

Abstract
A subset of nuclear-encoded RNAs has to be imported into mitochondria for the proper replication and transcription of the mitochondrial genome and, hence, for proper mitochondrial function. Polynucleotide phosphorylase (PNPase or PNPT1) is one of the very few components known to be involved in this poorly characterized process in mammals. At the organismal level, however, the effect of PNPase dysfunction and impaired mitochondrial RNA import are unknown. By positional cloning, we identified a homozygous PNPT1 missense mutation (c.1424A>G predicting the protein substitution p.Glu475Gly) of a highly conserved PNPase residue within the second RNase-PH domain in a family affected by autosomal-recessive nonsyndromic hearing impairment. In vitro analyses in bacteria, yeast, and mammalian cells showed that the identified mutation results in a hypofunctional protein leading to disturbed PNPase trimerization and impaired mitochondrial RNA import. Immunohistochemistry revealed strong PNPase staining in the murine cochlea, including the sensory hair cells and the auditory ganglion neurons. In summary, we show that a component of the mitochondrial RNA-import machinery is specifically required for auditory function.
Main Text
The integrity and function of mammalian mitochondria is critically dependent on the import of selected macromolecules that are encoded by the nuclear genome. The import of nuclear-encoded proteins into mitochondria is a well-studied phenomenon, and most of the proteins involved in this complex process of protein translocation and processing have been identified.1 Consistent with the importance of this process for mitochondrial function and the high energy demand of specific tissues, it has been shown that germline mutations in genes encoding members of this import machinery, as well as those encoding transported substrates, can cause a variety of human diseases characterized by the occurrence of diverse neurologic, cardiologic, and metabolic symptoms.2 In sharp contrast, little is known about the import of nuclear-encoded RNAs into mitochondria; such import has been shown to be required for proper replication and transcription of the mitochondrial genome.3,4 The known transported RNAs are small RNAs including 5S rRNA, RNase P RNA, MRP RNA, and different tRNAs.5 The only established protein components of this import machinery in mammals are rhodanese,6 MRP-L18,7 and polynucleotide phosphorylase (PNPase or PNPT1).8 At the cellular level, it has been demonstrated that downregulation of PNPT1 leads to mitochondrial dysfunction with an impairment of mitochondrial RNA processing and the accumulation of polycistronic transcripts.8 However, the consequence of PNPase dysfunction in complex tissues or whole organisms is unknown, and it is thus unclear whether different tissues might have differing degrees of dependence on PNPase function and mitochondrial RNA import.
We studied a consanguineous Moroccan family with three siblings (two males and one female) affected by a severe hearing impairment (Figure 1A), which was first noticed in early childhood and resulted in the inability to acquire normal speech. Three more siblings and both parents, who are first-degree cousins, are unaffected, confirming an autosomal-recessive inheritance pattern in this family. The study was approved by the ethics committees of the Institut Pasteur, Morocco and the Universities of Cologne and Ulm, Germany, and written informed consent was obtained from all participating individuals or their parents. The affected individuals were evaluated by detailed medical-history interviews and a physical examination, and they underwent an otological examination and pure-tone audiometry with air and bone conduction measurements. Taken when the affected children were between the ages of 12 (IV:6) and 17 (IV:4), pure-tone audiograms confirmed a hearing loss of at least 80 dB for all tested frequencies (Figure 1B). Clinical examinations revealed no obvious additional symptoms, such as vestibular dysfunction, cognitive or motor disturbances, or signs of metabolic diseases, in any of the affected individuals, leading to the classification of nonsyndromic hearing impairment (the oldest individual was 24 years of age at the last examination). We first excluded GJB2 (encoding connexin 26; MIM 121011) mutations, the most common cause of autosomal-recessive nonsyndromic hearing impairment (ARNSHI), by direct sequencing of genomic DNA of affected family members.
Figure 1.
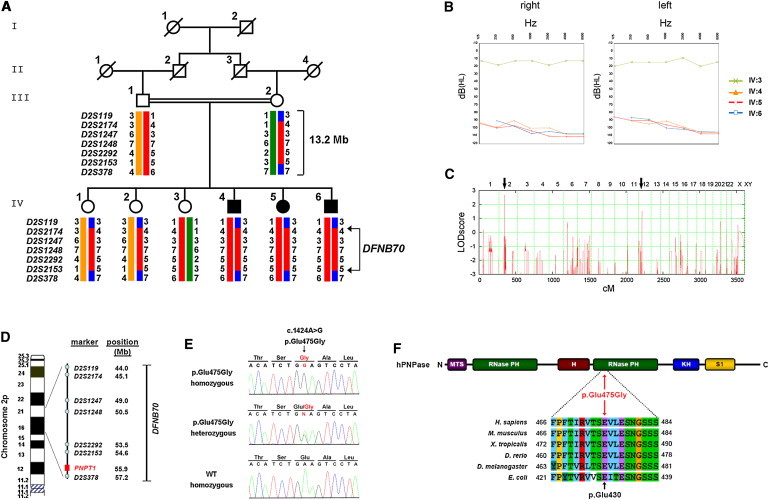
Identification of the Disease-Causing PNPT1 Mutation in a Consanguineous Family Affected by Nonsyndromic Hearing Loss
(A) Pedigree of the DFNB70-linked family. Microsatellite haplotypes indicate homozygosity between markers D2S119 and D2S378 in affected family members (filled symbols). According to the Genome Reference Consortium release h37, February 2009, the linked region spans approximately 13.2 Mb.
