

Abstract
Already 40 genes have been identified for autosomal-recessive nonsyndromic hearing impairment (arNSHI); however, many more genes are still to be identified. In a Dutch family segregating arNSHI, homozygosity mapping revealed a 2.4 Mb homozygous region on chromosome 11 in p15.1-15.2, which partially overlapped with the previously described DFNB18 locus. However, no putative pathogenic variants were found in USH1C, the gene mutated in DFNB18 hearing impairment. The homozygous region contained 12 additional annotated genes including OTOG, the gene encoding otogelin, a component of the tectorial membrane. It is thought that otogelin contributes to the stability and strength of this membrane through interaction or stabilization of its constituent fibers. The murine orthologous gene was already known to cause hearing loss when defective. Analysis of OTOG in the Dutch family revealed a homozygous 1 bp deletion, c.5508delC, which leads to a shift in the reading frame and a premature stop codon, p.Ala1838ProfsX31. Further screening of 60 unrelated probands from Spanish arNSHI families detected compound heterozygous OTOG mutations in one family, c.6347C>T (p.Pro2116Leu) and c. 6559C>T (p.Arg2187X). The missense mutation p.Pro2116Leu affects a highly conserved residue in the fourth von Willebrand factor type D domain of otogelin. The subjects with OTOG mutations have a moderate hearing impairment, which can be associated with vestibular dysfunction. The flat to shallow “U” or slightly downsloping shaped audiograms closely resembled audiograms of individuals with recessive mutations in the gene encoding α-tectorin, another component of the tectorial membrane. This distinctive phenotype may represent a clue to orientate the molecular diagnosis.
Main Text
Autosomal-recessive nonsyndromic hearing impairment (arNSHI, MIM 220700) is genetically extremely heterogeneous. So far more than 70 loci have been described and for 41 of these also the mutated genes have been identified (The Hereditary Hearing loss Homepage).1 Studies on mice with spontaneous or chemically induced mutations have not only significantly contributed to the discovery of deafness genes but also to the understanding of the molecular mechanisms of hearing and the pathogenesis of deafness.2,3 The discovery of mutations in PTPRQ (MIM 603317) and LOXHD1 (MIM 613072) to underlie hearing impairment nicely demonstrates the importance of mouse mutants in this process.2,4–6
To identify novel deafness genes, we selected 102 familial cases from 44 families and 40 isolated cases with putative arNSHI, almost exclusively of Dutch descent, for homozygosity mapping. There were neither indications for nongenetic causes of the hearing impairment in these subjects nor for mutations in GJB2 (MIM 121011) or deletions in GJB6 (MIM 604418). The study was approved by the local medical ethics committee and adhered to the tenets of the Declaration of Helsinki. Also, written informed consent was obtained from all subjects or from both parents in case of children. The approach of homozygosity mapping in this cohort already led to the identification of four novel deafness genes.6–9 Here we report on our studies in a family of this cohort, W00-384, with four affected sibs. The parents were not aware of any consanguinity. Genomic DNA isolated from peripheral blood samples of all four affected subjects was used for high-resolution SNP genotyping with Affymetrix Genechip Genome-Wide Human Arrays 5.0. Data were analyzed with the Genotyping Console software (Affymetrix) for allele calling and the Partek Genomics Suite (Partek Incorporated) for calculation of the regions of homozygosity. In total, five homozygous regions larger than 1 Mb were found to be shared by all affected individuals, which varied in size from 1.19 to 2.40 Mb. The largest of these shared homozygous regions was located on chromosome 11 in p15.1-15.2 (see Figure S1 available online). This region was flanked by SNP_A-4222425 (rs10741672) and SNP_A-1805451 (rs4756919), and homozygosity was confirmed by genotyping small tandem repeat (STR) markers as shown in Figure S2. Thirteen genes have been annotated in this region that partially overlapped with the previously described DFNB18 locus (MIM 602092). The other four shared homozygous regions in family W00-384 neither contained a known human or mouse deafness gene nor did they overlap with a known deafness locus.
