

Abstract
Pure hair and nail ectodermal dysplasia (PHNED) is a congenital condition characterized by hypotrichosis and nail dystrophy. Autosomal-recessive PHNED has previously been mapped to chromosomal region 12q12-q14.1, which contains the type II hair keratin and HOXC clusters. Hoxc13-null mice are known to develop hair and nail defects very similar to those seen in human PHNED. We performed whole-exome sequencing in a consanguineous Chinese family affected by PHNED and identified a homozygous nonsense mutation (c.390C>A [p.Tyr130∗]) in HOXC13 in all affected individuals. In an additional affected female from a consanguineous Afghan family, we found a 27.6 kb homozygous microdeletion involving the first exon of HOXC13. We examined HOXC13 expression in scalp specimen obtained from the index individual of the Chinese family and detected dramatically reduced mRNA levels in skin tissue and nearly absent protein staining in hair follicles, suggesting a mechanism of nonsense-mediated mRNA decay. We also observed markedly decreased expression of four HOXC13 target genes in the specimen. Taken together, our results demonstrate that loss-of-function mutations in HOXC13 cause autosomal-recessive PHNED and further highlight the importance of HOXC13 in hair and nail development.
Main Text
Ectodermal dysplasias (EDs) are a large and complex group of congenital disorders characterized by abnormal development in two or more ectodermal structures (hair, nails, teeth, and sweat glands).1 It is estimated that EDs include 170–200 distinct conditions, although molecular bases were identified in only about 30% of them.1,2 Pure hair and nail ectodermal dysplasia (PHNED [MIM 602032]) is a rare congenital condition characterized by hypotrichosis and nail dystrophy without nonectodermal or other ectodermal manifestations.3 Hypotrichosis usually occurs after birth with varying degrees of severity, ranging from mild hair loss to complete atrichia, including the loss of scalp hair, beard, eyebrows, eyelashes, axillary hair, and pubic hair. Nail dystrophy affects all twenty digits by causing short fragile nails or spoon nails (koilonychias). At least five clinical forms of PHNED with unique manifestations have been described3–8 with either autosomal-recessive or autosomal-dominant patterns of inheritance. In 2006, Naeem and colleagues performed linkage analysis in a consanguineous Pakistani family affected by PHNED and mapped the locus to chromosomal region 12q12-q14.1, which contains the type II hair keratin and HOXC gene clusters. They identified a homozygous missense mutation, c.233G>A (p.Arg78His), in the affected individuals in KRT85 (MIM 602767; RefSeq accession number NM_002283.3),4 which encodes one of the important structural components in the matrix of hair follicles and nails.9,10 However, follow-up studies in additional consanguineous Pakistani families affected by PHNED revealed KRT85 mutations in some but not all cases.5,11,12 Furthermore, a second PHNED locus was mapped to chromosomal region 17p12-q21.2 in a consanguineous Pakistani family.13 Taken together, these results suggest the existence of genetic heterogeneity for autosomal-recessive PHNED. Here, we report loss-of-function mutations in HOXC13 (MIM 142976) in two consanguineous families with different ethnic origins of PHNED.
We first ascertained a five-generation consanguineous Chinese Hui family affected by PHNED. All three affected individuals in the family were born to healthy first-cousin parents (Figure 1A), which is consistent with an autosomal-recessive mode of inheritance. The three affected individuals (V2, V3, and IV10 in Figure 1A) showed congenital atrichia and severe nail dystrophy. All vellus, lanugo, and terminal hairs were absent on the scalp and body, and axillary and pubic hairs never developed during puberty (Figures 1B and 1C). Soft and small keratinized structures developed later on all digits and presented as micronychia (Figure 1D). The affected individuals described that they never need their nails trimmed. Affected individuals V2 and IV10 underwent surgical management for unilateral cryptorchidism and inguinal hernia, and urologists suspected a diagnosis of persistent Müllerian duct syndrome (MIM 261550). No genital anomalies were observed in the affected female individual V3. No abnormalities of the nervous system, skeleton, sweat glands, sebaceous glands, eyes, or teeth were observed. Hair and nails appeared normal for obligate carriers and other family members. Histopathological examination of scalp skin of the index individual showed an apparently reduced number of hair follicles with disorganized hair shafts lacking the normal layered structure (Figure 1E).