(B) Pure-tone audiograms of the three affected children with severe hearing loss; an audiogram of an unaffected sibling is shown for comparison. The following abbreviations are used: dB(HL), decibel hearing loss; and Hz, frequency in Hertz.
(C) Genome-wide homozygosity mapping identified two putative loci (black arrows) with multipoint LOD scores of 2.7 and 1.5 on chromosomes 2 and 12, respectively (see also Figure S1).
(D) Illustration shows the position of the DFNB70 locus on the short arm of chromosome 2; PNPT1 is at the centromeric end (see also Figure S1 and Table S1).
(E) Direct sequencing identified a homozygous c.1424A>G mutation in PNPT1 in affected individuals (upper electropherogram) and in a heterozygous state in unaffected carriers (middle). A homozygous wild-type (WT) sequence is shown on the bottom. Amino acid abbreviations are shown above the sequence traces.
(F) A PNPase monomer consists of two RNase-PH domains, which are separated by an α-helix, and two RNA binding domains, KH and S1. Additionally, mammalian PNPase carries an N-terminal mitochondrial targeting sequence (MTS). The multiple-sequence alignment was generated with the ClustalW2 Multiple Sequence Alignment tool and depicts conservation of the crucial glutamic acid residue. Amino acid position 430 in E. coli corresponds to position 475 in humans.
To identify the chromosomal locus underlying the sensory disorder in this family, we performed genome-wide homozygosity mapping of seven family members (the parents, three affected siblings, and two unaffected siblings) by using the Affymetrix GeneChip Human Mapping 10K SNP array Xba142 (version 2.0). Data handling, evaluation, and statistical analysis were performed as described before.9 We identified two putative loci, one on chromosome 2 and a second of weaker significance on chromosome 12, with multipoint LOD scores of 2.7 (which is the maximum expected LOD score in this family and thus means perfect cosegregation of marker haplotypes with the phenotype) and 1.5, respectively (Figure 1C). We subsequently excluded the putative locus on chromosome 12 by genotyping microsatellite markers on the basis of nonsegregation of marker alleles with the phenotype (Figure S1, available online). By contrast, the putatively linked region on chromosome 2 was confirmed by microsatellite genotyping in all eight family members (Figure 1A). The linkage analysis defined a locus on the short arm of chromosome 2 (2p16.1), which is delineated by markers D2S119 and D2S378 and spans approximately 13.2 Mb of genomic sequence (Figure 1D) and which does not include or overlap with any known ARNSHI locus. The locus, named DFNB70 according to the official deafness loci nomenclature, harbors 49 annotated protein-coding genes (Figure S1 and Table S1).
We analyzed the coding exons and adjacent splice sites of all 49 positional candidate genes within the linked chromosomal interval by PCR amplification of genomic DNA from one affected individual and by subsequent direct Sanger sequencing. Primer sequences, which were obtained with Primer3, are available upon request. In the coding sequence, we identified just a single nonannotated nucleotide change (c.1424A>G [Figure 1E]) representing a missense mutation in PNPT1 (RefSeq accession number NM_033109.3), which codes for polynucleotide phosphorylase (PNPase or PNPT1).10–12 At the protein level, this transition at nucleotide position 1,424 predicts an exchange of a negatively charged glutamic acid to glycine, p.Glu475Gly, which is located within the second RNase-PH domain of the protein (Figure 1F). The alteration cosegregated with the hearing loss in the family—only affected individuals showed homozygosity for c.1424A>G—whereas both parents and the three unaffected siblings were heterozygous carriers. The missense alteration was not annotated in either dbSNP or the 1000 Genomes Project database; moreover, it was not present in over 10,000 Exome Variant Server alleles of European or African American descent, precluding that it represented a benign or more common polymorphism. Furthermore, we did not identify the missense alteration in 192 German, 160 Moroccan, or 57 Turkish control individuals as tested by a mutation-specific Hpy188III restriction digest. The glutamic acid is located in a functional domain of the protein and is strictly conserved throughout evolution (Figure 1F), compatible with an important functional role for this residue. All together, the genetic data strongly suggest a causative role for the PNPT1 missense alteration in hearing impairment.
PNPT1 has been reported to have a broad tissue expression, but expression in the inner ear has not been specifically analyzed until now. Using RT-PCR on total RNA isolated from postnatal day (P)2 wild-type (WT) mice (TRIzol reagent, Invitrogen, Darmstadt, Germany), we found Pnpt1 mRNA expression in the murine cochlea and various other tissues (Figure 2A), confirming the broad Pnpt1 expression pattern, including in the inner ear. In addition, immunoblot analysis with a commercial PNPase antibody (polyclonal rabbit anti-PNPase, Catalog No. 14487-1-AP, Proteintech, Manchester, UK) also revealed PNPase in extracts from murine total cochlea and brain (Figure 2B). To analyze the cellular localization of PNPase within the cochlea, we performed immunohistochemistry. Cochleae from P6 mice were dissected, and immunohistochemistry was performed according to standard procedures. The immunohistological staining pattern demonstrated a broad PNPase distribution in the inner ear, including the sensory hair cells (Figure 2C) and spiral ganglion neurons (Figure 2J), and was independently confirmed by the use of another commercial antibody (data not shown). Immunohistochemistry in organ-of-Corti whole-mount preparations13 from P19 mice further confirmed PNPase localization in inner and outer hair cells (Figure 2G). Heat-shock protein 60 (Hsp60) and apoptosis-inducing factor (Aif) were selected as mitochondrial markers (Figures 2D, 2H, and 2K) and colocalized with PNPase (Figures 2E, 2I, and 2L), thus confirming the predominant mitochondrial localization of PNPase also in the inner ear.