Mutations in USH1C (MIM 605242) have been described as the cause of DFNB18 related hearing impairment.10–12 Sequence analysis of all exons and exon-intron boundaries of this gene including the exons specifically expressed in the inner ear, did not reveal any potentially pathogenic variants. To determine whether compound heterozygous mutations in genes known to be involved in recessive and X-linked deafness might underlie the hearing impairment in this family, SNP genotypes of the affected males were compared for the corresponding regions. The four affected individuals carried the same genotype for the chromosomal region containing SMPX (MIM 300226) and for the region containing STRC (MIM 606440). Sequence analysis did not reveal any putatively causative variants in these genes.
Within the 2.4 Mb homozygous region, OTOG (MIM 604487) was a second excellent candidate gene. It encodes otogelin, a glycoprotein whose complementary DNA (cDNA) was initially identified from a cochlea-derived substracted cDNA library.13 Otogelin is a noncollagenous component of the acellular gelatinous structures that cover the sensory epithelia of the inner ear, i.e., the tectorial membrane (TM) in the cochlea, the otoconial membranes in the utricle and saccule, and the cupulae that cover the cristae ampullares of the semicircular canals in the vestibular organ.13 Defects in the orthologous gene in mouse result in hearing loss and severe imbalance.14,15 OTOG is not fully annotated in the UCSC Genome Browser (GRCh37/hg19). The UCSC gene uc001mnh.1 consists of 22 exons of which 21 are protein coding. However, the NCBI gene database reports the larger transcript XM_291816.8, which corresponds to the Ensemble transcript ENST00000399391 and encodes a protein of 2,925 amino acids. The orthologous gene in mouse encodes a protein of 2,911 amino acids. With RT-PCR we were able to detect transcripts with exons 26–27 and 54–55 in addition to exons 14–15 in fetal inner ear (Table S1; Figure S3). Sanger sequencing of all 55 exons represented in XM_291816.8 was performed as described.9 Primer sequences and PCR conditions are provided in Table S1. A homozygous 1 bp deletion was identified in exon 35, c.5508delC (reference sequence XM_291816.8), which is predicted to cause a frameshift and a premature stop codon, p.Ala1838ProfsX31 (Figure 1A). This variant was found in homozygous state in the four affected sibs and in heterozygous state in the parents. It was not found in 178 ethnically matched healthy controls by using restriction digestion with NlaIV of the exon 35_2 amplicon. The mutation removes an NlaIV restriction site. The c.5508delC variant was also not reported in the 1000 Genomes Project. To exclude mutations in any of the additional 11 genes within the homozygous region (according to UCSC Human Genome Database build hg19; Figure S1) as the cause of the hearing impairment in this family, the genes were analyzed by Sanger sequencing. No putative causative variants were identified, only variants also present in dbSNP as shown in Table S2.
Figure 1.
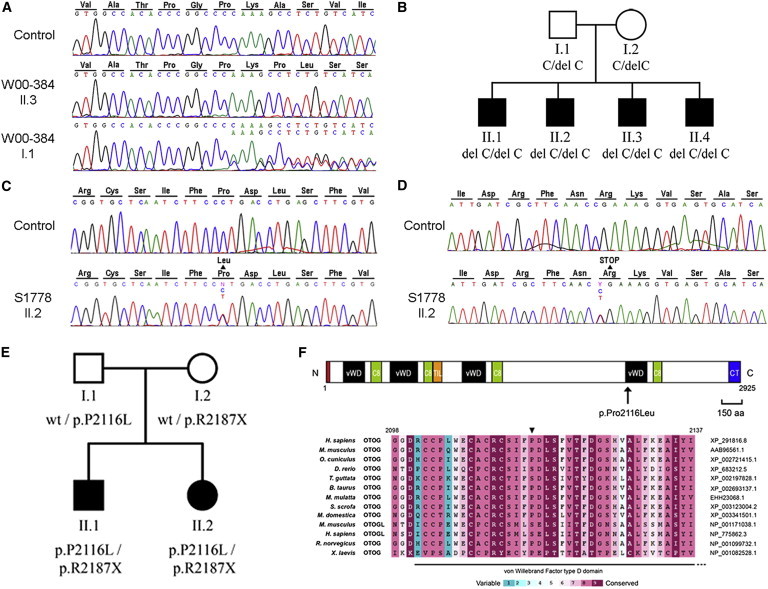
Mutation Analysis of OTOG
(A) Partial sequences are shown of OTOG exon 35 from an affected member and a parent of family W00-384 and a normal control. The amino acids are indicated above the sequence. The c.5508delC variant found is predicted to result in a frameshift and premature stop codon, p.Ala1838ProfsX31 (reference sequence XM_291816.8).
