

Abstract
Carpenter syndrome is an autosomal-recessive multiple-congenital-malformation disorder characterized by multisuture craniosynostosis and polysyndactyly of the hands and feet; many other clinical features occur, and the most frequent include obesity, umbilical hernia, cryptorchidism, and congenital heart disease. Mutations of RAB23, encoding a small GTPase that regulates vesicular transport, are present in the majority of cases. Here, we describe a disorder caused by mutations in multiple epidermal-growth-factor-like-domains 8 (MEGF8), which exhibits substantial clinical overlap with Carpenter syndrome but is frequently associated with abnormal left-right patterning. We describe five affected individuals with similar dysmorphic facies, and three of them had either complete situs inversus, dextrocardia, or transposition of the great arteries; similar cardiac abnormalities were previously identified in a mouse mutant for the orthologous Megf8. The mutant alleles comprise one nonsense, three missense, and two splice-site mutations; we demonstrate in zebrafish that, in contrast to the wild-type protein, the proteins containing all three missense alterations provide only weak rescue of an early gastrulation phenotype induced by Megf8 knockdown. We conclude that mutations in MEGF8 cause a Carpenter syndrome subtype frequently associated with defective left-right patterning, probably through perturbation of signaling by hedgehog and nodal family members. We did not observe any subject with biallelic loss-of function mutations, suggesting that some residual MEGF8 function might be necessary for survival and might influence the phenotypes observed.
Main Text
The multiple-congenital-anomalies disorder Carpenter syndrome (acrocephalopolysyndactyly type II [MIM 201000]) was first described by George Carpenter in 1901 in two siblings with craniosynostosis and polysyndactyly;1 in 1909 he added a case report of a third affected sibling in the family.2 Over 70 further cases have been described in the clinical literature, and a large number of additional phenotypic features have been noted and include high birth weight and obesity in later life, congenital heart disease, umbilical hernia, cryptorchidism in males, and genu valgum.3–5 Although often arising sporadically, consanguinity is frequent, and autosomal-recessive mutations are presumed to underlie most cases.
In 2007 the majority of individuals with Carpenter syndrome were shown by Jenkins et al. to have biallelic mutations in RAB23 (MIM 606144), encoding a member of the family of small guanosine triphosphatases (GTPases) that regulate intracellular trafficking of membrane-associated proteins.6 All together, Jenkins et al.6 and two subsequent studies7,8 have reported RAB23 mutations in a total of 24 families and 31 cases. Affected individuals in 13 families have been homozygous for the identical c.434T>A (p.Leu145∗) mutation, indicating a major founder effect in subjects of north European origin. However, an additional ten distinct mutant alleles have been described, consistent with complete loss of RAB23 function. The role of RAB23 as a negative regulator of hedgehog (HH) signaling was originally defined by the analysis of mutant phenotypes affecting the orthologous murine Rab23 (open brain mutant);9 homozygous mutants are more severely affected than humans in that they die in mid gestation because of neural-tube defects.9–11 Abrogation of RAB23 activity in human retinal cells prevents formation of the primary cilium,12 suggesting a likely mechanism linking to HH signaling,13 but the biochemical details are poorly understood. One study of canine cells proposed that Rab23 facilitates ciliary recycling of Smoothened (Smo), a HH-signaling mediator,14 and genetic evidence in mice places the Rab23 mutation downstream of Smo.15 Compatible with the latter observation, transcription of Gli1, a HH target, requires a physical association between RAB23 and the homolog of Drosophila suppressor of fused (SUFU), another HH-signaling component.16
Notwithstanding these gaps in knowledge, the origins of the cardinal physical features of Carpenter syndrome are better understood. In the limbs, reduced proteolytic processing of the transcription factor GLI3 to its shorter repressor form is the likely cause of polydactyly;15,17 in the skull, a similar mechanism might operate because Gli3-loss-of-function mouse mutants exhibit fusions of the lambdoid and interfrontal sutures.18,19 The abnormalities probably reflect disturbance of HH signaling, principally mediated by sonic hedgehog (SHH) in the limbs17 and by Indian hedgehog (IHH) in the cranial sutures.20,21 Also supporting the close links with HH signaling, we found that three subjects who had a clinical diagnosis of Carpenter syndrome and in whom RAB23 mutations were not present instead harbored heterozygous deletions of GLI3 (MIM 165240).22 These individuals had several features (polysyndactyly, congenital heart disease, and learning disability) overlapping those of RAB23 mutations, but craniosynostosis was restricted to the midline sutures (metopic or sagittal), and other somatic features (high birth weight, umbilical hernia, and hypogenitalism in males) were absent.
Our starting point for this work was a simplex case (subject 1)—the offspring of first-cousin parents from Turkey—who was diagnosed with Carpenter syndrome. The study was approved by Oxfordshire Research Ethics Committee B (reference C02.143) and the Riverside Research Ethics Committee (reference 09/H0706/20); written informed consent for gathering samples for genetics research was obtained from the parents. Although subject 1 exhibited the cardinal features (including bicoronal craniosynostosis with trigonocephaly and polysyndactyly) required for diagnosing Carpenter syndrome, he also had dextrocardia, which has not been described in association with RAB23 mutations (Figures 1A–1G and Table 1). DNA sequencing of RAB23 was normal and showed that he was heterozygous for a common intragenic SNP, rs1040461, excluding an occult autozygous mutation. Analysis of GLI3 by DNA sequencing and multiplex-ligation-dependent probe amplification analysis (MLPA) was also normal. These data excluded known genetic causes of Carpenter syndrome. On the basis of the consanguinity, we hypothesized that this individual had a different, autosomal-recessive basis for his condition.
