

Abstract
Shprintzen-Goldberg syndrome (SGS) is characterized by severe marfanoid habitus, intellectual disability, camptodactyly, typical facial dysmorphism, and craniosynostosis. Using family-based exome sequencing, we identified a dominantly inherited heterozygous in-frame deletion in exon 1 of SKI. Direct sequencing of SKI further identified one overlapping heterozygous in-frame deletion and ten heterozygous missense mutations affecting recurrent residues in 18 of the 19 individuals screened for SGS; these individuals included one family affected by somatic mosaicism. All mutations were located in a restricted area of exon 1, within the R-SMAD binding domain of SKI. No mutation was found in a cohort of 11 individuals with other marfanoid-craniosynostosis phenotypes. The interaction between SKI and Smad2/3 and Smad 4 regulates TGF-β signaling, and the pattern of anomalies in Ski-deficient mice corresponds to the clinical manifestations of SGS. These findings define SGS as a member of the family of diseases associated with the TGF-β-signaling pathway.
Main Text
Shprintzen-Goldberg syndrome (SGS [MIM 182212]) has been described as being associated with intellectual disability (ID), marfanoid habitus (including arachnodactyly, pectus deformity, scoliosis, and pes planus with foot deformity), camptodactyly, and facial dysmorphism (including hypertelorism, exophthalmos, downslanting palpebral fissures, and maxillary and mandibular hypoplasia). The hallmark of this syndrome, although inconsistent, is the presence of craniosynostosis1,2 (see Web Resources). Other findings include mitral valve prolapse, recurrent hernias, loss of subcutaneous tissue, and thin translucent skin. Infantile hypotonia, severe scoliosis, and obstructive apnea are common features as well. It is not known whether individuals with SGS display an aortic risk because some rare cases have been described with aortic dilatation1,2 (see Web Resources). We assumed that SGS is an autosomal-dominant disorder on the basis of previous descriptions of simplex cases (although recurrence in siblings has been reported).3 Because of the clinical overlap with Marfan syndrome (MFS [MIM 154700]) and Loeys-Dietz syndrome (LDS1A [MIM 609192], LDS1B [MIM 610168], LDS2A [MIM 608967], LDS2B [MIM 610380], LDS3 [MIM 613795], and LDS4 [MIM 190220]), mutations in FBN1 (MIM 134797), TGFBR1 (MIM 190181), and TGFBR2 (MIM 190182) should be excluded.4 We hypothesized that SGS is a clinically distinct entity resulting from heterozygous mutations of other gene(s) involved in the TGF-β-signaling pathway.
We recruited a cohort of 19 SGS-affected individuals originating from six European countries and Australia. The cohort included five related individuals from a family consistent with autosomal-dominant inheritance (family 3), another family with recurrence in siblings (family 4)3 (Figure 1 and Table 1), ten simplex cases (including one previously published individual),5 and one probable autosomal-dominant case. We also additionally assembled a second cohort of 11 individuals with marfanoid habitus and craniosynostosis; these individuals did not present with the dysmorphic features of SGS (Table S1, available online). Informed consent for research investigations was obtained from the affected individuals, legal representatives, or relatives. The research protocol was approved by the local ethics committees. The 30 individuals were first screened for FBN1, TGFBR1, and TGFBR2 mutations by direct sequencing and multiplex ligation-dependent probe amplification and for chromosomal rearrangements by 180K or 244K Agilent array comparative genomic hybridization. We identified simplex heterozygous missense mutations in FBN1 (c.3761G>A [p.Cys1254Tyr]; RefSeq accession number NM_000138.4), TGFBR1 (c.734A>G [p.Glu245Gly]; RefSeq NM_004612.2) and TGFBR2 (c.1583G>A [p.Arg528His]; RefSeq NM_003242.5) in three individuals from the second cohort (Table S1 and Figure S1).
Figure 1.