Figure 1.
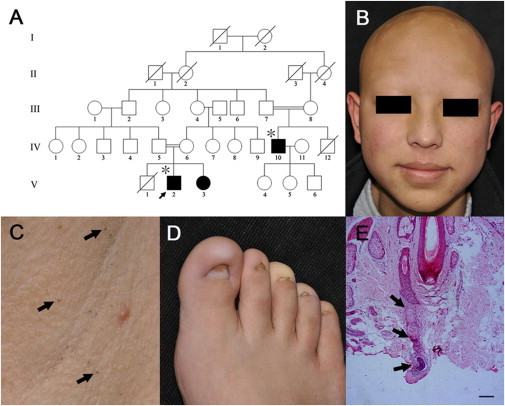
Clinical Findings of PHNED
(A) Pedigree of a Chinese Hui family affected by PHNED. The arrow indicates the index individual. Solid symbols indicate individuals with PHNED, and asterisks above pedigree symbols refer to individuals with persistent Müllerian duct syndrome.
(B) Congenial alopecia involving the scalp, beard, eyebrows, and eyelashes in individual V2.
(C) Close view of axillary area of individual V2 shows that hair shafts break off at the skin surface. Arrows show remnants of unprotruding hair.
(D) Severe nail dystrophy affecting all toenails in the index individual (V2).
(E) Scalp skin-biopsy section demonstrating normal epidermis and dermis. The arrow shows disorganization in a hair shaft without normal layered structure in the longitudinal section of a hair follicle. Hematoxylin-eosin staining was used. The scale bar represents 30 μm.
We collected blood samples from 18 individuals (3 affected, 12 unaffected, and 3 spouses) of the Chinese family after obtaining informed consent and approval of the Clinical Research Ethics Committee of the Peking University First Hospital. Genomic DNA was extracted by standard methods. Skin biopsies were performed on the scalp of the index individual (V2). After the exclusion of pathogenic mutations in KRT85 in the index individual by conventional Sanger sequencing, we performed whole-exome sequencing (WES) in the index individual (V2), his affected sister (V3), and their parents (IV5 and IV6). For WES, 10 μg of genomic DNA was used per individual. Exomes were captured with the SureSelect Human All Exon Kit (Agilent) and were then sequenced on an Illumina Hiseq2000 according to the manufacturer’s protocol. Variants were filtered against dbSNP134, the 1000 Genomes Project, HapMap8, and the YanHuang Project and Beijing Genomics Institute inner databases, as described in our previous study.14 Under the assumption of an autosomal-recessive pattern of inheritance, only variants that were homozygous in the two affected siblings and heterozygous in their parents were selected as candidates for further prediction by SIFT software.15 Among those variants predicted to be damaging, only three—c.390C>A (p.Tyr130∗) in HOXC13 (RefSeq NM_017410.2), c.539C>G (p.Pro180Arg) in DNAJC14 (MIM 606092; RefSeq NM_032364.5), and c.1410T>A (p.Asn470Lys) in GLI1 (MIM 165220; RefSeq NM_005269.2)—occurred in genes showing expression in skin according to the UniGene expression database (Table S1, available online). By Sanger sequencing, we verified all three of these variants and confirmed their cosegregation with the PHNED phenotype in the pedigree (Figure 2A) (primers for HOXC13 are listed in Table S2). By sequencing 200 ethnically matched normal controls, we detected the variant in DNAJC14 in two individuals but did not detect the variants in GLI1 or HOXC13. GLI1 is a target of sonic hedgehog, which functions as a key developmental regulator of hair follicles. However, Gli1-null mice did not show any observable hair or nail abnomalies,16 and Gli1-overexpressing grafts in a mouse model developed skin tumors without hair loss.17,18 The Asn470 residue in GLI1 is conserved in some but not all mammalian species (Figure S1). It is unlikely that mutations in GLI1 alone are responsible for the PHNED phenotype. Given that Hoxc13-null mice develop hair and nail defects very similar to those seen in human PHNED,19 the nonsense variant in HOXC13 was likely to be the pathogenic mutation underlying the PHNED phenotype in the Chinese family. Notably, in the three individuals (V2, V3, and IV10) affected by PHNED, our WES and subsequent Sanger sequencing also identified a homozygous nonsense mutation, c.238C>T (p.Arg80∗), in AMHR2 (MIM 600956; RefSeq NM_001164691.1), a gene which is localized 507 kb centromeric to HOXC13 and which has been associated with the male-limited persistent Müllerian duct syndrome (Figure S2).20
Figure 2.