Figure 1.

Clinical Features of Individuals with MEGF8 Mutations
(A–G) Subject 1 showing facial features at the age of 4.5 years (A), chest and side view at the age of 9 years (B), a chest radiograph at the age of 9 years (C), clinical appearance of hands at the age of 4.5 years (D) and feet at the age of 9 years (E), and radiographs of hands and feet at the age of 10 years (F and G). Note the tongue-like projections at the base of the proximal phalanges (F and G, arrowheads); these are frequently noted in Carpenter syndrome.
(H and I) Facial features of subject 2 at the age of 6.4 years.
Table 1.
Clinical Features of Subjects with Mutations in MEGF8
| Subject | Gender | Birth Weight | Craniosynostosis | Craniofacial Features | Digit Length and Position | Cutaneous Syndactyly | Polydactyly | Cardiac Features | Genitalia | Other | Height | Weight | Developmental Attainment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 4,500 g (+1.9 SDs) | RC and LC trigono-cephaly |
hypertelorism, epicanthus, upslanted palpebral fissures, blue sclerae, sparse eyebrows, low-set ears with increased posterior angulation, narrow nares, long philtrum, retrognathia, malocclusion, high narrow palate, and multiple dental caries | camptodactyly (H), broad radially deviated thumbs, short digits (F) | 1–5 (F) | bilateral postaxial polydactyly type B (H), preaxial polydactyly (F) | dextrocardia | small penis, shawl scrotum, bilateral cryptorchidism | short webbed neck, umblical hernia, pectus carinatum on left, wide-spaced hypoplastic nipples, accessory nipples, flexion contracture at knees | 102 cm at 4.5 years of age (−1.0 SD) | 18.2 kg at 4.5 years of age (+0.3 SD) | DQ 78 at 4.5 years of age |
| 2 | F | 3,620 g at 38 weeks of age (+1.5 SDs) | Me | hypertelorism, epicanthus, upslanted palpebral fissures, depressed and wide nasal bridge, high palate | broad radially deviated thumbs, short digits (H and F), equinovarus right foot | 2/3 (H); 1/2, 4/5 (F) | none | transposition of great arteries, atrial septal defect, patent ductus arteriosus, tricuspid insufficiency | normal | coxa vara, mild sensorineural hearing loss, hypoplastic wide-spaced nipples | 132 cm at 6.5 years of age (+2.1 SDs) | 47 kg at 6.5 years of age (+3.5 SDs) | IQ 109 at 5.8 years of age (verbal 124, nonverbal 88) |
| 3 | M | 4,000 g (+1.3 SDs) | Me and S | prominent forehead | fifth-finger clinodactyly | 3/4 (H), 1/2 (F) | preaxial polydactyly (F) | dextrocardia associated with situs inversus totalis | bilateral cryptorchidism | wide-spaced nipples | |||
| 4 | M | 4,739 g (+2.3 SDs) | Me | highly arched eyebrows, hypertelorism, epicanthus, ectropion of lower eyelids with bilateral nasolacrimal duct obstruction, depressed nasal bridge, enlarged anteverted nares, midface retrusion, narrow palate, low-set ears with increased posterior angulation | camptodactyly (H), short digits (H), absent middle phalanges (H and F), single transverse palmar creases (H), talipes equinovarus (F) | partial 1–5 (H), complete 1–5 (F) | none | patent ductus arteriosus | bilateral undescended testes | generally loose skin with excessive folds, broad short neck, wide-spaced nipples, pectus excavatum, bilateral eventration of diaphragm with central position of liver, no structural brain anomalies, normal karyotype | 77 cm at 10 months of age (+1.4 SDs) | 11.8 kg at 10 months of age (+2.0 SDs) | hypotonia and delayed milestones; sat with support at 10 months of age |
| 5a | M | 4,170 g (+1.7 SDs) | Me | brachycephaly, low anterior hair line, underdeveloped supraorbital ridges, highly arched eyebrows, hypertelorism, epicanthus, depressed and wide nasal bridge, anteverted nares, midface retrusion, narrow palate, low-set protruding ears | short digits (H and F), absent middle phalanges of digits 1–5 (H) and 3–6 (F), varus deformity of feet | 1–5 (H), 1–6 (F) | partial preaxial polydactyly (H), preaxial polydactyly (F) | normal echocardiogram | bilateral undescended testes | saggy skin, pectus carinatum, elevated right hemidiaphragm, 46, XY,t(2;6)(p12;q23) | 122 cm at 6 years of age (+1.3 SDs) | 26 kg at 6 years of age (+1.6 SDs) | persistent hypotonia and severe delay; walked at 2.5 years of age; single words at 6 years of age; EEG suggestive of absence seizures |
The following abbreviations are used: M, male; F, female; LC, left coronal; RC, right coronal; SD, standard deviation; DQ, developmental quotient; Me, metopic; S, sagittal; H, hands; F, feet; IQ, intelligence quotient; and EEG, electroencephalogram.