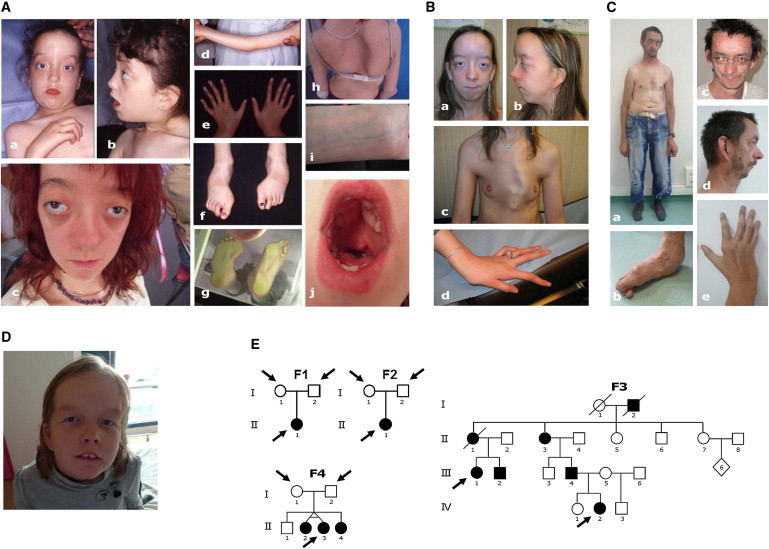
Clinical Presentations and Pedigrees of Subjects with SGS and Mutations in SKI
(A) Photographs of affected individual II-1 (from family 2), who has a SKI de novo c.94C>G variant. Note the hypertelorism, proptosis, downslanting palpebral fissures, maxillary and mandibular hypoplasia, low-set ears (Aa–Ac), joint contractures (Ad), arachnodactyly and camptodactyly (Ae), deformed feet (Af–Ag), severe scoliosis (Ah), translucent skin (Ai), and hypertrophy of the palatal shelves (Aj).
(B) Photographs of affected individual 14 (family 8), who has a SKI de novo c.103C>T variant. Note the dysmorphic features in favor of SGS (Ba–Bb), severe pectus carinatum (Bc), arachnodactyly, and camptodactyly (Bd).
(C) Photographs of affected individual III-4 (from family 3), who has a c.280_291delTCCGACCGCTCC variant in exon 1 of SKI. Note the dysmorphic features and habitus in favor of SGS (Ca, Cc, and Cd), foot deformity (Cb), and hand deformity with camptodactyly (Ce).
(D) Photographs of affected individual IV-2 from family 3 (child of individual III-4 in C).
(E) Pedigrees of families 1 (F1), 2 (F2), 3 (F3), and 4 (F4) studied by exome sequencing. Individuals studied are shown by an arrow.
Table 1.
Detailed Clinical Features of SGS Individuals and Summary of the Detected Mutations in SKI
|
Family 1 |
Family 2 |
Family 3 |
Family 4 |
Family 5 |
Family 6 |
Family 7 |
Family 8 |
Family 9 |
Family 10 |
Family 11 |
Family 12 |
Family 13 |
Total | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| II-1 | II-1 | III-4 | IV-2 | II-1 | III-1 | III-2 | II-2 | II-3 | II-4 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | ||
| Sex | F | F | M | F | F | F | M | F | F | F | M | M | F | F | F | F | M | M | M | 12F and 7M |
| Age (years) | 21 | 20 | 42 | 11 | 44a | 13 | 14 | 22 | 22 | 20 | 18 | 16 | 5 | 21 | 10a | 11 | 32 | 20 | 26 | − |
| Craniosynostosis | + | + | − | − | − | − | − | + | + | + | + | + | − | + | + | + | − | + | + | 12/19 |
| Arachnodactyly | − | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | 18/19 |
| Pectus deformity | + | + | + | − | − | − | − | + | + | + | + | + | + | + | + | + | + | + | + | 15/19 |
| Scoliosis | − | + | + | + | + | − | + | + | + | + | + | + | − | + | + | + | + | + | + | 16/19 |
| Joint contractures | + | + | + | + | + | + | + | + | + | + | − | + | − | + | + | + | − | + | − | 15/19 |
| Camptodactyly | + | + | − | − | − | − | − | − | − | − | − | + | − | + | + | + | + | + | + | 9/19 |
| Foot malposition | + | + | + | + | + | + | + | + | + | + | + | + | − | + | + | + | + | + | − | 17/19 |
| Scaphocephaly or dolichocephaly | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | − | + | + | + | 18/19 |
| Hypertelorism | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | − | + | + | + | 18/19 |
| Proptosis | + | + | + | − | + | + | − | + | + | − | + | − | + | + | + | + | + | + | + | 15/19 |
| Downslanting palpebral fissures | + | + | + | + | + | + | + | + | + | + | + | + | + | + | − | + | + | − | − | 16/19 |
| Micrognathia or retrognathia | + | + | + | − | + | − | − | + | + | + | + | + | + | + | + | − | + | + | + | 15/19 |
| Intellectual disability | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | 19/19 |
| Hernias | − | − | − | − | − | − | − | + | + | + | + | − | + | + | + | + | + | + | + | 11/19 |
| Loss of subcutaneous fat | + | − | − | − | − | − | − | + | + | − | − | + | − | + | + | − | − | + | − | 7/19 |
| Valvular anomalies | − | − | − | − | − | − | − | − | − | − | MVP | − | − | MVP | MVP, MI | MVP | MVP, MI | − | − | 5/19 |
| Aortic root dilatation | − | − | − | − | − | − | − | − | − | − | +b | − | − | − | UNL | + | + | − | − | 3/19 |
| Myopia | − | − | + | + | + | + | − | + | + | N/A | − | − | − | − | + | − | + | − | − | 8/18 |
