20KO hippocampal slices relative to the initial pre-tetanus level (P < 0.01), its amplitude was significantly reduced in comparison with controls (Fig. 6B and C). The mean fEPSPs slope at 60–90 min after LTP induction was enhanced to only 142.1 ± 8.2% of its pre-tetanus level in the CA1-ZB20KO slices, which is significantly less than that observed in control slices (194.8 ± 9.1%, P < 0.01) (Fig. 6D). These results suggest that Zbtb20 plays an important role in regulating LTP capacity.
Figure 6. Impaired LTP and NMDAR activity in control and CA1-ZB20KO mouse hippocampal slices.
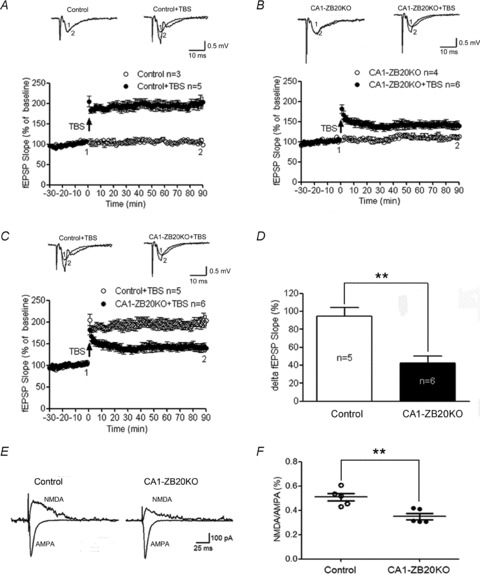
A, LTP elicited by TBS in control slices (Control, 5 slices, 3 mice; Control+TBS, 8 slices, 5 mice). B, LTP elicited by TBS in CA1-ZB20KO slices (CA1-ZB20KO, 7 slices, 4 mice; CA1-ZB20KO+TBS, 8 slices, 6 mice). C, Zbtb20 knockout impairs TBS-induced LTP in CA1-ZB20KO slices (panel C is a composite of A and B). D, mean ΔfEPSP slope measured 60–90 min after induction of LTP in control and CA1-ZB20KO slices (Control, 8 slices, 5 mice; CA1-ZB20KO, 8 slices, 6 mice). E, decreased NMDAR-mediated responses in the hippocampal CA1 of CA1-ZB20KO mice. Lower traces show representative AMPAR-mediated EPSCs recorded at −70 mV in the control solution. Upper traces show representative NMDAR-mediated EPSCs recorded at +40 mV in the presence of 10 μm CNQX, an AMPAR blocker. F, the NMDA/AMPA ratio in CA1 neurons of CA1-ZB20KO mice (34.2 ± 4.0%, 10 neurons, 10 slices, 5 mice) was significantly lower than in control mice (50.8 ± 4.4%, 9 neurons, 9 slices, 5 mice). The dotplots display the actual ratios for each animal in the control and CA1-ZB20KO groups **P < 0.01.
Decreased NMDAR-mediated EPSCs in CA1-ZB20KO mice
The induction of LTP in hippocampal CA1 region requires NMDAR activation, and NMDAR-dependent LTP has been considered a leading candidate for a cellular locus for some aspects of learning and memory (Zorumski & Izumi, 1998; Winder & Schramm, 2001). Thus, we further investigated whether NMDAR function was altered in adult CA1-ZB20KO mice by obtaining whole-cell recordings from CA1 pyramidal neurons. In order to examine NMDAR-mediated EPSCs in relation to synaptic activation, we recorded both NMDAR- and AMPAR-mediated EPSCs evoked by electric stimulation on Schaffer collaterals in CA1 neurons from control and CA1-ZB20KO hippocampal slices. As shown in Fig. 6E and F, the NMDA/AMPA ratio was significantly lower in CA1-ZB20KO neurons compared with control mice. Since AMPAR mediate basal synaptic transmission and basal synaptic transmission in CA1-ZB20KO neurons was not different from control (Fig. 5), we suspect AMPAR-mediated EPSC was not impaired and the decrease in the NMDA/AMPA ratio could be the result of reduced synaptic NMDAR currents.