Figure 2.
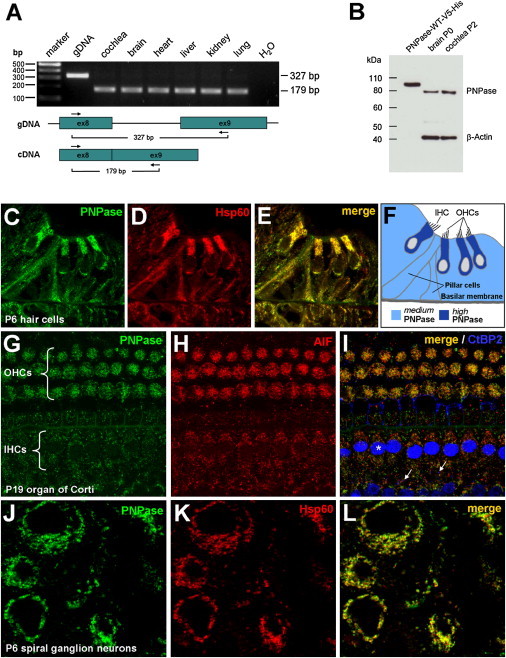
Expression of Pnpt1 and Detection of PNPase in the Murine Inner Ear
(A) Total RNA from selected tissues was isolated from P2 WT mice. RT-PCR using a forward primer in exon 8 and a reverse primer in exon 9 was performed. Amplification of cDNA leads to a 179 bp product, whereas amplification of genomic DNA (gDNA) results in a 327 bp product as a result of the inclusion of intron 8. Pnpt1 expression was observed in all tissues studied. “H2O” indicates the water control.
(B) In vitro translated and purified recombinant PNPase (PNPase-WT-V5-His) is recognized by PNPase antibody in immunoblot analysis. Endogenous murine PNPase was detected in brain and cochlea at P0 and P2, respectively. The higher molecular weight of the recombinant PNPase (+10 kDa) is due to the additional V5-His tag and the nonprocessing of the MTS. Protein mass was calculated with the PeptideMass online calculation tool.
(C–L) Immunohistochemistry of PNPase in the murine inner ear.
(C) Cross-section of P6 mouse cochlea reveals PNPase staining in hair cells and surrounding tissue.
(D and E) Heat-shock protein 60 (Hsp60) was used as a mitochondrial marker (D) and colocalizes with PNPase (E).
(F) Sketch illustrates PNPase levels in the organ of Corti and surrounding cells. The following abbreviations are used: IHC, inner hair cell; and OHCs, outer hair cells.
(G–I) Organ-of-Corti whole-mount preparation from P19 mice. Maximum intensity confocal projections of three Z-planes (∼1 μm depth) at the level of OHC apexes and IHC somata are shown.
(G) PNPase staining in OHCs and IHCs.
(H) AIF (apoptosis inducing factor) antibody was used as a positive control for mitochondria.
(I) PNPase and AIF merged. CtBP2 (also known as Ribeye) antibody was used for better orientation in the sample because it stained most brightly the IHC nuclei (∗) and the smaller presynaptic densities of the afferent synapses (arrows). Both AIF and PNPase displayed a staining pattern consistent with that of mitochondria.
(J) Cross-section showing PNPase staining in spiral ganglion neurons.
(K) Hsp60 was used for staining mitochondria.
(L) PNPase and Hsp60 merged.
To independently assess inner-ear expression of Pnpt1, we analyzed its expression in Danio rerio. Because the zebrafish ortholog of PNPT1 (pnpt1) was not annotated in public genome databases, we performed RT-PCR (OneStep RT-PCR Kit, QIAGEN, Hilden, Germany) and 5′ and 3′ RACE (rapid amplification of cDNA ends) PCR (5′–3′ RACE Kit 2nd generation, Roche, Mannheim, Germany) experiments based on the sequence of expressed sequence tags (ESTs; GenBank accession numbers EE694402.1 and CT592833.2) representing parts of pnpt1; these ESTs were identified by TBLASTN analysis with the human PNPase protein sequence. The coding sequence of pnpt1 (Figure S2) consisted of a 2,328 bp open reading frame encoding a protein of 776 amino acids. Human PNPase and the zebrafish ortholog show a high degree of conservation with approximately 70% amino acid sequence identity (Figure S3), which includes the conserved glutamic acid at human position 475 corresponding to Glu469 in Danio rerio. Also, the zebrafish ortholog is predicted to have a mitochondrial targeting signal (MTS), and transient overexpression of pnpt1 in mouse embryonic fibroblasts (MEFs) indeed showed a mitochondrial localization of the encoded protein (data not shown). For in situ hybridization, a zebrafish EST clone (GenBank accession number EE702826) harboring approximately 900 bp of pnpt1 was linearized for obtaining antisense and sense strands. In vitro transcription was carried out with the DIG RNA Labeling Kit (Roche, Mannheim, Germany) according to the manufacturer’s instructions. In situ hybridization was performed as previously described14 and revealed strong pnpt1 expression (maternal expression) at the two-cell stage (Figure 3A) and a rather broad staining pattern during early development (Figures 3B–3D); this pattern became more restricted later in development and showed pronounced expression in the tectum, the gill arches, and the developing ear (Figures 3E and 3F). In summary, we showed conserved expression of Pnpt1/pnpt1 in the inner ear in mice and in the ear in zebrafish, as well as high PNPase levels in the murine cochlea, which is compatible with a role for polynucleotide phosphorylase in auditory function.