Older brother of subject OX4865; no DNA sample was available for confirmation of the diagnosis.
We performed homozygosity mapping in the proband, his unaffected sister, and both parents by using the Illumina HumanCytoSNP-12 BeadArray (300K) and identified nine >5 Mb regions, composing ∼5% of the genome and containing 1,030 genes, that were uniquely homozygous in the proband and that were thus likely to harbor a recessive disease-causing mutation on the basis of the assumption of autozygosity.23,24 We then performed exome sequencing25 by using an Agilent SureSelect Human All Exon Kit (v.1; 38 Mb) to capture exonic DNA from a library prepared from 3 μg of the proband’s DNA. The enriched DNA was sequenced on an Illumina GAIIx platform with 51 bp paired-end reads. We generated 4.1 Gb of sequence that, after mapping with Bowtie software26 to the hg19 genome and removal of artifacts, resulted in an average coverage of 43×. Variants were called with the SAMtools program.27 We identified 100,695 variants in the exome sequence, which we prioritized by excluding (1) variants present either in dbSNP131 or five other exomes from subjects with unrelated clinical conditions, leaving 8,452; (2) variants not mapping to regions previously shown to be uniquely homozygous in subject 1, leaving 897; and (3) variants located in introns (except splice sites) or untranslated regions or that encoded synonymous substitutions, leaving 166 variants for consideration. Manual examination of this final list highlighted a homozygous missense change (c.4496G>A [p.Arg1499His]) within multiple epidermal-growth-factor-like-domains 8 (MEGF8 [MIM 604267]), which was confirmed by dideoxy sequencing (Figure S1, available online). We considered this variant to be a strong candidate as the causative mutation because an ethylnitrosourea (ENU)-induced homozygous missense mutation (c.577T>C [p.Cys193Arg]) in the orthologous mouse Megf8 was previously described in a mutant with very similar phenotypic features, including abnormalities of organ laterality in both the thoracic and abdominal cavities and preaxial duplication of all four limbs, to those of the proband.28,29
To gather further genetic evidence that MEGF8 mutations could be causative of human phenotypes, we undertook DNA sequencing of the coding region of MEGF8 in additional unrelated subjects. MEGF8, located in chromosomal region 19q13.2, is transcribed into two major splice forms, differing by the inclusion (Ensembl transcript ENST00000251268.5) or exclusion of a single 201 nt exon (12A, following exon 12) that does not alter the reading frame. The reference cDNA (RefSeq accession number NM_001410.2) constitutes the shorter splice form, which comprises 41 exons and encodes a 2,778 amino acid protein (Figures 2A and 2B). We selected two categories of samples to study: (1) samples from individuals who were referred because of a suspected diagnosis of Carpenter syndrome but who had a normal RAB23 DNA sequence (22 samples, including a recently published subject31 with Carpenter syndrome and complete situs inversus); and (2) samples from individuals with complex forms of congenital heart disease suggestive of heterotaxy but without craniosynostosis or limb anomalies (15 samples; see Table S1 for clinical details). Table S2 lists the primer sequences and PCR conditions that we used to amplify all 42 coding exons. We found no mutations in the isolated heterotaxy group, indicating that MEGF8 mutations are not a common cause of this phenotype. However, we did identify plausible mutations in 3 of 22 samples (subjects 2, 3, and 4) in the Carpenter syndrome group, supporting our hypothesis that the MEGF8 mutation was causative in the original subject.
Figure 2.

Structure of MEGF8, Domain Organization of the Encoded Protein, and Position of Mutations Identified in Carpenter Syndrome
(A) Cartoon shows organization of the 41 exon MEGF8. The reference sequence omits the alternatively spliced exon 12A (red box).
(B) Domain organization of encoded protein shows motifs identified by SMART analysis.30
The DNA sequence changes found are illustrated in Figure S1, and the mutations identified are summarized in Table 2. In subject 2, we found compound heterozygosity for a missense substitution (c.7099A>G [p.Ser2367Gly]) and a nonsense mutation (c.1342C>T [p.Arg448∗]); in subject 3 (offspring of first-cousin parents),31 there was homozygosity for a missense substitution (c.595G>C [p.Gly199Arg]); and in subject 4, there was heterozygosity for two different splice-site mutations (c.3349+3_3349+4dupAA and c.7069-2A>G). None of the mutations identified are listed in either dbSNP135 or in the Exome Variant Server; in addition, we excluded them from ethnically matched normal control samples (192 samples for c.7099A>G in northern European controls; 189 samples, kindly analyzed by E. Taskiran and N. Akarsu, for c.1342C>T and c.595G>C in Turkish controls; and 41 samples for c.3349+3_3349+4dupAA and c.7069-2A>G in Saudi Arabian and Jordanian controls). Analysis of the splice-site mutations present in subject 4 with the use of Splice Site Prediction by Neural Network showed that the score for the c.7069-2A>G acceptor splice site of exon 41 had changed from 0.71 to not recognized; this change indicated that it would abolish splicing. For c.3349+3_3349+4dupAA, the score for the splice donor site of exon 19 was altered from 1.0 to 0.84, indicating that the mutation would diminish the efficiency of splicing but probably not abolish it. Unfortunately, we were unable to demonstrate that the two mutations in subject 4 were present on different alleles or that they affected splicing because no sources of mRNA from the proband, or DNA from other family members, were available for analysis. Although the evidence that MEGF8 mutations are causative in this family is insufficient for constituting formal proof, we have included the clinical details of the two affected cases because they resemble subjects 1–3 in many aspects. The clinical features of subject 2 are illustrated in Figure 1B, those of subject 3 were recently reported,31 and those of subjects 4 and 5 (an older sibling of subject 4, from whom no DNA was available) are illustrated in Figure S2. Table 1 summarizes the clinical features of all cases.