| SKI mutation | c.100G>T | c.94C>G | c.280_291delTCCGACCGCTCC | c.280_291delTCCGACCGCTCC | c.280_291delTCCGACCGCTCC | c.280_291delTCCGACCGCTCC | c.280_291delTCCGACCGCTCC | c.101G>T | c.101G>T | c.101G>T | c.104C>A | c.94C>G | c.283_291delGACCGCTCC | c.103C>T | c.95T>C | c.100G>A | c.94C>G | c.92C>T | − | − |
| Amino acid substitution | p.Gly34Cys | p.Leu32Val | p.Ser94_Ser97del | p.Ser94_Ser97del | p.Ser94_Ser97del | p.Ser94_Ser97del | p.Ser94_Ser97del | p.Gly34Val | p.Gly34Val | p.Gly34Val | p.Pro35Gln | p.Leu32Val | p.Asp95_Ser97del | p.Pro35Ser | p.Leu32Pro | p.Gly34Ser | p.Leu32Val | p.Ser31Leu | − | 18/19 |
| Inheritance | de novo | de novo | AD | AD | AD | AD | AD | AD, SM | AD, SM | AD, SM | de novo | father N/A | de novo | de novo | de novo | parents N/A | parents N/A | parents N/A | AD | − |
The following abbreviations are used: AD, autosomal dominant; F, female; SM, somatic mosaicism; M, male; MVP, mitral valve prolapse; MI, mitral insufficiency; N/A, not available; and UNL, upper normal limit.
Affected individual 5 died of respiratory insufficiency. Affected individual 15 died suddenly, and an autopsy showed severe mitral valve dysplasia with calcifications of the mitral annulus.
Aortic dilatation requiring surgery at 16 years of age (aortic root dilatation with Z score = 7.014). He also has vertebrobasilar and internal carotid tortuosity and a dilated pulmonary-artery root.
First, we used the Nimblegen SeqCap EZ Exome v.2.0 kit to perform exome sequencing in two trios (families 1 and 2; Figure 1) with simplex SGS according to standard procedures; we used 8 μg of DNA from affected individuals and both parents (Figure S1). The resulting exome-capture libraries underwent two 75 bp paired-end sequencing runs on an Illumina HiSeq 2000. Reads were aligned to the human reference genome (GRCh37/hg19) with the Burrows-Wheeler Aligner,6 and potential duplicate paired-end reads were removed with Picard v.1.22 (see Web Resources). The Genome Analysis Toolkit (GATK) v.1.0.57 was used for base quality-score recalibration and indel realignment,7 as well as for single-nucleotide-variant and indel discovery and genotyping with the use of standard hard-filtering parameters.7 Variants with a quality score < 30, allele balance > 0.75, sequencing depth < 4, quality-to-depth ratio < 5.0, length of homopolymer run > 5.0, and strand bias > –0.10 were flagged and excluded from subsequent analyses. We used the GATK Depth of Coverage tool to assess coverage by ignoring reads with a mapping quality < 20 and by ignoring bases with a base quality < 30. In total, 92% of the primary target was covered at least four times in all individuals (Table S2). All variants identified in the affected individuals were annotated with SeattleSeq SNP annotation (see Web Resources). We focused on de novo heterozygous exonic variants (missense, nonsense, and splice-site variants and coding indels). Candidate mutational events were then inspected with the Integrative Genomics Viewer (see Web Resources).8 The resulting variants were excluded when the frequency was over 1/1,000 in the National Heart, Lung, and Blood Institute (NHLBI) Exome Variant Server (EVS) (see Web Resources). After applying variant calling filters, we failed to identify any candidate de novo mutations. Indeed, none of the variants identified in family 1 were confirmed by Sanger sequencing, and two de novo variants were confirmed in family 2. The first de novo variant was a COL4A4 (RefSeq NM_000092.4) missense mutation (c.4423G>T [p.Asp1475Tyr]) that is likely to cause benign hematuria only. The second de novo variant was a C1ORF54 (RefSeq NM_024579.3) frameshift mutation (c.272delT [p.Val91Glufs∗3]). Because C1ORF54 is predicted to encode a brain-, smooth-muscle-, and skin-secreted extracellular protein, the gene was sequenced in the rest of the cohort, but no pathogenic mutation was identified (conditions are available on request).