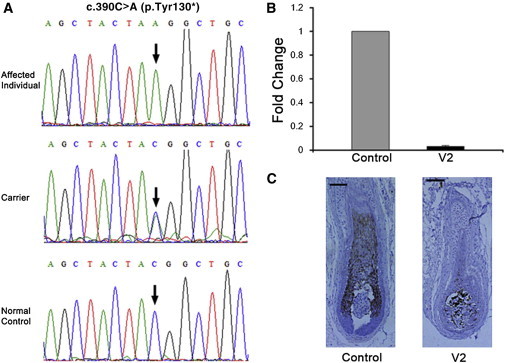
Homozygous Nonsense Mutation in HOXC13 in the Chinese Family
(A) Sequence chromatograms show the homozygous c.390C>A mutation (resulting in nonsense change p.Tyr130∗) in an affected individual (top panel). The middle panel shows heterozygous alleles in a carrier, and the bottom panel shows alleles in a normal control.
(B) qRT-PCR results of HOXC13 mRNA in an affected individual (V2) and a normal control. Only trace expression of HOXC13 was detected in the skin tissue of V2 compared to the normal control. The error bar represents the standard error of the mean (SEM).
(C) Immunohistochemistry shows HOXC13 deficiency in a scalp hair follicle of an affected individual (right panel) and expression in the cortex region of a control follicle (left panel). The scale bars represent 30 μm.
To determine whether HOXC13 is also mutated in an additional individual affected by autosomal-recessive PHNED, we amplified and sequenced the coding regions of the two exons, as well as their intron junctions in a 21-year-old Afghan female. This affected individual was born to consanguineous first-cousin parents and presented with severe congenital hypotrichosis and nail dystrophy (Figure S3). She had her nails trimmed once a month and had never cut her hair. The female’s parents and siblings (two sisters and three brothers), as well as other relatives, were described to have no hair or nail changes. DNA was not available from the relatives. After we excluded KRT85 mutations in the affected female, our PCR amplification produced a specific fragment of expected size for HOXC13 exon 2, and subsequent Sanger sequencing revealed no mutation. On the contrary, we were unable to generate amplicons of the expected size for exon 1 of HOXC13 by using genomic DNA from the female, suggesting the presence of a homozygous partial deletion of HOXC13. To confirm and delineate the deletion, we designed 18 PCR assays (primers are listed in Table S3) to cover a 39.4 kb genomic region containing HOXC13 (Figure 3). Our genomic PCR assays identified a 27.6 kb homozygous microdeletion (chr12: 54,308,194–54,335,815) involving all of exon 1 and part of the HOXC13 intron (Figure 3). We also sequenced the coding regions of GLI1 in the female and did not find any mutation (primers are listed in Table S4).
Figure 3.
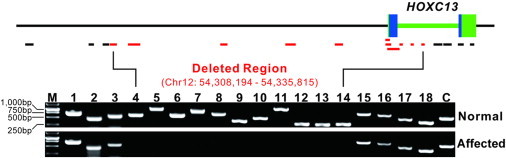
DNA Electrophoresis Demonstrating a Homozygous Partial Deletion of HOXC13 in an Afghan Female with PHNED
A ∼40 kb genomic region containing HOXC13 is shown. The solid bars indicate the positions and sizes of the 18 amplicons, and the deleted ones are highlighted in red. The deleted region is given. The following abbreviation is used: C, control amplicon.