Impaired ERK and CREB phosphorylation in CA1-ZB20KO mice
A great deal of evidence indicates that LTP and memory are dependent on the activation ERK, which leads ultimately to activation of transcription factors such as CREB and translation. (Lynch, 2004; Benito & Barco, 2010), so we further examined whether phosphorylation of ERK and CREB was impaired in CA1-ZB20KO mice.
We used Western blotting to detect ERK, phosphorylated ERK (pERK), CREB and phosphorylated CREB (CREB). There was no significant difference in basal ERK or CREB levels between sham shocked control (animals placed in the fear conditioning apparatus as in the normal training protocol, but without the shocks) and CA1-ZB20KO mice (Fig. 7 and Supplemental Fig. S7). Upon stimulation with contextual fear conditioning protocols (Dai et al. 2008), pERK (Fig. 7A and B) and pCREB (Fig. 7C and D) levels in whole-cell lysates from the CA1 region of control and CA1-ZB20KO hippocampi were significantly increased 1 h and 24 h after foot shock (P < 0.01); however, the level of increase in CA1-ZB20KO mice at 1 h was significantly less than that observed in controls (P < 0.01; Fig. 7).
Figure 7. Impaired ERK and CREB phosphorylation in the hippocampal CA1 of CA1-ZB20KO mice.
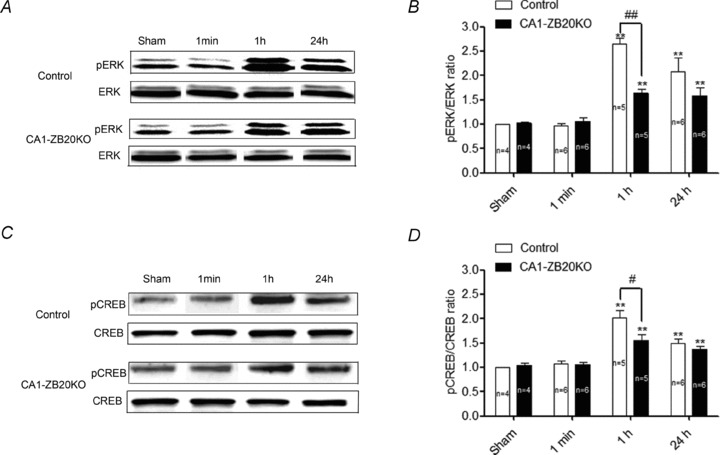
A and C, representative western blots of ERK (A) and CREB (C) protein concentrations and phosphorylation levels in the tissue lysates from hippocampal CA1 of sham shocked and shocked animals. No significant changes in overall ERK or CREB levels were observed between sham shocked and shocked control and CA1-ZB20KO mice. B and D, densitometric analysis of ERK (B) and CREB (D) activation 1 min, 1 h and 24 h after shock (both isoforms of pERK were together analysed). Levels of pERK and pCREB in control and CA1-ZB20KO mice were significantly increased 1 h and 24 h after shock, but this increase was significantly less in CA1-ZB20KO mice at 1 h compared with that in control counterparts. No significant changes in ERK or CREB activation were observed 1 min after shock. #P < 0.05, ##P < 0.01 vs. control mice 1 h after shock, **P < 0.01 vs. sham shocked mice.
We next assessed expression of CREB and phosphorylated CREB (pCREB) by immunohistochemistry. As shown in Supplemental Fig. S8 and Fig. 8A and B, CREB and pCREB expression were not different between sham foot shocked control and CA1-ZB20KO mice, and pCREB expression in hippocampi of control and CA1-ZB20KO mice was low before shock. Upon stimulation with contextual fear conditioning protocols (Dai et al. 2008), a substantial increase in pCREB was detected in both control and mutant hippocampi 1 h after foot shock, especially in the CA1 and DG regions, but phosphorylation of CREB was much lower in the mutant CA1 pyramidal cells relative to control counterparts (Fig. 8C and D). On the other hand, CREB phosphorylation in the mutant DG region was indistinguishable from that of control, suggesting that Zbtb20 deficiency in mature CA1 pyramidal cells specifically impaired CREB activation in CA1 neurons. Meanwhile, CREB expression was not different between control and CA1-ZB20KO mice 1 h after foot shock. These results suggest that CREB phosphorylation induced by contextual fear conditioning protocols is impaired in CA1-ZB20KO mice.