Table 2.
MEGF8 Mutations Identified in Individuals Presenting with Carpenter Syndrome
| Subject | Subject ID | Consanguineous Parents | Country of Origin | Allele 1 (Paternal) | Allele 2 (Maternal) |
|---|---|---|---|---|---|
| 1 | OX4128 | first cousins | Turkey | c.4496G>A (p.Arg1499His) | c.4496G>A (p.Arg1499His) |
| 2 | OX4170 | no | Germany | c.1342C>T (p.Arg448∗) | c.7099A>G (p.Ser2367Gly) |
| 3 | OX5151 | first cousins | Turkey | c.595G>C (p.Gly199Arg) | c.595G>C (p.Gly199Arga) |
| 4 | OX4865 | first cousins | Saudi Arabia | c.3349+3_3349+4dupAAb | c.7069-2A>Gb |
Sample not available from the mother, but MLPA analysis in subject 3 showed that the mutation was present in two copies.
Samples from subject 5 (sibling) and the parents were not available, so parental origins shown here are arbitrary, and compound heterozygosity of the two mutations was not formally established.
The positions of the mutations in the gene and encoded protein are shown in Figure 2. The three missense substitutions localize to three different conserved domains of MEGF8: p.Arg1499His (in subject 1) is in a kelch domain, p.Ser2367Gly (in subject 2) in an epidermal-growth-factor (EGF)-like laminin domain, and p.Gly199Arg (in subject 3) in an EGF domain. MEGF8 is highly conserved in metazoans; for example, linear homology between the human and Drosophila orthologs can be traced over >2,400 amino acids, including 33% identities. With the use of SMART profiling,30 homologs including each of the domains in which mutations were found can be identified in many diverged animal species, including sea squirts (Ciona), lancelets (Branchiostoma), sea urchins (Strongylocentrotus), insects (Drosophila), and sea anemones (Nematostella). Homologies can also be detected for ATTRACTIN, a protein that modulates hair pigmentation (mahogany mutation) in mice.32
To assess the pathogenic significance of the missense substitutions, we examined their deep evolutionary sequence conservation and mapped the mutations onto crystallographic structures of related family members (Figure S3). Gly199 (in subject 3) locates to a highly conserved position within an EGF domain and is between two cross-linked cysteine residues; only glycine at this position can permit the required bending in the main chain (Figure S3A).33 Also of note is that the mouse Megf8 ENU mutation locates just six amino acids away at one of these cysteine residues (Cys193).29 Arg1499 (in subject 1) locates at a highly conserved position of a kelch domain within the second blade of a predicted six-bladed propeller; arginine at this position is highly conserved, and in the related human kelch-like ECH-associated protein 1 (KEAP1), it forms hydrogen bonds both with the carbonyl group of a highly conserved glycine residue and with glutamate or aspartate residues and thus stabilizes the intrablade structure (Figure S3B).34,35 Mutations at one (Arg413) of these residues in KEAP1 have been reported in lung cancer.36 Ser2367 (in subject 2) in an EGF laminin domain is less highly conserved—threonine and asparagine (but never glycine) are observed in homologous proteins. The structure of the related laminin-γ1 chain37 shows that this residue is surface exposed, making it difficult to predict the consequence of substitution at this position. From this analysis, we considered the evidence for pathogenicity to be strong for the p.Gly199Arg and p.Arg1499His substitutions (in subjects 3 and 1, respectively), both of which were present in a homozygous state, but weaker for the p.Ser2367Gly substitution (in subject 2), present in trans with a predicted null allele. To further explore the significance of the p.Ser2367Gly substitution, we heterologously expressed and purified the corresponding EGF laminin domain (residues Asp2326 to Lys2399) in E.coli38 and analyzed the domain structure by using far-UV circular-dichroism spectroscopy. However, we found that the normal protein exhibits a low degree of secondary structure, as seen for other EGF (-like) domains,39 limiting the ability to detect potential perturbations induced by the mutation (Figure S4).