(B) Pedigree of family W00-384 and segregation of the c.5508delC variant.
(C) Partial sequences of OTOG exon 37 from a normal-hearing control and affected subject S1778 II.2, showing the c.6347C>T (p.Pro2116Leu) substitution.
(D) Partial sequences of OTOG exon 38 from a normal-hearing control and affected subject S1778 II.2, showing the c.6559C>T (p.Arg2187X) nonsense mutation.
(E) Pedigree of Spanish family S1778 and segregation analysis of the c.6347C>T and c.6559C>T mutations.
(F) Localization and conservation of the Pro-2116 residue. Above are shown domains of human otogelin. Red box, N-terminal signal peptide; vWD, von Willebrand factor type D domain; C8, domain containing eight conserved cysteine residues; TIL, trypsin inhibitor-like domain; CT, cystine knot-like domain. The localization of the p.Pro2116Leu missense mutation is indicated by an arrow. Below are shown alignment of diverse vertebrate otogelin (OTOG) and otogelin-like (OTOGL) amino acid sequences (accession numbers are shown on the right). ConSeq conservation scores are shown by the color scale. Residue Pro-2116 (arrowhead), located in the fourth von Willebrand factor type D domain, is highly conserved (score 8).
In a parallel and independent approach, OTOG was investigated in a cohort of 60 Spanish families with arNSHI, which was moderate (24 families), severe (9 families), or profound (27 families). All of them had at least two affected siblings, and the presence of GJB2 mutations or GJB6 deletions had been excluded by routine testing. After approval of the study by the local medical ethics committee, written informed consent was obtained from all participants or their parents (in case of children). Siblings and parents were genotyped for STR-markers D11S902, D11S4138, and D11S4130, which flank OTOG. Haplotype analysis revealed compatibility with genetic linkage to these markers in seven families. Sequencing of all 55 OTOG exons and exon/intron boundaries in the index case of each pedigree revealed five already known polymorphic variants and two novel mutations: c.6347C>T (p.Pro2116Leu) in exon 37 and c.6559C>T (p.Arg2187X) in exon 38 (reference sequence XM_291816.8). These two mutations were in compound heterozygous state in the two affected subjects of nonconsanguineous family S1778 (Figures 1C–1E). Parents were heterozygous carriers, confirming that these mutations were in trans in the affected subjects. These variants were not found in 315 normal-hearing Spanish subjects with sequence analysis of exons 37 and 38, and they were not reported in the 1000 Genomes Project. The missense mutation p.Pro2116Leu affects a highly conserved residue (ConSeq score of 8, The ConSeq Server) in the fourth von Willebrand factor type D domain of otogelin (Figure 1F). Pathogenicity of this mutation is also supported by predictions from specialized software that classified it as very likely damaging (score 0 in SIFT, score 0.97 in PolyPhen-2). A hypothetical pathogenic role of the adjacent USH1C gene was excluded by sequencing all its exons and exon-intron boundaries in one affected subject of family S1778. Also, all other known arNSHI genes were excluded in family S1778 by haplotype analysis of flanking VNTR markers or by Sanger sequencing for those genes that could not be excluded based on the VNTR haplotypes. The latter set of genes included TMC1 (MIM 606706), OTOF (MIM 603681), CDH23 (MIM 605516), PCDH15 (MIM 605514), GRXCR1 (MIM 613283), TRIOBP (MIM 609761), MYO3A (MIM 606808), WHRN (MIM 607928), ESRRB (MIM 602167), MARVELD2 (MIM 610572), PJVK (MIM 610219), LRTOMT (MIM 612414), BSND (MIM 606412), LOXHD1, TPRN (MIM 613354), and GJB3 (MIM 603324). No potentially pathogenic variants were found.