Figure 3.
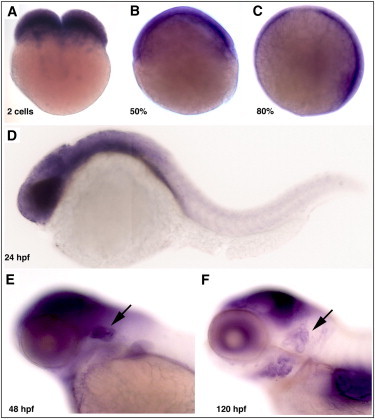
In Situ Hybridization of the PNPT1 Ortholog in Zebrafish
(A) Maternal expression of pnpt1 in 2-cell-stage fertilized zebrafish eggs.
(B–C) At 50% and 80% epiboly, pnpt1 mRNA is broadly expressed in the whole embryo.
(D) 24 hr postfertilization (hpf) expression is highest in the head area and disappears toward the tail.
(E–F) 48 hpf and 120 hpf expression becomes restricted to the tectum, the gill arches, and the developing ear (indicated by arrows).
A possible functional consequence of the missense alteration could be a decrease in protein stability or an altered subcellular localization. We therefore subcloned PNPT1 WT cDNA in a mammalian expression vector (pcDNA3.1/V5-His-TOPO Invitrogen, Darmstadt, Germany) and introduced the c.1424A>G point mutation by in vitro mutagenesis (QuikChange II Site-Directed Mutagenesis Kit, Agilent, Waldbronn, Germany). However, by transient overexpression in human embryonic kidney (HEK) 293T cells, we did not observe either potential effect (Figure S4). The unaltered mitochondrial localization of the protein encoded by the mutant cDNA was confirmed in transfected COS7 cells and transformed yeast (Figure S4). Because of the mitochondrial function of PNPase, we also analyzed protein stability after induction of oxidative stress. HEK 293T cells expressing either WT or mutant PNPT1 were incubated with various substances, but, again, we could not demonstrate a significant difference between the WT and mutant protein stability (Figure S4).
Glu475 is located in the second RNase-PH domain of PNPase and is strictly conserved in orthologs from over 500 published prokaryotic and eukaryotic species for which protein sequences are available in the UCSC multiple-species sequence-alignment database (Figure S5 and data not shown). To possibly predict a functional consequence of the p.Glu475Gly alteration, we modeled human PNPase to the published structure of the E. coli ortholog15 by using the PHYRE2 server.16 The three-dimensional modeling of PNPase revealed that Glu475 is not located close to the central canal of the PNPase trimer, which is involved in RNA processing and possibly transport, and that p.Glu475Gly is not predicted to significantly change the functional properties of the PNPase monomer. After analysis of the quaternary structure of the PNPase trimer, it appears that the glutamic acid residue p.Glu430 in E. coli (corresponding to p.Glu475 in humans) is located at the trimerization interface (Figures 4A and 4B) and that the negatively charged glutamic acid is predicted to build a salt bridge with the positively charged arginine residue at position 66 (corresponding to p.Lys105 in humans) of the neighboring monomer (Figures 4A and 4B). More than 95% of the over 500 published PNPase orthologs have a positively charged residue at this position (data not shown). Changing Glu475 to glycine, as found in the family with ARNSHI, is predicted to disrupt the salt bridge and will thus most likely interfere with PNPase function by compromising trimerization.
Figure 4.
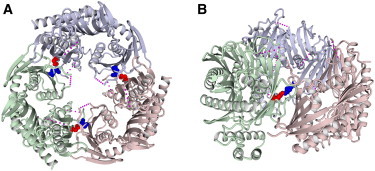
Three-Dimensional Modeling of Human PNPase
The structure of bacterial PNPase from E.coli (Protein Data Bank accession number 3CDI) is a homotrimer; the subunits are indicated by different colors (light red, green, and blue). The glutamate residue corresponding to Glu475 of human PNPase (red space-filling representation) forms a salt bridge to an arginine residue (blue spacefill) of the neighboring subunit (see also Figure S5). The pink dotted lines indicate unresolved parts of the structure.
(A) Top view along the trimer symmetry axis; all three salt bridges are indicated.
(B) Side view; only one of the salt bridges is shown.