Subsequently, we performed exome sequencing by using Nimblegen Exome v.3.0 capture in the two most distant affected relatives (individuals III-1 and IV-2, who share 1/32 of their genomes) from family 3 and in one (individual II-3) of the three affected siblings from family 4, as well as in all of their unaffected parents (Figure 1), in accordance with the manufacturer’s recommendations. After mapping the raw sequencing reads against the reference genome and applying the same variant calling filters, we searched for heterozygous variants present in both distant relatives in family 3 and for heterozygous variant calls present in one sibling but absent in both parents in family 4 according to the gonadal-mosaicism hypothesis (Table S2). We first identified 314 variants shared by the two distant affected individuals from family 3. Considering the hypothesis of the implication of the TGF-β pathway, we set up a biological filter with the EMBL-EBI reactome,9 and 42 partners were listed (Table S3). When we filtered against EVS data and the TGF-β-signaling biological filter, only SKI was revealed (Table S2). Indeed, we found a 12 bp deletion (c.280_291delTCCGACCGCTCC [p.Ser94_Ser97del]) in a highly conserved region of exon 1 of SKI (RefSeq NM_003036.3) (Figure 2). Remarkably, in the first attempt of exome analysis, no SKI mutation was detected in family 1 or 2, but we noted that exon 1 (and therefore the mutational hotspot) was not covered by v.2.0 of Nimblegen SeqCap EZ Exome capture. By reducing the quality filters and the number of reads in family 4, we detected suggestive evidence of a missense mutation (c.101G>T [p.Gly34Val]; two reads) in SKI (RefSeq NM_003036.3). Sanger sequencing confirmed this mutation and the segregation in favor of a somatic mosaicism given that we found a lower level of the mutant allele in the asymptomatic mother. We performed PCR reactions on genomic DNA by using primers designed to amplify all seven exons and intron-exon boundaries of SKI in the remaining individuals from the SGS and non-SGS marfanoid-craniosynostosis cohorts (Table S5). After a variant was identified, the parents were secondarily studied when available. It was difficult to set up PCR conditions for the amplification of SKI as a result of GC-rich regions, particularly in exon 1. Because of the size of exon 1, it was necessary to design three pairs of primers (E1-1, E1-2, and E1-3), and a new pair of primers was necessary for achieving the sequencing of the hot-spot region (named E1-ATG, Table S5). PCR fragments were purified with the multiscreen Vacuum Manifold system (Millipore). Sequencing was performed with the ABI BigDye Terminator Cycle Sequencing kit (v.3.1) (Applied Biosystems) in ABI 3130 sequencer 7 (Applied Biosystems) according to the manufacturer’s instructions. Sequence data were analyzed with SeqScape v.2.7 (Applied Biosystems). The pathogenicity of missense mutations was tested with PolyPhen-2 and SIFT online software (see Web Resources).10 Screening of our entire cohort of SGS individuals revealed a total of ten de novo missense mutations, including somatic mosaicism in a family with recurrence in siblings and two overlapping in-frame deletions (one of them was dominantly inherited in a large family) (Table 1 and Tables S4 and S5), accounting for 18 of 19 cases tested. All mutations were found in the R-SMAD binding domain, affecting five conserved residues. Familial segregation and in silico prediction models were in favor of their pathogenicity (Table S4). A three-dimensional protein modeling was realized with Phyre2 software (see Web Resources). An automatic modeling script with standard parameters in the Phyre2 pipeline was used for generating the Protein Data Bank file of the protein. Overall, 83% of residues were modeled at >90% confidence, and 104 residues were modeled ab initio. A detailed description of the protein-modeling results is provided in Figure 3. We also sequenced SKI in the second cohort of individuals with a marfanoid-craniosynostosis phenotype incompatible with SGS but found no variant, thus further highlighting the phenotypic and genetic specificity of SGS.