Figure 8. Decreased expression of pCREB in the CA1 of hippocampus of CA1-ZB20KO mice.
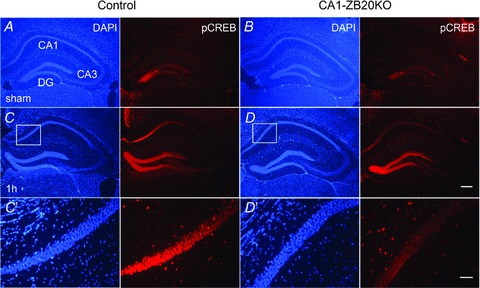
Immunohistochemistry was used to detect pCREB expression on coronal forebrain sections. A and B, pCREB expression was low and not different between sham foot shocked control and CA1-ZB20KO mice. C and D, pCREB expression was significantly increased in the hippocampus 1 h after foot shock. In the CA1 marked by box, pCREB expression in CA1-ZB20KO mice was significantly lower than control mice. C′ and D′, High-magnification views of the boxed areas in C and D, respectively. (Scale bars: 200 μm for A-D; 50 μm for C′ and D′).
Discussion
Our previous work established Zbtb20 as a critical transcription factor for cell fate determination of CA1 pyramidal neurons in the developing hippocampus (Xie et al. 2010). In the present study, we generated CA1-ZB20KO mice in order to determine the function of this molecule in mature CA1 neurons. The CaMKII-Cre transgenic line T29-1 that we used mediates Cre/LoxP recombination in highly differentiated CA1 pyramidal neurons (Tsien et al. 1996a,b), and tends to expand into the forebrain in an age-dependent manner (Fukaya et al. 2003; Brigman et al. 2010). Therefore, this line has also been used to generate forebrain-specific knockout mice (Chan et al. 2007; Gould et al. 2008; Brigman et al. 2010). In the adult CA1-ZB20KO mice we generated, Zbtb20 knockout was restricted to CA1 pyramidal cells, with no obvious effects on Zbtb20 expression in the CA3 and DG regions of the hippocampus until at least 4 months of age. As Zbtb20 is not expressed in adult forebrain neurons (only in forebrain glia) (Mitchelmore et al. 2002), we do not expect that Cre-mediated recombination has an effect on Zbtb20 expression outside of the hippocampal CA1 region. One possibility for this discrepancy of recombination specificity is that Cre recombination at the floxed alleles largely depends on the gene alleles where loxP sites are inserted. Therefore, we obtained the mouse model selectively lacking Zbtb20 in CA1 pyramidal neurons.
CA1-ZB20KO mice exhibited normal hippocampal architecture, and Zbtb20 deletion in mature CA1 neurons did not alter their cellular identity, as evidenced by the apparently normal expression of the mature CA1 markers Man1α and Calbindin-D28k. This is in contrast to the severe defects in hippocampal development and loss of CA1 identity observed in the whole-body knockout mice of Zbtb20 (Xie et al. 2010). CA1 differentiation begins at E15.5, well after the activation of Zbtb20 in the hippocampal anlage at E12.5 (Xie et al. 2010). By P7, CA1 pyramidal cells are already well differentiated, with fully established synaptic connections (Stanfield & Cowan, 1979; Pokorny & Yamamoto, 1981a,b). In CA1-ZB20KO mice, Zbtb20 deletion in CA1 neurons occurs after the third postnatal week, beyond the critical stages of CA1 development. Thus, we reason that Zbtb20 is essential for the specification of CA1 field identity during early developmental stages, but once CA1 neurons are fully developed, Zbtb20 is not required to maintain CA1 differentiation and field identity.