To study this predicted functional deficit of the altered PNPase, we first performed a bacterial-growth assay. In E. coli, PNPase exists as a homotrimeric complex, associates solely with RhlB (an RNA helicase that facilitates degradation of double-stranded RNA), or exists within a multiprotein complex termed the RNA degradosome.10 Although the cellular function of PNPase obviously differs between bacteria and mammals, the ability of PNPase monomers to form trimers is important for both bacteria and mammals. The deletion of pnp in E. coli is known to cause a growth deficit under increased oxidative stress induced by hydrogen peroxide.17 We therefore analyzed bacterial growth in a pnp-knockout strain (Δpnp, KEIO collection clone JW5851, National BioResource Project, Japan). E. coli pnp was cloned into the inducible bacterial expression vector pBAD-DEST49 (Invitrogen, Darmstadt, Germany) with the use of plasmid pKAK718 as a template, and the Δpnp strain was transformed with constructs either expressing E. coli pnp WT (Δpnp/WT) or the analogous mutant (Δpnp/p.Glu430Gly) (Figure 1F). This was done because complementation of the bacterial phenotype with PNPT1, either with or without a MTS, was not efficient (data not shown). Whereas induced expression of E. coli pnp WT led to a nearly complete rescue of the H2O2-induced growth defect, expression of the pnp mutant (Δpnp/p.Glu430Gly) resulted in a significantly diminished ability to complement the phenotype both in spot tests and in liquid cultures (Figures 5A and 5B). The growth tests indicated that the amino acid substitution does not cause a complete loss of function but rather leads to a strongly impaired PNPase function, which is consistent with the finding that a constitutive knockout of murine Pnpt1 is lethal in the embryonic stage.8
Figure 5.
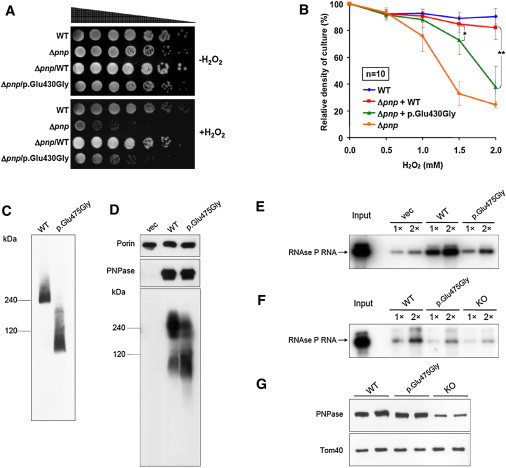
Functional Consequences of the Identified PNPT1 Mutation
(A–B) Complementation experiments in bacteria: E. coli pnp-knockout strain (Δpnp) was transformed with constructs expressing either bacterial WT or mutant (p.Glu430Gly) pnp. E. coli K-12 strain BW25113 was used as a WT reference.
(A) Spot tests. Strains were serially diluted and grown on lysogeny broth (LB) agar plates either with or without 0.6 mM hydrogen peroxide. Growth of Δpnp is suppressed by hydrogen peroxide and can be almost completely restored by WT Pnp but only partially by p.Glu430Gly Pnp.
(B) Indicated strains were grown in LB medium and exposed to increasing concentrations of hydrogen peroxide. Exogenous expression of WT pnp restores growth significantly better than does expression of the pnp mutant encoding p.Glu430Gly (Student’s two-tailed t test: ∗p = 7.06 × 10−3, ∗∗p = 2.56 × 10−6). Error bars indicate standard deviation of ten independent experiments.
(C–D) The assembly state of exogenous WT and p.Glu475Gly PNPase was determined by nondenaturing blue-native gel electrophoresis.
(C) In MEFs with floxed endogenous mouse Pnpt1 knocked down by CMV-CRE recombinase-induced partial knockout, the trimerization of exogenous p.Glu475Gly PNPase is severely impaired and shifted toward monomers.
(D) In the GA74-1A yeast strain, p.Glu475Gly PNPase colocalized with a mitochondrial marker, PORIN, and showed a protein level that was similar to that of exogenous WT PNPase. However, the assembly of p.Glu475Gly PNPase in a higher-order, trimeric PNPase complex was impaired compared to WT PNPase, as shown by a significant reduction of the trimer band at ∼240 kDa and increase of the monomer band at ∼85 kDa.
(E) RNA import assay in yeast. In-vitro-transcribed human RNase P RNA was incubated with yeast mitochondria expressing WT PNPT1, a PNPT1 mutant coding for p.Glu475Gly, or an empty vector (“vec”). Import reactions were repeated with 1× and 2× amounts of RNA. Abundance of imported RNase P RNA in mitochondria containing p.Glu475Gly PNPase is one quarter of the level of that in mitochondria containing WT PNPase.
(F) RNA-import assay in MEFs. In-vitro-transcribed human RNase P RNA was incubated with mitochondria isolated from different MEF lines: WT (mouse Pnpt1 partial knockout plus exogenous WT PNPT1 expression), p.Glu475Gly (Pnpt1 partial knockout plus exogenous PNPT1 expression encoding p.Glu475Gly), or knockout (Pnpt1 expression reduced by 75%). Import reactions were repeated with 1× and 2× amounts of RNA. Import of RNase P RNA is two times lower in PNPT1-mutant-expressing mitochondria than in WT-PNPT1-transduced mitochondria.
(G) Immunoblot of PNPase and mitochondria-localized TOM40 from different MEF lines.