Figure 2.
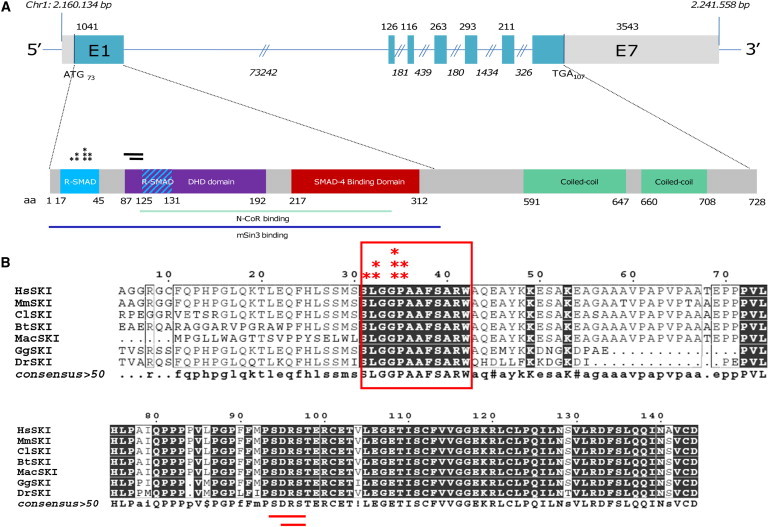
Location of SGS-Associated Mutations in SKI
(A) Schematic representation of the seven coding exons of SKI (top). The 5′ and 3′ UTRs are denoted in light gray. Exon 1 encodes the N-terminal R-SMAD- and SMAD- binding domains (blue and red box, respectively, at the bottom) and the DHD domain (purple box), and the remaining exons encode the C terminus with its two coiled-coil domains (green boxes at the bottom). Sites for interaction with N-CoR and mSin3 are also shown as light blue and dark blue lines, respectively. All mutations (asterisks for missense variants and lines for deletions) are located in the R-SMAD binding domain.
(B) Highly conserved amino acid residues (indicated in dark boxes) are conserved in vertebrates. All mutations affect highly conserved residues. The following abbreviations are used: Hs, Homo sapiens; Ms, Mus musculus; Cf, Canis familiaris; Bt, Bos Taurus; Mac, Macropus eugeneii; Gg, Gorilla gorilla; and Dr, Danio rerio.
Figure 3.
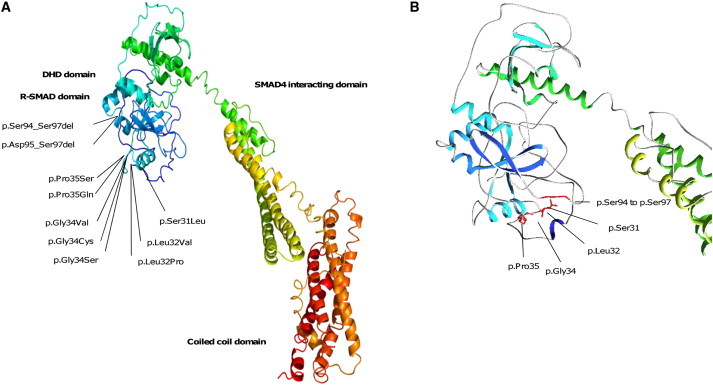
Three-Dimensional Modeling of SKI
(A) Functional domains of wild-type protein composed of an N-terminal DNA transcriptional regulating domain (dark blue) including R-SMAD (light blue) and DHD domains, a central SMAD4-interacting domain (greenish yellow), and a C-terminal coiled-coil domain (red).