As an alternative, biological test of pathogenicity, we explored the consequence of knocking down expression of the unique zebrafish megf8 ortholog by using two validated morpholinos.29 Experimental procedures were performed in accordance with UK Animals (Scientific Procedures) Act, 1986. In addition to reproducing the previously described heart-looping defects, we observed at 10 hr postfertilization (hpf) abnormal gastrulation cell movements, characterized by a short and wide notochord with discontinuous expression of no tail (ntl) and delayed closure of the blastopore (epiboly defects; Figure 3A). We observed similar defects after knockdown of rab23 (D.J., unpublished data). To investigate the consequences of the identified MEGF8 missense substitutions on protein function, we tested for genetic rescue of these defects after coinjection of MEGF8 RNA, which shares no significant sequence similarity with the morpholino. Whereas 83% of embryos injected with 6 ng of megf8 morpholino exhibited epiboly defects, only 12% of embryos coinjected with wild-type MEGF8 RNA were affected, indicating a significant rescue (p < 0.0001) and that the human gene can functionally compensate for the zebrafish ortholog (Figure 3B). We then introduced each of the nucleotide changes causing missense substitutions into the human cDNA by PCR-mediated mutagenesis and confirmed the sequence integrity of each construct. Coinjection of any of the three missense mutant versions of MEGF8 did not result in an effective rescue (Figure 3B); however, the reduced proportion of epiboly defects (60%–73%) compared to that caused by morpholino knockdown alone raises the possibility that the mutant proteins might harbor some residual function.
Figure 3.
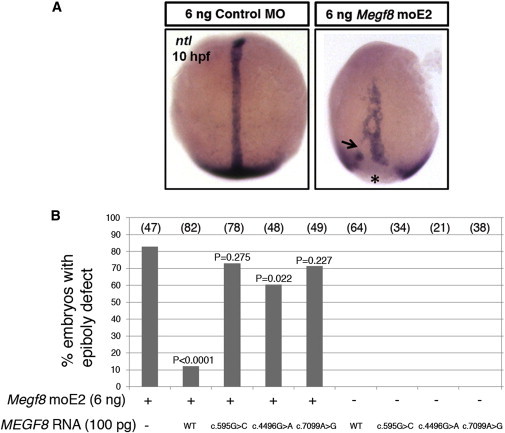
Functional Analysis of MEGF8 Missense Substitutions in Zebrafish Embryos
(A) In situ hybridization of ntl at 10 hpf for labeling the notochord and blastoderm margin, shown in dorsal view. Embryos injected with 6 ng of megf8 moE229 exhibited severe epiboly defects (right), characterized by the shortening and widening of the notochord, discontinuous staining (indicated by the arrow), and an open blastopore (asterisk). Embryos injected with control morpholino (MO) are shown on the left.
(B) Quantification of epiboly defects and genetic rescue. Shown are the proportions of embryos injected with the indicated dose of megf8 morpholino and different versions of MEGF8 RNA (wild-type [WT] or missense mutant forms [c.595G>C (p.Gly199Arg), c.4496G>A (p.Arg1499His), or c.7099A>G (p.Ser2367Gly)]) either in combination or alone. p values correspond to statistical differences compared to uninjected embryos according to a Fisher’s exact test (two-tailed). megf8 MOs and standard control MOs were purchased from Gene Tools. A full-length (41 exon) MEGF8 cDNA clone was obtained from Origene, and mutant versions encoding each missense mutation were generated with the QuikChange Site-Directed Mutagenesis Kit (Stratagene) (see Table S2 for oligonucleotides). All clones were sequence verified and subcloned into pcDNA3-DEST53 with Gateway cloning (Invitrogen). Capped RNA was synthesized with the mMESSAGE mMACHINE kit (Ambion). Wild-type zebrafish (Danio rerio) embryos were raised at 28.5°C, and microinjections (∼1 nl) were performed at the 1-cell stage. In situ hybridization was performed with RNA probes labeled with digoxigenin (Roche) and detected with BCIP/NBT (5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium) (Sigma).
Putting the genetic, structural, and functional evidence together, we conclude that biallelic mutations of MEGF8 result in a phenotype closely resembling classical Carpenter syndrome caused by RAB23 mutation. Detailed examination of the clinical features (Figure 1 and Table 1) suggests that the spectrum of limb anomalies associated with mutations in the two genes is very similar and includes brachydactyly, syndactyly, and preaxial polydactyly; radiographic examination of the limbs of subject 1 showed unusual epiphyseal spurs (Figures 1F and 1G) reminiscent of those described in classical Carpenter syndrome.3,4,6 Subjects with MEGF8 mutations show additional features resembling the RAB23 mutant phenotype, and these include a tendency toward high birth weight and childhood obesity and the occurrence of cryptorchidism or undescended testes in males. However, there are subtle differences in many other clinical features. Craniosynostosis tends to be less severe in the case of MEGF8 mutations and usually involves only the metopic suture. This is frequently accompanied by paradoxical hypertelorism, associated with a distinctive dysmorphic facies comprising a broad depressed nasal bridge, epicanthus, upslanted palpebral fissures, highly arched eyebrows, and low-set, posteriorly angulated ears (Figure 1 and Figure S2). Defects of lateralization were present in at least three subjects and ranged from transposition of the great arteries (in subject 2) to dextrocardia (in subject 1) to complete situs inversus (in subject 3). Subjects 4 and 5 were both diagnosed with diaphragmatic eventration and a central position of the liver, although the clinical information was incomplete. The range of cardiac defects closely mirrors those associated with the previously described mouse mutation28,29 but is rare with RAB23 mutations, although two cases of polysplenia (another manifestation of defective lateralization) have been recorded.7,8 However, we excluded the possibility that MEGF8 mutations commonly cause cardiac malformations related to disturbed laterality in the absence of additional syndromic features (Table S1). Wide-spaced, hypoplastic, and/or supernumerary nipples and thoracic skeletal abnormalities also seem more characteristic of MEGF8 mutations; conversely, umbilical hernia and genu valgum, two prominent features of the RAB23-mutant phenotype, appear less characteristic of MEGF8 mutation.