The homotrimerization of PNPase is necessary for the RNA processing and transporting function of the protein, and the identified missense exchange was predicted to interfere with oligomerization. We therefore continued by investigating the ability of p.Glu475Gly PNPase to assemble into functional trimers by blue-native gel electrophoresis, which was performed as previously described.19 When the PNPT1 mutation encoding for PNPase p.Glu475Gly was exogenously expressed in MEFs with downregulated endogenous Pnpt1,8 its ability to assemble into higher-order trimeric PNPase complexes was severely impaired, as shown by a significant drift of the trimer band at ∼240 kDa to the monomer band at ∼85 kDa (Figure 5C). To a lesser extent, this drift toward monomers was also seen in yeast cells artificially expressing the mutated PNPT1 (Figure 5D), thereby confirming the prediction of the three-dimensional modeling.
Finally, we studied the genuine mammalian function, mitochondrial RNA import, of WT and p.Glu475Gly PNPase in yeast and mammalian cells. To analyze mitochondrial RNA-import capacity, we isolated mitochondria from three strains of yeast: WT yeast (which does not have a PNPT1 ortholog), yeast expressing WT PNPT1, and yeast expressing the PNPT1 missense mutant coding for p.Glu475Gly PNPase. This was followed by incubation with in-vitro-transcribed human RNase P RNA, a known substrate of PNPase. As expected, the amount of RNase P RNA import was much higher in mitochondria expressing WT PNPT1 than in yeast mitochondria containing an empty vector.8 In contrast, mitochondria expressing the PNPT1 missense mutant construct coding for p.Glu475Gly PNPase showed two times more RNase P RNA than did empty-vector-containing yeast mitochondria but showed 75% less RNA than did mitochondria expressing WT PNPT1 (Figure 5E), suggesting that in yeast cells p.Glu475Gly PNPase is only partially functional for RNA import. Furthermore, to provide mammalian cell relevance, we also expressed WT or mutant PNPT1 in MEFs with floxed endogenous mouse Pnpt1 knocked down by CMV-CRE recombinase-induced partial knockout. The level of p.Glu475Gly PNPase in MEF mitochondria was similar to exogenous protein levels of WT PNPase (Figure 5G). PNPT1-mutant-transduced mitochondria showed two times less RNase P RNA import than did exogenous WT-PNPT1-expressing mitochondria (Figure 5F). Compared with the RNase P RNA import in the yeast assay, the less-pronounced reduction in RNase P RNA import in MEFs is probably related to the amount of endogenous WT PNPase that is required for maintaining cell viability in the MEFs. The import of RNase P RNA into p.Glu475Gly-PNPase-containing mitochondria, however, was slightly higher than that into MEF mitochondria containing ∼33% of the endogenous level of PNPase, consistent with a role for p.Glu475Gly PNPase as a hypomorph in mammalian cells. Both the yeast and the mammalian RNA-import assays were performed as previously described.8 All together, our functional analyses in bacteria, yeast, and mammalian cells show a severe functional PNPase impairment caused by the identified PNPT1 missense mutation.
The inner ear is one of the tissues with a relatively high energy demand, which is supported by the finding that selected mtDNA mutations—such as the m.1555A>G mutation in the mitochondrial 12S rRNA gene (MIM 500008) or mutations in the tRNASer gene (MIM 590080)—cause nonsyndromic hearing impairment.20 Other mtDNA mutations are found in syndromic types of hearing impairment often characterized by additional neurologic, cardiologic, or metabolic symptoms.20 In many cases, mtDNA mutations do not cause an overt hearing phenotype at all, demonstrating the variable expressivity and incomplete penetrance of diseases arising from mtDNA mutations.21 In addition, mutations in nuclear genes encoding for proteins necessary for mitochondrial function can cause secondary types of mitochondrial diseases.22 This group of disorders could, in principle, manifest as syndromic or nonsyndromic types of hearing impairment, but most of them do not show a prominent hearing involvement or, even less, an exclusive disturbance of the inner ear. Indeed, and despite the extreme locus heterogeneity of autosomal-recessive nonsyndromic hearing impairment (see Hereditary Hearing Loss Homepage and Dror and Avraham23), just recently the first mutations in a nuclear gene (MSRB3) encoding a protein with a possible mitochondrial function have been described in ARNSHI (MIM 613718).24 MSRB3 encodes the zinc-containing methionine sulfoxide reductase B3, which has been reported to have human isoforms with MTSs, although the mouse ortholog does not have a functional MTS and is exclusively localized in the endoplasmic reticulum.25 Whether or not this form of ARNSHI is indeed mitochondrial in origin, it is evident that nonsyndromic types of hearing impairment are rarely caused by mutations in nuclear genes encoding proteins with a mitochondrial function, indicating that most mitochondrial processes and functions are not specifically important for the function of the inner ear. In this report, we add autosomal-recessive, congenital, nonsyndromic hearing impairment type DFNB70 to the growing list of secondary mitochondrial diseases, and it belongs to the rare group of early-onset but seemingly monosymptomatic mitochondrial disorders. Although we cannot exclude the future development of additional late-onset symptoms in affected family members, the disease in all three affected individuals is clearly dominated by a congenital and profound hearing impairment, and, until now, there have been no obvious additional symptoms present in any of these individuals.