In spite of their normal gross hippocampal morphology, CA1-ZB20KO mice exhibited significant deficits in hippocampus-dependent memory formation, and impairments in LTP and NMDAR-mediated EPSCs. The contextual conditioning phenotype is a specific impairment in long-term memory, but not short-term memory, which is consistent with the fact that Zbtb20 is a transcription factor and transcription is required for long-term memory formation. Hippocampal LTP of synaptic transmission is a crucial cellular mechanism underlying learning and memory (Lynch, 2004). LTP of excitatory synaptic transmission can be induced by a variety of different stimulation protocols such as high frequency stimulation, TBS, or pairing postsynaptic depolarization with low frequency stimulation (Gustafsson et al. 1987; Tang et al. 1999; Jeon et al. 2003). Potentiation induced by each of these methods requires the activation of NMDARs and postsynaptic Ca2+ during LTP induction. In the present study, LTP was induced by TBS. TBS-induced LTP is believed to involve both presynaptic and postsynaptic mechanisms (Malinow & Tsien, 1991; Malenka & Nicoll, 1999; Zakharenko et al. 2001). Our results showed no significant difference between control and CA1-ZB20KO mice in the density of dendritic spines, input–output relations of the fEPSPs and PPF. Synaptic transmission efficiency and presynaptic release probability thus appear normal in CA1-ZB20KO mice, indicating that the decrease in LTP is most likely due to postsynaptic defects. Consistent with the critical role of postsynaptic NMDARs in LTP (Sakimura et al. 1995; Liu et al. 2004), we found that NMDAR-mediated EPSCs were significantly decreased in the hippocampus of CA1-ZB20KO mice relative to control. NMDAR function can be regulated at multiple levels, including subunit composition and expression, subcellular localization, trafficking, phosphorylation status, and interactions with cofactors and scaffolding molecules (Cull-Candy & Leszkiewicz, 2004; Lau & Zukin, 2007; Fourgeaud et al. 2010). The exact mechanism by which Zbtb20 deletion affects NMDAR function in the CA1 remains to be determined.
The facilitation of hippocampus-based, long-lasting synaptic plasticity involves some key cellular events: Ca2+ influx through the NMDAR, activation of critical protein kinases (CaMKII, PKA, PKC and ERK) and transcription factor CREB, and subsequent transcription of plasticity-associated genes (Roberson et al. 1999; Waltereit & Weller, 2003). ERK act as critical points of convergence for many signalling cascades from PI-3K, PKA, PKC and CaMKII, and mediates phosphorylation of CREB (Roberson et al. 1999; Lynch, 2004). A great deal of evidence indicates that the activation of ERK and CREB plays a vital role in LTP and memory (English & Sweatt, 1997; Atkins et al. 1998; Benito & Barco, 2010). Consistent with the previous reports (Atkins et al. 1998; Stanciu et al. 2001), we detected activity-induced phosphorylation of ERK and CREB in hippocampus after contextual fear conditioning. Importantly, phosphorylation of ERK and CREB was impaired in the CA1 of CA1-ZB20KO hippocampus, which might, at least partly, explain the LTP phenotype and impaired long-term memory. It is also worth noting that Zbtb20 in CA1 neurons is required for NMDAR activity. Considering that NMDAR can act upstream of ERK, we reason that Zbtb20 may modulate NMDAR activity, which in turn regulates ERK and CREB signalling pathways. It is also likely that Zbtb20 regulates ERK activation through other molecules and pathways beyond NMDAR, which simultaneously affect NMDAR activity. As Zbtb20 is a novel transcription factor, very limited is known about its targets. So far, only two direct target genes have been identified, alpha-fetoprotein (Afp) in liver (Xie et al. 2008), and fructose-1,6,-bisphosphotase 1 (Fbp1) in pancreatic β cells (Zhang et al. 2012). We speculate that identification of the targets of Zbtb20 in CA1 neurons will help to understand the mechanisms underlying its role in hippocampus-dependent memory.