Despite these differences, the many overlapping features of the mutant phenotype suggest that RAB23 and MEGF8 might be closely related in their developmental role and cellular actions. An apparent paradox raised by this conclusion is that although MEGF8 (2,778 amino acids) represents a far larger target for mutation than does RAB23 (237 amino acids), mutations in RAB23 seem to outnumber those in MEGF8 by ∼8:1. One explanation might be the lack of a major founder effect for MEGF8 mutations given that no mutant allele was identified independently more than once (Table 2). Another possibility is that the Carpenter syndrome phenotype associated with MEGF8 mutations arises only in a narrow window of residual protein activity (hypomorphic mutation); no affected individual had two alleles predicting complete loss of function, compatible with the notion that homozygosity for the null state might be lethal (as occurs in other recessive disorders).40 Variation in the amount of residual activity might cause clinical variability (for example, in the degree of intellectual disability, which did not show a consistent pattern) between individuals with different combinations of mutations.
Given the deep evolutionary origins of MEGF8, which was first identified in 1998 in a screen for proteins containing EGF motifs,41 surprisingly little is currently known about its functions. The presence of multiple domains with recognized protein-binding potential and a predicted transmembrane domain near the C terminus (Figure 2B) might suggest actions in cell-surface recognition. However, previous analyses using an antibody directed to an N-terminal epitope did not observe cell-surface expression and instead described intracellular localization, both cytoplasmic and nuclear.29 In zebrafish, the megf8 ortholog was found to be ubiquitously expressed, and morpholino knockdown disturbed left-right patterning—75% of embryos were observed to have discordant situs.29 Although mouse embryos homozygous for the p.Cys193Arg substitution showed asymmetric expression of Nodal at the node (indicating that the earliest stages of symmetry breaking were intact), the fact that the normally asymmetric expression of Nodal in the lateral plate mesoderm was lost indicates a defect in transduction of the asymmetric signal from the node by an unknown mechanism.29 A clue to how Megf8 might be working biochemically is provided by recent studies on mouse Atrn (attractin), which appears to be the closest relative of Megf8 and shares several similar domains (Figure S3). Attractin was shown to affect the fate of membrane-bound receptor molecules by determining their trafficking to either the lysosome or to the cell surface;32 an analogous function for MEGF8 in intracellular trafficking could account for both the presence of the predicted transmembrane domain and the phenotypic similarity in the consequences of MEGF8 and RAB23 mutations.
Acknowledgments
We are very grateful to the families for their participation in this study. We thank John O’Sullivan and Anna Topf for help with sample recruitment; Tracy Lester for coordinating diagnostic genetic analysis; Lorna Gregory and the staff at the High-Throughput Genomics facility at the Wellcome Trust Centre for Human Genetics (Oxford) for exome sequencing; Sue Butler, John Frankland, and Tim Rostron for help with cell culture and DNA sequencing; and Ekim Taskiran and Nurten Akarsu (Hacettepe University Medical Faculty, Department of Medical Genetics, Ankara, Turkey) for analysis of Turkish control samples. This work was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre, Oxford and received funding from the Department of Health’s NIHR Biomedical Research Centre’s funding scheme (to S.J.L.K. and A.O.M.W.), the European Union (FP7-HEALTH grant 241955-2-SYSCILIA to P.L.B.), the Wellcome Trust (University College London School of Life and Medical Sciences funding to D.J., Senior Research Fellowship in Clinical Science to P.L.B., Core Award 090532/Z/09/Z to S.J.L.K., and Project Grant 093329 to A.O.M.W and S.R.F.T.), and the Newlife Foundation for Disabled Children (10-11/04 to A.O.M.W. and S.R.F.T.). The views expressed in this publication are those of the authors and are not necessarily those of the Department of Health.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Protein Data Bank, http://www.rcsb.org/pdb/home/home.do
SAMtools, http://samtools.sourceforge.net/
Splice Site Prediction by Neural Network, http://www.fruitfly.org/seq_tools/splice.html
References
- 1.Carpenter G. Two sisters showing malformations of the skull and other congenital abnormalities. Rep. Soc. Study Dis. Child. Lond. 1901;1:110–118. [Google Scholar]