On the basis of three-dimentional-modeling data, the identified p.Glu475Gly alteration is predicted to cause a disturbance of the quaternary structure of the PNPase trimer, which we verified experimentally in yeast and mammalian cells. This kind of structural trimerization disturbance, which impairs PNPase function, was shown before for the artificial protein variant p.Asp135Gly.26 Also, we demonstrated that impaired trimerization causes a severely reduced functionality of PNPase in cells of such diverse species as E. coli, S. cerevisiae, and M. musculus. Interestingly, the functional studies consistently showed that p.Glu475Gly PNPase behaves as a hypomorph, which is consistent with expectations given that a constitutive knockout of the murine gene is not compatible with life.8 Together with the genetic data and our detection of evolutionarily conserved Pnpt1 and pnpt1 expression in the ear, the functional studies have thus confirmed the pathogenic role of the identified missense mutation and the identification of PNPT1 as a gene whose dysfunction can cause autosomal-recessive hearing loss. If one were to think about other types of potential PNPT1 mutations, it seems possible that mutations causing an even more pronounced loss of function without being a null allele—which could well be lethal, like in the mouse—might manifest as a syndromic type of deafness with involvement of additional organ systems of high energy demand. On the other hand, PNPT1 mutations causing a functional deficit milder than that observed for p.Glu475Gly might, as a result of the importance of PNPase function in the inner ear, show a progressive late-onset type of hearing loss or an increased susceptibility toward complex inherited presbyacusis, which is speculated to have at least in part a mitochondrial origin.27
In summary, we elucidated a hereditary disturbance of mitochondrial RNA import by the identification of a hypofunctional mutation in PNPT1 in congenital severe hearing impairment, demonstrating the rather unexpected and specific importance of PNPase for auditory function.
Acknowledgments
We thank the family members for their participation and cooperation, K. Schnetz for providing E. coli K-12 wild-type strain BW25113, S.R. Kushner for providing plasmid pKAK7, and Thomas Langer and Rudolf Wiesner for discussions. The authors also thank the National Heart, Lung, and Blood Institute Grand Opportunity (GO) Exome Sequencing Project and its ongoing studies, which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the Women’s Health Initiative Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010). This work was supported by the European Commission FP6 Integrated Project EUROHEAR (LSHG-CT-20054-512063), the National Institutes of Health (GM061721, GM073981, CA90571, and CA156674), the Muscular Dystrophy Association (022398), the American Heart Association (0640076N), the California Institute for Regenerative Medicine (RS1-00313 and RB1-01397), the Wellcome Trust (087737), and the Alexander von Humboldt Foundation.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Browser, http://www.1000genomes.org/ensembl-browser
ClustalW2: Multiple Sequence Alignment, http://www.ebi.ac.uk/Tools/msa/clustalw2
Exome Variant Server, http://evs.gs.washington.edu/EVS/
Hereditary Hearing Loss Homepage, http://hereditaryhearingloss.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PeptideMass, http://www.expasy.ch/tools/peptide-mass.html
Primer3, http://biotools.umassmed.edu/bioapps/primer3_www.cgi
UCSC Genome Browser, http://genome.ucsc.edu
Accession Numbers
The GenBank accession number for the zebrafish pnpt1 cDNA sequence reported in this paper is JN381023.
References
- 1.Chacinska A., Koehler C.M., Milenkovic D., Lithgow T., Pfanner N. Importing mitochondrial proteins: Machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacKenzie J.A., Payne R.M. Mitochondrial protein import and human health and disease. Biochim. Biophys. Acta. 2007;1772:509–523. doi: 10.1016/j.bbadis.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Entelis N.S., Kolesnikova O.A., Martin R.P., Tarassov I.A. RNA delivery into mitochondria. Adv. Drug Deliv. Rev. 2001;49:199–215. doi: 10.1016/s0169-409x(01)00135-1. [DOI] [PubMed] [Google Scholar]
- 4.Sieber F., Placido A., El Farouk-Ameqrane S., Duchêne A.M., Maréchal-Drouard L. A protein shuttle system to target RNA into mitochondria. Nucleic Acids Res. 2011;39:e96. doi: 10.1093/nar/gkr380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang G., Shimada E., Koehler C.M., Teitell M.A. PNPASE and RNA trafficking into mitochondria. Biochim. Biophys. Acta. 2012;1819:998–1007. doi: 10.1016/j.bbagrm.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smirnov A., Comte C., Mager-Heckel A.M., Addis V., Krasheninnikov I.A., Martin R.P., Entelis N., Tarassov I. Mitochondrial enzyme rhodanese is essential for 5 S ribosomal RNA import into human mitochondria. J. Biol. Chem. 2010;285:30792–30803. doi: 10.1074/jbc.M110.151183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smirnov A., Entelis N., Martin R.P., Tarassov I. Biological significance of 5S rRNA import into human mitochondria: Role of ribosomal protein MRP-L18. Genes Dev. 2011;25:1289–1305. doi: 10.1101/gad.624711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang G., Chen H.W., Oktay Y., Zhang J., Allen E.L., Smith G.M., Fan K.C., Hong J.S., French S.W., McCaffery J.M. PNPASE regulates RNA import into mitochondria. Cell. 2010;142:456–467. doi: 10.1016/j.cell.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalay E., Li Y., Uzumcu A., Uyguner O., Collin R.W., Caylan R., Ulubil-Emiroglu M., Kersten F.F., Hafiz G., van Wijk E. Mutations in the lipoma HMGIC fusion partner-like 5 (LHFPL5) gene cause autosomal recessive nonsyndromic hearing loss. Hum. Mutat. 2006;27:633–639. doi: 10.1002/humu.20368. [DOI] [PubMed] [Google Scholar]