(B) Enlargement of the region affected by all the mutations. The in-frame deletions shorten a loop (between residues 92 and 97). The missense mutations disrupt a flexible region (residues 31–35). All the mutations are localized on the same surface of the R-SMAD-binding domain.
Here, we report the identification of heterozygous exon 1 SKI mutations in 18 cases presenting with the characteristic features of SGS. The identification of recurrent heterozygous mutations in a specific area of exon 1 will facilitate genetic screening and help genetic counseling. Our results also feature information useful in clinical care because three individuals of the SGS cohort presented with aortic dilation; one such individual had vertebrobasilar and internal carotid tortuosity and a dilated pulmonary-artery root, further highlighting the overlap between SGS and Loeys-Dietz syndrome (Table 1).11 Therefore, a transthoracic echocardiogram, as well as imaging by computed tomography or magnetic resonance imaging of the neck, thorax, abdomen, and pelvis, can be justified. All of the individuals with SKI mutations had intellectual disability, supporting the hypothesis that SGS and Furlong syndrome should be separate.12 Given the absence of SKI mutation from the second cohort with non-SGS marfanoid craniosynostosis, we can conclude that other gene(s) remain to be determined for other types of marfanoid-craniosynostosis syndromes.
Several lines of evidence implicate the TGF-β pathway in marfanoid habitus, and SGS-affected individuals present with severe marfanoid habitus, allowing us to apply a biological filter strategy to select variants in the TFG-β pathway.9 SKI is an outstanding candidate gene because it encodes a ubiquitous transcription factor with a precise pattern of spatiotemporal constitutional expression and is implicated in promoting differentiation and maturation of chondrocyte cells and inhibiting proliferation of cells. SKI is implicated in the repression of TGF-β signaling, mainly through inhibition of SMAD2 phosphorylation, and competes with pSMAD3-SMAD4 binding and recruiting transcriptional repressor proteins such as N-CoR and mSIN3 (Figures 2 and 3 and Figure S2).13–16 Mutations found in the reported individuals affect the SMADs interacting domain and the transcription regulation domain DHD. All the mutated residues induce polarity changes and are located on the same structural surface, suggesting modification of the binding properties of SKI to the SMADs. The identified mutations in the SMAD interacting domains could lead to abnormal transcriptional repression of the downstream TFG-β signaling.16 Furthermore, Ski−/− mice display a lethal phenotype with associated midline facial cleft, a depressed nasal bridge, eye anomalies, skeletal muscle defects, and digital anomalies.17 Besides the mouse knockout model, the other major argument linking the SGS phenotype with SKI mutations is the role of SKI in the TGF-β pathway, a role which has been implicated in marfanoid habitus. It has been shown that the regulation of TGF-β signaling by SKI plays an important role in chondrocyte differentiation and maturation.18 Because the SMAD4-SKI complex modulates the transcription of genes regulated by TGF-β signaling, missense mutations within SMAD-interacting domains could lead to abnormal transcriptional repression of the downstream TGF-β-signaling genes in SGS (Figure 2 and Figure S2).
Interestingly, the hallmark of most diseases with defects in the TGF-β pathway is the high risk of developing thoracic aortic aneurysms (TAAs), although aortic complications seem less frequent in the SGS cohort.1 However, this finding could be explained by the young age of the reported individuals. Recently, functional studies in SMAD3 mutants raised the hypothesis that the ERK noncanonical TGF-β pathway could be implicated in TAAs. A crucial pathophysiologic distinction between canonical and noncanonical pathway activation points to the importance of the chronic activation of the noncanonical TGF-β pathway in the development of vascular symptoms in marfanoid syndromes.19 In Myhre syndrome (MIM 139210), SMAD4 mutations in the mad homology 2 domain protect mutant SMAD4 complexes from ubiquitination and impair the expression of TGF-β-driven target genes.20,21 Accordingly, the increased accumulation of SMAD4 in Myhre syndrome results in developmental delay and short stature and has no known risk of TAAs.21 Further studies would be useful for better understanding this aspect of the disease.