The nonsense HOXC13 mutation identified in the Chinese family is predicted to encode a truncated protein without the C-terminal homeodomain and to thus lead to a complete loss of function. To investigate in vivo the functional consequences of the mutation, we examined the mRNA expression of HOXC13 in the scalp skin tissue from the index individual by using quantitative real-time PCR (qRT-PCR). Total RNA was isolated from the tissue with TRIZOL reagent (Invitrogen). After removal of DNA by RQ1 RNase-free DNase (Promega), single-stranded cDNA templates were synthesized with Superscript II (Invitrogen). qRT-PCR was then performed with specific primers (listed in Table S5) and Faststart universal SYBR green master (Roche) on a 7500 Real Time PCR System (Applied Biosystems). Three replicates were conducted per sample. HOXC13 mRNA expression was normalized to that of the 18S rRNA, and the relative expression level was determined on the basis of the comparative cycle-threshold (2-ΔΔCt) method with the use of cDNA templates generated from scalp skin tissues of healthy controls as the calibrator. The qRT-PCR experiments were repeated three times. As shown in Figure 2B, we could only detect trace expression of HOXC13 mRNA in the skin tissue of the index individual, suggesting a mechanism of nonsense-mediated mRNA decay. We also applied qRT-PCR to quantify the relative mRNA expression of FOXN1 (MIM 600838), DSG4 (MIM 607892), KRT85, and KRT35 (MIM 602764), all well-known examples of HOXC13 target genes, which are essential for hair differentiation and nail development. As expected, all these genes showed dramatically decreased or absent mRNA expression in the skin tissue of the index individual (Figure 4A).
Figure 4.
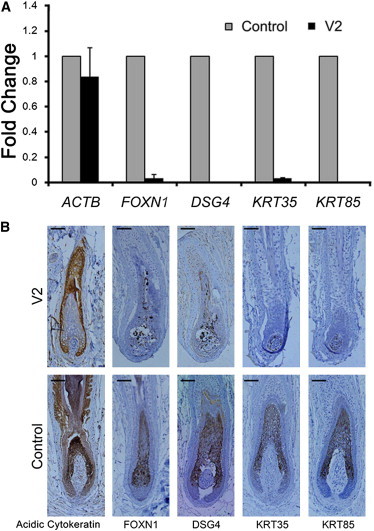
Expression Analysis of the HOXC13 Target Genes
(A) Expression of ACTB (β-actin), FOXN1, DSG4, KRT35, and KRT85 is detected by qRT-PCR and normalized to 18S rRNA. Error bars represent the SEM.
(B) Localization of acidic cytokeratin, FOXN1, DSG4, KRT35, and KRT85 in hair follicles from the index individual (V2) and normal control is investigated by immunohistochemistry. Scale bars represent 30 μm.
To validate these findings in mRNA-expression analysis, we further examined formalin-fixed paraffin sections of scalp skin tissue of the index individual by using monoclonal or polyclonal antibodies, including a mouse monoclonal antibody against HOXC13 (ab55251, Abcam), a rabbit polyclonal antibody against FOXN1 (ab113235, Abcam), a goat polyclonal antibody against DSG4 (sc-28067, Santa Cruz), a guinea pig polyclonal antibody against KRT85 (LS-C86153, LifeSpan Bioscience), a rabbit polyclonal antibody against KRT35 (ab103346, Abcam), and a mouse monoclonal antibody against acidic cytokeratin (AE1) (ab3117, Abcam). Consistent with the results of the qRT-PCR analysis, our immunohistochemical staining of skin-tissue sections showed weak or negative signals for the HOXC13 transcription factor and the four examined target proteins in the hair follicles of the index individual (Figures 2C and 4B). These contrast with the strong positive signals in the precortex and cortex region of hair follicles of a normal control (Figures 2C and 4B).
We have identified different homozygous loss-of-function mutations in HOXC13 for the rare skin disorder PHNED in two consanguineous families of different ethnic backgrounds. Our in vivo functional analysis in scalp skin tissue clearly shows that the nonsense HOXC13 mutation identified in the Chinese family results in a nearly complete loss of expression of the studied hair and nail keratins, the type I keratin KRT35 and the type II keratin KRT85. Furthermore, complete segregation of the homozygous nonsense mutation with the hair and nail phenotypes in the Chinese family provides additional supportive evidence toward its causal role. Taken together, our results suggest that loss-of-function mutations in HOXC13 cause a subset of autosomal-recessive PHNED.