- 10.Chen H.W., Koehler C.M., Teitell M.A. Human polynucleotide phosphorylase: Location matters. Trends Cell Biol. 2007;17:600–608. doi: 10.1016/j.tcb.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Sarkar D., Fisher P.B. Polynucleotide phosphorylase: An evolutionary conserved gene with an expanding repertoire of functions. Pharmacol. Ther. 2006;112:243–263. doi: 10.1016/j.pharmthera.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 12.Yehudai-Resheff S., Portnoy V., Yogev S., Adir N., Schuster G. Domain analysis of the chloroplast polynucleotide phosphorylase reveals discrete functions in RNA degradation, polyadenylation, and sequence homology with exosome proteins. Plant Cell. 2003;15:2003–2019. doi: 10.1105/tpc.013326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khimich D., Nouvian R., Pujol R., Tom Dieck S., Egner A., Gundelfinger E.D., Moser T. Hair cell synaptic ribbons are essential for synchronous auditory signalling. Nature. 2005;434:889–894. doi: 10.1038/nature03418. [DOI] [PubMed] [Google Scholar]
- 14.Hammerschmidt M., Pelegri F., Mullins M.C., Kane D.A., van Eeden F.J., Granato M., Brand M., Furutani-Seiki M., Haffter P., Heisenberg C.P. dino and mercedes, two genes regulating dorsal development in the zebrafish embryo. Development. 1996;123:95–102. doi: 10.1242/dev.123.1.95. [DOI] [PubMed] [Google Scholar]
- 15.Shi Z., Yang W.Z., Lin-Chao S., Chak K.F., Yuan H.S. Crystal structure of Escherichia coli PNPase: Central channel residues are involved in processive RNA degradation. RNA. 2008;14:2361–2371. doi: 10.1261/rna.1244308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelley L.A., Sternberg M.J. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 17.Wu J., Jiang Z., Liu M., Gong X., Wu S., Burns C.M., Li Z. Polynucleotide phosphorylase protects Escherichia coli against oxidative stress. Biochemistry. 2009;48:2012–2020. doi: 10.1021/bi801752p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donovan W.P., Kushner S.R. Polynucleotide phosphorylase and ribonuclease II are required for cell viability and mRNA turnover in Escherichia coli K-12. Proc. Natl. Acad. Sci. USA. 1986;83:120–124. doi: 10.1073/pnas.83.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H.W., Rainey R.N., Balatoni C.E., Dawson D.W., Troke J.J., Wasiak S., Hong J.S., McBride H.M., Koehler C.M., Teitell M.A., French S.W. Mammalian polynucleotide phosphorylase is an intermembrane space RNase that maintains mitochondrial homeostasis. Mol. Cell. Biol. 2006;26:8475–8487. doi: 10.1128/MCB.01002-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kokotas H., Petersen M.B., Willems P.J. Mitochondrial deafness. Clin. Genet. 2007;71:379–391. doi: 10.1111/j.1399-0004.2007.00800.x. [DOI] [PubMed] [Google Scholar]
- 21.Greaves L.C., Reeve A.K., Taylor R.W., Turnbull D.M. Mitochondrial DNA and disease. J. Pathol. 2012;226:274–286. doi: 10.1002/path.3028. [DOI] [PubMed] [Google Scholar]
- 22.Koopman W.J., Willems P.H., Smeitink J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012;366:1132–1141. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- 23.Dror A.A., Avraham K.B. Hearing impairment: A panoply of genes and functions. Neuron. 2010;68:293–308. doi: 10.1016/j.neuron.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 24.Ahmed Z.M., Yousaf R., Lee B.C., Khan S.N., Lee S., Lee K., Husnain T., Rehman A.U., Bonneux S., Ansar M. Functional null mutations of MSRB3 encoding methionine sulfoxide reductase are associated with human deafness DFNB74. Am. J. Hum. Genet. 2011;88:19–29. doi: 10.1016/j.ajhg.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim H.Y., Gladyshev V.N. Characterization of mouse endoplasmic reticulum methionine-R-sulfoxide reductase. Biochem. Biophys. Res. Commun. 2004;320:1277–1283. doi: 10.1016/j.bbrc.2004.06.078. [DOI] [PubMed] [Google Scholar]
- 26.Portnoy V., Palnizky G., Yehudai-Resheff S., Glaser F., Schuster G. Analysis of the human polynucleotide phosphorylase (PNPase) reveals differences in RNA binding and response to phosphate compared to its bacterial and chloroplast counterparts. RNA. 2008;14:297–309. doi: 10.1261/rna.698108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamasoba T., Someya S., Yamada C., Weindruch R., Prolla T.A., Tanokura M. Role of mitochondrial dysfunction and mitochondrial DNA mutations in age-related hearing loss. Hear. Res. 2007;226:185–193. doi: 10.1016/j.heares.2006.06.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.