Myhre syndrome and SGS are the only TGF-β-pathway-related syndromes associated with ID. This feature can be explained in SGS given that SKI is necessary for neuronal proliferation and maturation and has been designated as a critical gene for ID in 1p36 telomeric deletion. Indeed, expression of SKI has been reported to be regulated by axon-Schwann-cell interactions and to be a crucial signal in Schwann cell development and myelination.22 The SKI/SnoN domain of Drosophila melanogaster and Caenorhabditis elegans was shown to be necessary for the proper cellular differentiation of neuronal progenitors.23 Moreover, Baranek et al. also showed that SKI, as a repressor of the TGF-β pathway, modulates its action during cortical development through recruitment of the Sin3/HDAC complex to SMADs and thereby fine tunes the balance between proliferation and differentiation of progenitor cells.24
In conclusion, our findings show that in-frame mutations in exon 1 of SKI cause SGS. Additional studies are necessary for elucidating the region-specific and tissue-specific consequences of defective SKI-mediated TGF-β signaling. Furthermore, because SKI mutations could not be identified in a cohort with non-SGS marfanoid craniosynotosis, mutations in yet-to-be-identified genes are most likely responsible for other types of marfanoid-craniosynostosis syndromes.
Acknowledgments
The authors thank the GIS-Institut des Maladies Rares for funding of the high-throughput-sequencing approach of the targeted region, the French Ministry of Health (PHRC national 2008) and Regional Council of Burgundy for their financial support of the project, the Genoscope (especially Vincent Meyer) and IntegraGen for technical assistance, and the families. The authors also thank Valérie Serre for her helpful comments regarding protein modeling. B.C. and J.D.B. are, respectively, postdoctoral and senior clinical researchers from the Fund for Scientific Research, Flanders. A.D.P. is a holder of a Methusalem grant (BOF 08/01M01108) from Ghent University and the Flemish government. Finally, the authors would like to thank the National Heart, Lung, and Blood Institute Grand Opportunity (GO) Exome Sequencing Project and its ongoing studies, which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the Women’s Health Initiative Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010). The authors thank Julie Plaisancié for the phenotyping of family 3.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Broad Institute Integrated Genomics Viewer, http://www.broadinstitute.org/igv/
Ensembl, http://www.ensembl.org/tools.html
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
GeneDistiller 2, http://www.genedistiller.org/
GeneReviews, Greally, M.T. (1993). Shprintzen-Goldberg Syndrome, http://www.ncbi.nlm.nih.gov/books/NBK1277/
Genoscope, https://www.genoscope.cns.fr/
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Open Astex Viewer, http://openastexviewer.net/web/
Phyre2, www.sbg.bio.ic.ac.uk/phyre2/
Picard, http://picard.sourceforge.net/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Primer3, http://frodo.wi.mit.edu/primer3/
SeattleSeq Annotation 131, http://snp.gs.washington.edu/SeattleSeqAnnotation131/
References
- 1.Robinson P.N., Neumann L.M., Demuth S., Enders H., Jung U., König R., Mitulla B., Müller D., Muschke P., Pfeiffer L. Shprintzen-Goldberg syndrome: fourteen new patients and a clinical analysis. Am. J. Med. Genet. A. 2005;135:251–262. doi: 10.1002/ajmg.a.30431. [DOI] [PubMed] [Google Scholar]
- 2.Shprintzen R.J., Goldberg R.B. A recurrent pattern syndrome of craniosynostosis associated with arachnodactyly and abdominal hernias. J. Craniofac. Genet. Dev. Biol. 1982;2:65–74. [PubMed] [Google Scholar]
- 3.Adès L.C., Morris L.L., Power R.G., Wilson M., Haan E.A., Bateman J.F., Milewicz D.M., Sillence D.O. Distinct skeletal abnormalities in four girls with Shprintzen-Goldberg syndrome. Am. J. Med. Genet. 1995;57:565–572. doi: 10.1002/ajmg.1320570410. [DOI] [PubMed] [Google Scholar]
- 4.Kosaki K., Takahashi D., Udaka T., Kosaki R., Matsumoto M., Ibe S., Isobe T., Tanaka Y., Takahashi T. Molecular pathology of Shprintzen-Goldberg syndrome. Am. J. Med. Genet. A. 2006;140:104–108. doi: 10.1002/ajmg.a.31006. author reply 109–110. [DOI] [PubMed] [Google Scholar]