HOXC13 belongs to the homeobox (HOX) gene family, members of which encode a set of evolutionarily conserved transcription factors that serve as regulators in initiating developmental programs by activating transcription of downstream genes involved in cell proliferation and differentiation.21 In humans, as in mice, there are 39 HOX genes arranged in four clusters (HOXA–HOXD) on separate chromosomes. By now, four HOX genes, HOXA1 (MIM 142955), HOXB1 (MIM 142968), HOXA13 (MIM 142959), and HOXD13 (MIM 142989) have been demonstrated to be associated with human developmental disorders.22–26 Our present studies expand the phenotypic spectrum of mutations in human HOX genes.
The mammalian organization of the HOX genes appears to follow a spatiotemporal colinearity during embryonic development of the anterior-posterior axis.27 HOXC13 is an exception from the principle of colinearity. In mice, Hoxc13 is expressed in both vibrissae and all body hair follicles at later developmental stage.19 In postnatal hair follicles, Hoxc13 is expressed in the bulb region, extending into the medulla, cortex, and cuticles, as well as the cuticles of the inner root sheath and the portion of the outer root sheath surrounding the upper portion of the dermal papilla.19,28,29 Both Hoxc13-null mice and transgenic Hoxc13-overexpressing mice presented with alopecia, indicating that Hoxc13 is a vital regulator in hair morphogenesis and postnatal cycling.19,28 The Hoxc13-null mice were characterized by the lack of external hair, abnormal nail development, and an increased number of caudal vertebrae with lateral processes and more rounded filiform papillae.19,30 Consistent with those of the Hoxc13-null mice, hair follicles of our affected individuals are identical to follicles in the anagen phase in that the disorganized keratinocytes of the hair shaft lack a normal layered structure. As a result of insufficient structural rigidity, these abnormal hair shafts are unable to penetrate the epidermis, leading to a completely nude phenotype.19,30,31 The appearances of nail dystrophy in affected individuals and Hoxc13-deficient mice were similar too. Potter et al. revealed that Hoxc13-null mice had nail-development defects, including a reduced nail matrix, extended stratum granulosum reaching into the matrix, and a cornified squamous epithelium structure instead of a normal translucent nail plate.31 Interestingly, unlike Hoxc13-null mice, affected individuals with PHNED showed normal growth and no obvious defect in vertebral development.
HOXC13 can regulate, directly or via the FOXN1 transcription factor, the expression of a number of hair and nail keratins, keratin-associated proteins, and desmosomal cadherins,27,28,31–34 thereby contributing to hair differentiation and nail development. In accordance with the previous reports, our present in vivo analysis in skin tissue from an individual with PHNED shows that complete functional loss of HOXC13 could lead to a markedly reduced or absent expression of the target genes FOXN1, KRT35, and KRT85. Using a luciferase reporter-gene assay, Bazzi et al. demonstrated that HOXC13 could function as a repressor of DSG4 transcription.35 On the contrary, we did not observe increased expression of DSG4 in scalp skin tissue of the HOXC13-null individual. This contradiction might be explained by more complicated regulatory networks in vivo. Clearly, further studies are needed for clarifying how HOXC13 regulates DSG4 transcription.
Two genes in the HOXC13-FOXN1-KRT85 regulatory cascade have previously been associated with hypotrichosis and nail dystrophy in humans. Mutations in FOXN1 cause T cell immunodeficiency, congenital alopecia, and nail dystrophy (MIM 601705).36 In three Pakistani families affected by PHNED, homozygous mutations in KRT85 were detected. Our present studies demonstrated mutations in HOXC13 as a cause of PHNED in humans. HOXC13 and KRT85 are located on the same region on chromosome 12. In an additional two Pakistani families affected by PHNED, linked to chromosomal regions 12p11.1-q21.111 and 12p11.1-q14.3,12 no mutation in KRT85 was found. Conceivably, HOXC13 might be mutated in these PHNED-affected families. The discovery of the molecular basis of PHNED might contribute to a better understanding of the complex regulatory pathway of HOXC13 and to finding new clues for the treatment of hair and nail disorders.