- 2.Carpenter G. Acrocephaly, with other congenital malformations. Proc. R. Soc. Med. 1909;2(Sect Study Dis Child):45–53. doi: 10.1177/003591570900201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Temtamy S.A. Carpenter’s syndrome: Acrocephalopolysyndactyly. An autosomal recessive syndrome. J. Pediatr. 1966;69:111–120. doi: 10.1016/s0022-3476(66)80368-2. [DOI] [PubMed] [Google Scholar]
- 4.Cohen D.M., Green J.G., Miller J., Gorlin R.J., Reed J.A. Acrocephalopolysyndactyly type II—Carpenter syndrome: Clinical spectrum and an attempt at unification with Goodman and Summit syndromes. Am. J. Med. Genet. 1987;28:311–324. doi: 10.1002/ajmg.1320280208. [DOI] [PubMed] [Google Scholar]
- 5.Gorlin R.J., Cohen M.M., Jr., Hennekam R.C.M. Syndromes of the Head and Neck. 4th ed. Oxford University Press; New York: 2001. Carpenter syndrome (acrocephalopolysyndactyly) pp. 666–668. [Google Scholar]
- 6.Jenkins D., Seelow D., Jehee F.S., Perlyn C.A., Alonso L.G., Bueno D.F., Donnai D., Josifova D., Mathijssen I.M.J., Morton J.E.V. RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am. J. Hum. Genet. 2007;80:1162–1170. doi: 10.1086/518047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alessandri J.L., Dagoneau N., Laville J.M., Baruteau J., Hébert J.C., Cormier-Daire V. RAB23 mutation in a large family from Comoros Islands with Carpenter syndrome. Am. J. Med. Genet. A. 2010;152A:982–986. doi: 10.1002/ajmg.a.33327. [DOI] [PubMed] [Google Scholar]
- 8.Jenkins D., Baynam G., De Catte L., Elcioglu N., Gabbett M.T., Hudgins L., Hurst J.A., Jehee F.S., Oley C., Wilkie A.O.M. Carpenter syndrome: Extended RAB23 mutation spectrum and analysis of nonsense-mediated mRNA decay. Hum. Mutat. 2011;32:E2069–E2078. doi: 10.1002/humu.21457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eggenschwiler J.T., Espinoza E., Anderson K.V. Rab23 is an essential negative regulator of the mouse Sonic hedgehog signalling pathway. Nature. 2001;412:194–198. doi: 10.1038/35084089. [DOI] [PubMed] [Google Scholar]
- 10.Günther T., Struwe M., Aguzzi A., Schughart K. Open brain, a new mouse mutant with severe neural tube defects, shows altered gene expression patterns in the developing spinal cord. Development. 1994;120:3119–3130. doi: 10.1242/dev.120.11.3119. [DOI] [PubMed] [Google Scholar]
- 11.Eggenschwiler J.T., Anderson K.V. Dorsal and lateral fates in the mouse neural tube require the cell-autonomous activity of the open brain gene. Dev. Biol. 2000;227:648–660. doi: 10.1006/dbio.2000.9918. [DOI] [PubMed] [Google Scholar]
- 12.Yoshimura S., Egerer J., Fuchs E., Haas A.K., Barr F.A. Functional dissection of Rab GTPases involved in primary cilium formation. J. Cell Biol. 2007;178:363–369. doi: 10.1083/jcb.200703047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim Y.S., Chua C.E., Tang B.L. Rabs and other small GTPases in ciliary transport. Biol. Cell. 2011;103:209–221. doi: 10.1042/BC20100150. [DOI] [PubMed] [Google Scholar]
- 14.Boehlke C., Bashkurov M., Buescher A., Krick T., John A.K., Nitschke R., Walz G., Kuehn E.W. Differential role of Rab proteins in ciliary trafficking: Rab23 regulates smoothened levels. J. Cell Sci. 2010;123:1460–1467. doi: 10.1242/jcs.058883. [DOI] [PubMed] [Google Scholar]
- 15.Eggenschwiler J.T., Bulgakov O.V., Qin J., Li T., Anderson K.V. Mouse Rab23 regulates hedgehog signaling from smoothened to Gli proteins. Dev. Biol. 2006;290:1–12. doi: 10.1016/j.ydbio.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 16.Chi S., Xie G., Liu H., Chen K., Zhang X., Li C., Xie J. Rab23 negatively regulates Gli1 transcriptional factor in a Su(Fu)-dependent manner. Cell. Signal. 2012;24:1222–1228. doi: 10.1016/j.cellsig.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Litingtung Y., Dahn R.D., Li Y., Fallon J.F., Chiang C. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature. 2002;418:979–983. doi: 10.1038/nature01033. [DOI] [PubMed] [Google Scholar]
- 18.Rice D.P.C., Connor E.C., Veltmaat J.M., Lana-Elola E., Veistinen L., Tanimoto Y., Bellusci S., Rice R. Gli3Xt-J/Xt-J mice exhibit lambdoid suture craniosynostosis which results from altered osteoprogenitor proliferation and differentiation. Hum. Mol. Genet. 2010;19:3457–3467. doi: 10.1093/hmg/ddq258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Veistinen L., Takatalo M., Tanimoto Y., Kesper D.A., Vortkamp A., Rice D.P.C. Loss-of-function of Gli3 in mice causes abnormal frontal bone morphology and premature synostosis of the interfrontal suture. Front Physiol. 2012;3:121. doi: 10.3389/fphys.2012.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lenton K., James A.W., Manu A., Brugmann S.A., Birker D., Nelson E.R., Leucht P., Helms J.A., Longaker M.T. Indian hedgehog positively regulates calvarial ossification and modulates bone morphogenetic protein signaling. Genesis. 2011;49:784–796. doi: 10.1002/dvg.20768. [DOI] [PubMed] [Google Scholar]