Taken together, our study shows that ablating Zbtb20 in the hippocampal CA1 leads to impaired LTP and memory in CA1-ZB20KO mice. These phenotypes appear to be due at least in part to decreased NMDAR activity, ERK and CREB phosphorylation. In combination with our previous work (Xie et al. 2010), these findings demonstrate that Zbtb20 is not only essential for the specification of CA1 field identity in the developing hippocampus, but is also critical for hippocampus-dependent long-term memory formation in mature CA1 pyramidal cells.
Acknowledgments
This work was supported by the National Natural Science Foundation of China grants 31140090 (to Z.X.), 31025013 (to W.Z.), 30970589 (to Z.X.), 31128008 (to D.H.) and 30800375 (to A.R.). W.Z. was supported in part by National Key Basic Research Program of China (2012CB524904) and Shanghai Municipal Science and Technology Commission (11XD1406500). X.Z. was supported in part by the SA-SIBS Scholarship Program. A.R. was supported in part by Shanghai Postdoctoral Scientific Program (12R21418000).
Glossary
- AFP
α-fetoprotein
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AMPAR
AMPA receptor
- CaMK
calmodulin-dependent kinases
- CREB
cAMP response element binding protein
- CS
conditioned stimulus
- DG
dentate gyrus
- EPSC
excitatory postsynaptic current
- ERK
extracellular signal-regulated kinase
- fEPSP
field excitatory postsynaptic potential
- LTP
long-term potentiation
- Man1α
mannosidase 1 α
- NMDA
N-methyl-d-aspartate
- NMDAR
NMDA receptor
- PKA
protein kinase A
- PKC
protein kinase C
- PPF
paired-pulse facilitation
- PPR
paired-pulse ratio
- TBS
theta burst stimulation
- US
unconditioned stimulus
- Zbtb20
zinc finger and BTB domain-containing protein 20
Author contributions
A.R., Z.X., X.Z. and W.Z. designed research. A.R., H.Z., Z.X., X.M. and W.J performed research; A.R., D.H., W.Y., Y.D., X.Z. and W.Z. analysed data; and A.R. and W.Z. wrote the paper.
Supplementary material
Supplemental Fig. S1
Supplemental Fig. S2
Supplemental Fig. S3
Supplemental Fig. S4
Supplemental Fig. S5
Supplemental Fig. S6
Supplemental Fig. S7
Supplemental Fig. S8
References
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Benito E, Barco A. CREB's control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33:230–240. doi: 10.1016/j.tins.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Brigman JL, Wright T, Talani G, Prasad-Mulcare S, Jinde S, Seabold GK, Mathur P, Davis MI, Bock R, Gustin RM, Colbran RJ, Alvarez VA, Nakazawa K, Delpire E, Lovinger DM, Holmes A. Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J Neurosci. 2010;30:4590–4600. doi: 10.1523/JNEUROSCI.0640-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Levenson JM, Mukhopadhyay PS, Zong L, Bradley A, Sweatt JD, Davis RL. α3-Integrins are required for hippocampal long-term potentiation and working memory. Learn Mem. 2007;14:606–615. doi: 10.1101/lm.648607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- Dai JX, Han HL, Tian M, Cao J, Xiu JB, Song NN, Huang Y, Xu TL, Ding YQ, Xu L. Enhanced contextual fear memory in central serotonin-deficient mice. Proc Natl Acad Sci U S A. 2008;105:11981–11986. doi: 10.1073/pnas.0801329105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19 103–19 106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Fourgeaud L, Davenport CM, Tyler CM, Cheng TT, Spencer MB, Boulanger LM. MHC class I modulates NMDA receptor function and AMPA receptor trafficking. Proc Natl Acad Sci U S A. 2010;107:22278–22283. doi: 10.1073/pnas.0914064107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukaya M, Kato A, Lovett C, Tonegawa S, Watanabe M. Retention of NMDA receptor NR2 subunits in the lumen of endoplasmic reticulum in targeted NR1 knockout mice. Proc Natl Acad Sci U S A. 2003;100:4855–4860. doi: 10.1073/pnas.0830996100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga K, Stoppini L, Miyamoto E, Muller D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1993;268:7863–7867. [PubMed] [Google Scholar]