Acknowledgments
We are grateful to the family members for participation in this study. This work was mainly supported by the National Natural Science Foundation of China (81071289 to Y.Y.) and the Program for Changjiang Scholars and Innovative Research Team in University (LRT1006 to X. Z.). R.C.B. is a recipient of a Heisenberg Professorship of the German Research foundation. K.A.G. would like to thank Professor Hans Wolff for his clinical support.
Contributor Information
Xue Zhang, Email: xuezhang@pumc.edu.cn.
Yong Yang, Email: dryongyang@bjmu.edu.cn.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
NCBI RefSeq, http://www.ncbi.nlm.nih.gov/RefSeq/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu/
UniGene, http://www.ncbi.nlm.nih.gov/unigene
References
- 1.Visinoni A.F., Lisboa-Costa T., Pagnan N.A., Chautard-Freire-Maia E.A. Ectodermal dysplasias: Clinical and molecular review. Am. J. Med. Genet. A. 2009;149A:1980–2002. doi: 10.1002/ajmg.a.32864. [DOI] [PubMed] [Google Scholar]
- 2.Irvine A.D. Towards a unified classification of the ectodermal dysplasias: Opportunities outweigh challenges. Am. J. Med. Genet. A. 2009;149A:1970–1972. doi: 10.1002/ajmg.a.32852. [DOI] [PubMed] [Google Scholar]
- 3.Calzavara-Pinton P., Carlino A., Benetti A., De Panfilis G. Pili torti and onychodysplasia. Report of a previously undescribed hidrotic ectodermal dysplasia. Dermatologica. 1991;182:184–187. [PubMed] [Google Scholar]
- 4.Naeem M., Wajid M., Lee K., Leal S.M., Ahmad W. A mutation in the hair matrix and cuticle keratin KRTHB5 gene causes ectodermal dysplasia of hair and nail type. J. Med. Genet. 2006;43:274–279. doi: 10.1136/jmg.2005.033381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimomura Y., Wajid M., Kurban M., Sato N., Christiano A.M. Mutations in the keratin 85 (KRT85/hHb5) gene underlie pure hair and nail ectodermal dysplasia. J. Invest. Dermatol. 2010;130:892–895. doi: 10.1038/jid.2009.341. [DOI] [PubMed] [Google Scholar]
- 6.Pinheiro M., Freire-Maia N. Hair-nail dysplasia—A new pure autosomal dominant ectodermal dysplasia. Clin. Genet. 1992;41:296–298. doi: 10.1111/j.1399-0004.1992.tb03401.x. [DOI] [PubMed] [Google Scholar]
- 7.Barbareschi M., Cambiaghi S., Crupi A.C., Tadini G. Family with “pure” hair-nail ectodermal dysplasia. Am. J. Med. Genet. 1997;72:91–93. doi: 10.1002/(sici)1096-8628(19971003)72:1<91::aid-ajmg19>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 8.Harrison S., Sinclair R. Hypotrichosis and nail dysplasia: A novel hidrotic ectodermal dysplasia. Australas. J. Dermatol. 2004;45:103–105. doi: 10.1111/j.1440-0960.2004.00055.x. [DOI] [PubMed] [Google Scholar]
- 9.Langbein L., Rogers M.A., Winter H., Praetzel S., Schweizer J. The catalog of human hair keratins. II. Expression of the six type II members in the hair follicle and the combined catalog of human type I and II keratins. J. Biol. Chem. 2001;276:35123–35132. doi: 10.1074/jbc.M103305200. [DOI] [PubMed] [Google Scholar]
- 10.Perrin C., Langbein L., Schweizer J. Expression of hair keratins in the adult nail unit: An immunohistochemical analysis of the onychogenesis in the proximal nail fold, matrix and nail bed. Br. J. Dermatol. 2004;151:362–371. doi: 10.1111/j.1365-2133.2004.06108.x. [DOI] [PubMed] [Google Scholar]