- 5.Van Lierde K.M., Mortier G., Loeys B., Baudonck N., De Ley S., Marks L.A., Van Borsel J. Overall intelligibility, language, articulation, voice and resonance characteristics in a child with Shprintzen-Goldberg syndrome. Int. J. Pediatr. Otorhinolaryngol. 2007;71:721–728. doi: 10.1016/j.ijporl.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matthews L., Gopinath G., Gillespie M., Caudy M., Croft D., de Bono B., Garapati P., Hemish J., Hermjakob H., Jassal B. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009;37(Database issue):D619–D622. doi: 10.1093/nar/gkn863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loeys B.L., Chen J., Neptune E.R., Judge D.P., Podowski M., Holm T., Meyers J., Leitch C.C., Katsanis N., Sharifi N. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 12.Mégarbané A., Hokayem N. Craniosynostosis and marfanoid habitus without mental retardation: Report of a third case. Am. J. Med. Genet. 1998;77:170–171. [PubMed] [Google Scholar]
- 13.Wilson J.J., Malakhova M., Zhang R., Joachimiak A., Hegde R.S. Crystal structure of the dachshund homology domain of human SKI. Structure. 2004;12:785–792. doi: 10.1016/j.str.2004.02.035. [DOI] [PubMed] [Google Scholar]
- 14.Denissova N.G., Liu F. Repression of endogenous Smad7 by Ski. J. Biol. Chem. 2004;279:28143–28148. doi: 10.1074/jbc.M404961200. [DOI] [PubMed] [Google Scholar]
- 15.Nomura T., Khan M.M., Kaul S.C., Dong H.D., Wadhwa R., Colmenares C., Kohno I., Ishii S. Ski is a component of the histone deacetylase complex required for transcriptional repression by Mad and thyroid hormone receptor. Genes Dev. 1999;13:412–423. doi: 10.1101/gad.13.4.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deheuninck J., Luo K. Ski and SnoN, potent negative regulators of TGF-beta signaling. Cell Res. 2009;19:47–57. doi: 10.1038/cr.2008.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colmenares C., Heilstedt H.A., Shaffer L.G., Schwartz S., Berk M., Murray J.C., Stavnezer E. Loss of the SKI proto-oncogene in individuals affected with 1p36 deletion syndrome is predicted by strain-dependent defects in Ski-/- mice. Nat. Genet. 2002;30:106–109. doi: 10.1038/ng770. [DOI] [PubMed] [Google Scholar]
- 18.Kim K.O., Sampson E.R., Maynard R.D., O’Keefe R.J., Chen D., Drissi H., Rosier R.N., Hilton M.J., Zuscik M.J. Ski inhibits TGF-β/phospho-Smad3 signaling and accelerates hypertrophic differentiation in chondrocytes. J. Cell. Biochem. 2012;113:2156–2166. doi: 10.1002/jcb.24089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holm T.M., Habashi J.P., Doyle J.J., Bedja D., Chen Y., van Erp C., Lindsay M.E., Kim D., Schoenhoff F., Cohn R.D. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358–361. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Goff C., Cormier-Daire V. From tall to short: The role of TGFβ signaling in growth and its disorders. Am. J. Med. Genet. C. Semin. Med. Genet. 2012;160C:145–153. doi: 10.1002/ajmg.c.31337. [DOI] [PubMed] [Google Scholar]
- 21.Le Goff C., Mahaut C., Abhyankar A., Le Goff W., Serre V., Afenjar A., Destrée A., di Rocco M., Héron D., Jacquemont S. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat. Genet. 2012;44:85–88. doi: 10.1038/ng.1016. [DOI] [PubMed] [Google Scholar]
- 22.Atanasoski S., Notterpek L., Lee H.Y., Castagner F., Young P., Ehrengruber M.U., Meijer D., Sommer L., Stavnezer E., Colmenares C., Suter U. The protooncogene Ski controls Schwann cell proliferation and myelination. Neuron. 2004;43:499–511. doi: 10.1016/j.neuron.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 23.Anderson J., Salzer C.L., Kumar J.P. Regulation of the retinal determination gene dachshund in the embryonic head and developing eye of Drosophila. Dev. Biol. 2006;297:536–549. doi: 10.1016/j.ydbio.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baranek C., Atanasoski S. Modulating epigenetic mechanisms: The diverse functions of Ski during cortical development. Epigenetics. 2012;7:676–679. doi: 10.4161/epi.20590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.