- 21.Klopocki E., Lohan S., Brancati F., Koll R., Brehm A., Seemann P., Dathe K., Stricker S., Hecht J., Bosse K. Copy-number variations involving the IHH locus are associated with syndactyly and craniosynostosis. Am. J. Hum. Genet. 2011;88:70–75. doi: 10.1016/j.ajhg.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hurst J.A., Jenkins D., Vasudevan P.C., Kirchhoff M., Skovby F., Rieubland C., Gallati S., Rittinger O., Kroisel P.M., Johnson D. Metopic and sagittal synostosis in Greig cephalopolysyndactyly syndrome: Five cases with intragenic mutations or complete deletions of GLI3. Eur. J. Hum. Genet. 2011;19:757–762. doi: 10.1038/ejhg.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods C.G., Cox J., Springell K., Hampshire D.J., Mohamed M.D., McKibbin M., Stern R., Raymond F.L., Sandford R., Malik Sharif S. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am. J. Hum. Genet. 2006;78:889–896. doi: 10.1086/503875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hildebrandt F., Heeringa S.F., Rüschendorf F., Attanasio M., Nürnberg G., Becker C., Seelow D., Huebner N., Chernin G., Vlangos C.N. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet. 2009;5:e1000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aune C.N., Chatterjee B., Zhao X.Q., Francis R., Bracero L., Yu Q., Rosenthal J., Leatherbury L., Lo C.W. Mouse model of heterotaxy with single ventricle spectrum of cardiac anomalies. Pediatr. Res. 2008;63:9–14. doi: 10.1203/PDR.0b013e31815b6926. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Z., Alpert D., Francis R., Chatterjee B., Yu Q., Tansey T., Sabol S.L., Cui C., Bai Y., Koriabine M. Massively parallel sequencing identifies the gene Megf8 with ENU-induced mutation causing heterotaxy. Proc. Natl. Acad. Sci. USA. 2009;106:3219–3224. doi: 10.1073/pnas.0813400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Letunic I., Doerks T., Bork P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012;40(Database issue):D302–D305. doi: 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altunhan H., Annagur A., Ors R. The association of Carpenter syndrome and situs inversus totalis: First case report. Turkiye Klinikleri J. Med. Sci. 2011;31:464–467. [Google Scholar]
- 32.Overton J.D., Leibel R.L. Mahoganoid and mahogany mutations rectify the obesity of the yellow mouse by effects on endosomal traffic of MC4R protein. J. Biol. Chem. 2011;286:18914–18929. doi: 10.1074/jbc.M111.224592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu H.S., Chai J.J., Li M., Huang B.R., He C.H., Bi R.C. Crystal structure of human epidermal growth factor and its dimerization. J. Biol. Chem. 2001;276:34913–34917. doi: 10.1074/jbc.M102874200. [DOI] [PubMed] [Google Scholar]
- 34.Li X., Zhang D., Hannink M., Beamer L.J. Crystal structure of the Kelch domain of human Keap1. J. Biol. Chem. 2004;279:54750–54758. doi: 10.1074/jbc.M410073200. [DOI] [PubMed] [Google Scholar]
- 35.Beamer L.J., Li X., Bottoms C.A., Hannink M. Conserved solvent and side-chain interactions in the 1.35 Angstrom structure of the Kelch domain of Keap1. Acta Crystallogr. D Biol. Crystallogr. 2005;61:1335–1342. doi: 10.1107/S0907444905022626. [DOI] [PubMed] [Google Scholar]
- 36.Hayes J.D., McMahon M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009;34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 37.Stetefeld J., Mayer U., Timpl R., Huber R. Crystal structure of three consecutive laminin-type epidermal growth factor-like (LE) modules of laminin gamma1 chain harboring the nidogen binding site. J. Mol. Biol. 1996;257:644–657. doi: 10.1006/jmbi.1996.0191. [DOI] [PubMed] [Google Scholar]
- 38.Gileadi O., Burgess-Brown N.A., Colebrook S.M., Berridge G., Savitsky P., Smee C.E., Loppnau P., Johansson C., Salah E., Pantic N.H. High throughput production of recombinant human proteins for crystallography. Methods Mol. Biol. 2008;426:221–246. doi: 10.1007/978-1-60327-058-8_14. [DOI] [PubMed] [Google Scholar]
- 39.Periz J., Gill A.C., Knott V., Handford P.A., Tomley F.M. Calcium binding activity of the epidermal growth factor-like domains of the apicomplexan microneme protein EtMIC4. Mol. Biochem. Parasitol. 2005;143:192–199. doi: 10.1016/j.molbiopara.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 40.Mougou-Zerelli S., Thomas S., Szenker E., Audollent S., Elkhartoufi N., Babarit C., Romano S., Salomon R., Amiel J., Esculpavit C. CC2D2A mutations in Meckel and Joubert syndromes indicate a genotype-phenotype correlation. Hum. Mutat. 2009;30:1574–1582. doi: 10.1002/humu.21116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakayama M., Nakajima D., Nagase T., Nomura N., Seki N., Ohara O. Identification of high-molecular-weight proteins with multiple EGF-like motifs by motif-trap screening. Genomics. 1998;51:27–34. doi: 10.1006/geno.1998.5341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.