- 11.Naeem M., John P., Ali G., Ahmad W. Pure hair-nail ectodermal dysplasia maps to chromosome 12p11.1-q21.1 in a consanguineous Pakistani family. Clin. Exp. Dermatol. 2007;32:502–505. doi: 10.1111/j.1365-2230.2007.02413.x. [DOI] [PubMed] [Google Scholar]
- 12.Rasool M., Nawaz S., Azhar A., Wajid M., Westermark P., Baig S.M., Klar J., Dahl N. Autosomal recessive pure hair and nail ectodermal dysplasia linked to chromosome 12p11.1-q14.3 without KRTHB5 gene mutation. Eur. J. Dermatol. 2010;20:443–446. doi: 10.1684/ejd.2010.0962. [DOI] [PubMed] [Google Scholar]
- 13.Naeem M., Jelani M., Lee K., Ali G., Chishti M.S., Wali A., Gul A., John P., Hassan M.J., Leal S.M., Ahmad W. Ectodermal dysplasia of hair and nail type: Mapping of a novel locus to chromosome 17p12-q21.2. Br. J. Dermatol. 2006;155:1184–1190. doi: 10.1111/j.1365-2133.2006.07509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin Z., Chen Q., Lee M., Cao X., Zhang J., Ma D., Chen L., Hu X., Wang H., Wang X. Exome sequencing reveals mutations in TRPV3 as a cause of Olmsted syndrome. Am. J. Hum. Genet. 2012;90:558–564. doi: 10.1016/j.ajhg.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 16.Park H.L., Bai C., Platt K.A., Matise M.P., Beeghly A., Hui C.C., Nakashima M., Joyner A.L. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000;127:1593–1605. doi: 10.1242/dev.127.8.1593. [DOI] [PubMed] [Google Scholar]
- 17.Nilsson M., Undèn A.B., Krause D., Malmqwist U., Raza K., Zaphiropoulos P.G., Toftgård R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc. Natl. Acad. Sci. USA. 2000;97:3438–3443. doi: 10.1073/pnas.050467397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oro A.E., Higgins K. Hair cycle regulation of Hedgehog signal reception. Dev. Biol. 2003;255:238–248. doi: 10.1016/s0012-1606(02)00042-8. [DOI] [PubMed] [Google Scholar]
- 19.Godwin A.R., Capecchi M.R. Hoxc13 mutant mice lack external hair. Genes Dev. 1998;12:11–20. doi: 10.1101/gad.12.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imbeaud S., Belville C., Messika-Zeitoun L., Rey R., di Clemente N., Josso N., Picard J.Y. A 27 base-pair deletion of the anti-müllerian type II receptor gene is the most common cause of the persistent müllerian duct syndrome. Hum. Mol. Genet. 1996;5:1269–1277. doi: 10.1093/hmg/5.9.1269. [DOI] [PubMed] [Google Scholar]
- 21.Pick L., Heffer A. Hox gene evolution: Multiple mechanisms contributing to evolutionary novelties. Ann. N Y Acad. Sci. 2012;1256:15–32. doi: 10.1111/j.1749-6632.2011.06385.x. [DOI] [PubMed] [Google Scholar]
- 22.Tischfield M.A., Bosley T.M., Salih M.A., Alorainy I.A., Sener E.C., Nester M.J., Oystreck D.T., Chan W.M., Andrews C., Erickson R.P., Engle E.C. Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat. Genet. 2005;37:1035–1037. doi: 10.1038/ng1636. [DOI] [PubMed] [Google Scholar]
- 23.Webb B.D., Shaaban S., Gaspar H., Cunha L.F., Schubert C.R., Hao K., Robson C.D., Chan W.M., Andrews C., MacKinnon S. HOXB1 founder mutation in humans recapitulates the phenotype of Hoxb1-/- mice. Am. J. Hum. Genet. 2012;91:171–179. doi: 10.1016/j.ajhg.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mortlock D.P., Innis J.W. Mutation of HOXA13 in hand-foot-genital syndrome. Nat. Genet. 1997;15:179–180. doi: 10.1038/ng0297-179. [DOI] [PubMed] [Google Scholar]
- 25.Muragaki Y., Mundlos S., Upton J., Olsen B.R. Altered growth and branching patterns in synpolydactyly caused by mutations in HOXD13. Science. 1996;272:548–551. doi: 10.1126/science.272.5261.548. [DOI] [PubMed] [Google Scholar]