- Gould TD, O’Donnell KC, Picchini AM, Dow ER, Chen G, Manji HK. Generation and behavioral characterization of β-catenin forebrain-specific conditional knock-out mice. Behav Brain Res. 2008;189:117–125. doi: 10.1016/j.bbr.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresack JE, Schafe GE, Orr PT, Frick KM. Sex differences in contextual fear conditioning are associated with differential ventral hippocampal extracellular signal-regulated kinase activation. Neuroscience. 2009;159:451–467. doi: 10.1016/j.neuroscience.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Gustafsson B, Wigstrom H, Abraham WC, Huang YY. Long-term potentiation in the hippocampus using depolarizing current pulses as the conditioning stimulus to single volley synaptic potentials. J Neurosci. 1987;7:774–780. doi: 10.1523/JNEUROSCI.07-03-00774.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon D, Yang YM, Jeong MJ, Philipson KD, Rhim H, Shin HS. Enhanced learning and memory in mice lacking Na+/Ca2+ exchanger 2. Neuron. 2003;38:965–976. doi: 10.1016/s0896-6273(03)00334-9. [DOI] [PubMed] [Google Scholar]
- Kolb B, Buhrmann K, McDonald R, Sutherland RJ. Dissociation of the medial prefrontal, posterior parietal, and posterior temporal cortex for spatial navigation and recognition memory in the rat. Cereb Cortex. 1994;4:664–680. doi: 10.1093/cercor/4.6.664. [DOI] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Lein ES, Callaway EM, Albright TD, Gage FH. Redefining the boundaries of the hippocampal CA2 subfield in the mouse using gene expression and 3-dimensional reconstruction. J Comp Neurol. 2005;485:1–10. doi: 10.1002/cne.20426. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation – a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW. Long-term potentiation: postsynaptic activation of Ca2+-dependent protein kinases with subsequent presynaptic enhancement. Prog Brain Res. 1991;89:271–289. doi: 10.1016/s0079-6123(08)61728-8. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Mitchelmore C, Kjaerulff KM, Pedersen HC, Nielsen JV, Rasmussen TE, Fisker MF, Finsen B, Pedersen KM, Jensen NA. Characterization of two novel nuclear BTB/POZ domain zinc finger isoforms. Association with differentiation of hippocampal neurons, cerebellar granule cells, and macroglia. J Biol Chem. 2002;277:7598–7609. doi: 10.1074/jbc.M110023200. [DOI] [PubMed] [Google Scholar]
- Mizuno K, Giese KP. Towards a molecular understanding of sex differences in memory formation. Trends Neurosci. 2010;33:285–291. doi: 10.1016/j.tins.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Kandel ER. Brief theta-burst stimulation induces a transcription-dependent late phase of LTP requiring cAMP in area CA1 of the mouse hippocampus. Learn Mem. 1997;4:230–243. doi: 10.1101/lm.4.2.230. [DOI] [PubMed] [Google Scholar]
- Nielsen JV, Blom JB, Noraberg J, Jensen NA. Zbtb20-induced CA1 pyramidal neuron development and area enlargement in the cerebral midline cortex of mice. Cereb Cortex. 2010;20:1904–1914. doi: 10.1093/cercor/bhp261. [DOI] [PubMed] [Google Scholar]
- Nielsen JV, Nielsen FH, Ismail R, Noraberg J, Jensen NA. Hippocampus-like corticoneurogenesis induced by two isoforms of the BTB-zinc finger gene Zbtb20 in mice. Development. 2007;134:1133–1140. doi: 10.1242/dev.000265. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Pokorny J, Yamamoto T. Postnatal ontogenesis of hippocampal CA1 area in rats. I. Development of dendritic arborisation in pyramidal neurons. Brain Res Bull. 1981a;7:113–120. doi: 10.1016/0361-9230(81)90075-7. [DOI] [PubMed] [Google Scholar]