- 26.Zhao X., Sun M., Zhao J., Leyva J.A., Zhu H., Yang W., Zeng X., Ao Y., Liu Q., Liu G. Mutations in HOXD13 underlie syndactyly type V and a novel brachydactyly-syndactyly syndrome. Am. J. Hum. Genet. 2007;80:361–371. doi: 10.1086/511387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Awgulewitsch A. Hox in hair growth and development. Naturwissenschaften. 2003;90:193–211. doi: 10.1007/s00114-003-0417-4. [DOI] [PubMed] [Google Scholar]
- 28.Tkatchenko A.V., Visconti R.P., Shang L., Papenbrock T., Pruett N.D., Ito T., Ogawa M., Awgulewitsch A. Overexpression of Hoxc13 in differentiating keratinocytes results in downregulation of a novel hair keratin gene cluster and alopecia. Development. 2001;128:1547–1558. doi: 10.1242/dev.128.9.1547. [DOI] [PubMed] [Google Scholar]
- 29.Jave-Suarez L.F., Winter H., Langbein L., Rogers M.A., Schweizer J. HOXC13 is involved in the regulation of human hair keratin gene expression. J. Biol. Chem. 2002;277:3718–3726. doi: 10.1074/jbc.M101616200. [DOI] [PubMed] [Google Scholar]
- 30.Godwin A.R., Capecchi M.R. Hair defects in Hoxc13 mutant mice. J. Investig. Dermatol. Symp. Proc. 1999;4:244–247. doi: 10.1038/sj.jidsp.5640221. [DOI] [PubMed] [Google Scholar]
- 31.Potter C.S., Pruett N.D., Kern M.J., Baybo M.A., Godwin A.R., Potter K.A., Peterson R.L., Sundberg J.P., Awgulewitsch A. The nude mutant gene Foxn1 is a HOXC13 regulatory target during hair follicle and nail differentiation. J. Invest. Dermatol. 2011;131:828–837. doi: 10.1038/jid.2010.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Potter C.S., Peterson R.L., Barth J.L., Pruett N.D., Jacobs D.F., Kern M.J., Argraves W.S., Sundberg J.P., Awgulewitsch A. Evidence that the satin hair mutant gene Foxq1 is among multiple and functionally diverse regulatory targets for Hoxc13 during hair follicle differentiation. J. Biol. Chem. 2006;281:29245–29255. doi: 10.1074/jbc.M603646200. [DOI] [PubMed] [Google Scholar]
- 33.Peterson R.L., Tkatchenko T.V., Pruett N.D., Potter C.S., Jacobs D.F., Awgulewitsch A. Epididymal cysteine-rich secretory protein 1 encoding gene is expressed in murine hair follicles and downregulated in mice overexpressing Hoxc13. J. Investig. Dermatol. Symp. Proc. 2005;10:238–242. doi: 10.1111/j.1087-0024.2005.10114.x. [DOI] [PubMed] [Google Scholar]
- 34.Pruett N.D., Tkatchenko T.V., Jave-Suarez L., Jacobs D.F., Potter C.S., Tkatchenko A.V., Schweizer J., Awgulewitsch A. Krtap16, characterization of a new hair keratin-associated protein (KAP) gene complex on mouse chromosome 16 and evidence for regulation by Hoxc13. J. Biol. Chem. 2004;279:51524–51533. doi: 10.1074/jbc.M404331200. [DOI] [PubMed] [Google Scholar]
- 35.Bazzi H., Demehri S., Potter C.S., Barber A.G., Awgulewitsch A., Kopan R., Christiano A.M. Desmoglein 4 is regulated by transcription factors implicated in hair shaft differentiation. Differentiation. 2009;78:292–300. doi: 10.1016/j.diff.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frank J., Pignata C., Panteleyev A.A., Prowse D.M., Baden H., Weiner L., Gaetaniello L., Ahmad W., Pozzi N., Cserhalmi-Friedman P.B. Exposing the human nude phenotype. Nature. 1999;398:473–474. doi: 10.1038/18997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.