- Pokorny J, Yamamoto T. Postnatal ontogenesis of hippocampal CA1 area in rats. II. Development of ultrastructure in stratum lacunosum and moleculare. Brain Res Bull. 1981b;7:121–130. doi: 10.1016/0361-9230(81)90076-9. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Delaney KR, Tank DW. The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapse. J Neurosci. 1994;14:523–537. doi: 10.1523/JNEUROSCI.14-02-00523.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Sweatt JD. Transient activation of cyclic AMP-dependent protein kinase during hippocampal long-term potentiation. J Biol Chem. 1996;271:30436–30441. doi: 10.1074/jbc.271.48.30436. [DOI] [PubMed] [Google Scholar]
- Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H, et al. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature. 1995;373:151–155. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- Stanciu M, Radulovic J, Spiess J. Phosphorylated cAMP response element binding protein in the mouse brain after fear conditioning: relationship to Fos production. Brain Res Mol Brain Res. 2001;94:15–24. doi: 10.1016/s0169-328x(01)00174-7. [DOI] [PubMed] [Google Scholar]
- Stanfield BB, Cowan WM. The development of the hippocampus and dentate gyrus in normal and reeler mice. J Comp Neurol. 1979;185:423–459. doi: 10.1002/cne.901850303. [DOI] [PubMed] [Google Scholar]
- Sutherland AP, Zhang H, Zhang Y, Michaud M, Xie Z, Patti ME, Grusby MJ, Zhang WJ. Zinc finger protein Zbtb20 is essential for postnatal survival and glucose homeostasis. Mol Cell Biol. 2009;29:2804–2815. doi: 10.1128/MCB.01667-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996a;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996b;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Vaccarino F, Guidotti A, Costa E. Ganglioside inhibition of glutamate-mediated protein kinase C translocation in primary cultures of cerebellar neurons. Proc Natl Acad Sci U S A. 1987;84:8707–8711. doi: 10.1073/pnas.84.23.8707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltereit R, Weller M. Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol Neurobiol. 2003;27:99–106. doi: 10.1385/MN:27:1:99. [DOI] [PubMed] [Google Scholar]
- Winder DG, Schramm NL. Plasticity and behavior: new genetic techniques to address multiple forms and functions. Physiol Behav. 2001;73:763–780. doi: 10.1016/s0031-9384(01)00514-5. [DOI] [PubMed] [Google Scholar]
- Xie Z, Ma X, Ji W, Zhou G, Lu Y, Xiang Z, Wang YX, Zhang L, Hu Y, Ding YQ, Zhang WJ. Zbtb20 is essential for the specification of CA1 field identity in the developing hippocampus. Proc Natl Acad Sci U S A. 2010;107:6510–6515. doi: 10.1073/pnas.0912315107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Zhang H, Tsai W, Zhang Y, Du Y, Zhong J, Szpirer C, Zhu M, Cao X, Barton MC, Grusby MJ, Zhang WJ. Zinc finger protein ZBTB20 is a key repressor of alpha-fetoprotein gene transcription in liver. Proc Natl Acad Sci U S A. 2008;105:10 859–10 864. doi: 10.1073/pnas.0800647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci. 2001;4:711–717. doi: 10.1038/89498. [DOI] [PubMed] [Google Scholar]
- Zhang W, Mi J, Li N, Sui L, Wan T, Zhang J, Chen T, Cao X. Identification and characterization of DPZF, a novel human BTB/POZ zinc finger protein sharing homology to BCL-6. Biochem Biophys Res Commun. 2001;282:1067–1073. doi: 10.1006/bbrc.2001.4689. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xie Z, Zhou L, Li L, Zhang H, Zhou G, Ma X, Herrera PL, Liu Z, Grusby MJ, Zhang WJ. The zinc finger protein ZBTB20 regulates transcription of fructose-1,6-bisphosphatase 1 and β cell function in mice. Gastroenterology. 2012;142:1571–1580. doi: 10.1053/j.gastro.2012.02.043. [DOI] [PubMed] [Google Scholar]
- Zorumski CF, Izumi Y. Modulation of LTP induction by NMDA receptor activation and nitric oxide release. Prog Brain Res. 1998;118:173–182. doi: 10.1016/s0079-6123(08)63207-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.