To further study the involvement of OTOG in arNSHI we screened a panel of 85 Dutch subjects with presumably arNSHI for presence of the c.5508delC variant. These subjects were not preselected based on type or severity of their hearing impairment. Mutations in GJB2 and GJB6 deletions were excluded in these individuals. No carriers of the c.5508delC variant were found in this panel. Also, screening of OTOG in 12 index patients of Dutch origin with a similar audiogram as seen in subjects with OTOG mutations did not reveal any putatively causative variants. The same was true for probands of 13 families of Turkish origin (1–3 affected subjects per family) in which the affected individuals were homozygous for STR-markers D11S902 and D11S4138 that flank OTOG.
In mice, Otog is specifically expressed in inner ear.13 We studied the expression of OTOG in human tissues by performing quantitative PCR (qPCR) on cDNA from various fetal and adult human tissues as described (Figure 2).9 Among the tested fetal tissues, the transcript level was highest in inner ear, followed by kidney, lung, spleen, thymus, and liver. Transcript levels were below detection level in fetal heart, skeletal muscle, brain, colon, and stomach. In cDNA derived from tested adult tissues, OTOG expression was detected with the highest level in retina followed by testis and heart. These levels were lower than in fetal inner ear, which was included in the same experiment for comparison. Although OTOG transcription is detected in several tissues, subjects with mutations in OTOG only reported symptoms associated with inner ear defects.
Figure 2.
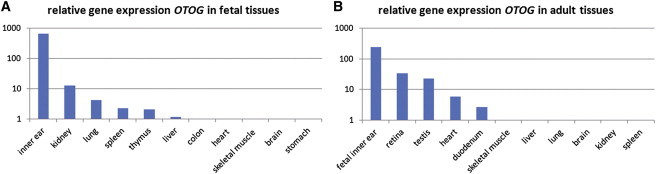
OTOG Expression Profile in Human Tissues
Relative OTOG mRNA levels as determined by qPCR in fetal (A) and adult (B) tissues. Because this was performed for adult and fetal tissues in two separate experiments, fetal inner ear was included in both to be able to compare the expression levels. The delta Ct method was used to calculate the expression levels. To calculate the relative expression, we set the tissue with the lowest detectable expression (Ct < 35) to 1. In the adult tissues, the expression in colon was set to 1 and in fetal tissues the expression in kidney. RNA was isolated from inner ear of an embryo at 8 weeks gestation; the other fetal RNA samples were commercially available (all adult tissues from Stratagene; fetal heart, brain, and kidney from Clontech, fetal skeletal muscle, liver, lung, spleen, thymus, colon, and stomach from Stratagene) and derived from embryos at 20–21 weeks of gestation.
The four affected subjects of W00-384 all had a flat to shallow U-shaped audiogram (Figure 3A). Three out of these four affected individuals (II.1, II.2, and II.4) exhibited a delayed speech development, which suggests a prelingual onset of the hearing impairment. Subject II.1 was referred for pure tone audiometry at the age of 7.5 years, and a hearing loss of 40–45 dB was measured. Subsequently, the three younger children were tested, which revealed thresholds of 35–40 dB (subject II.2, 5.5 yrs), 40 dB (subject II.3, 3.5 yrs), and 50 dB (subject II.4, 2 yrs). Representative audiograms of all four hearing impaired subjects of W00-384 are presented in Figure 3A. For subjects II.2 and II.4 of family W00-384, a delayed motor development was reported, suggesting vestibular problems at young age. Subject II.2 started to crawl and sit only at 12 months of age, to stand at 14 months, and to walk at 18 months. Subject II.4 had delayed milestones for crawling (11 months), rolling over (12 months), sitting (12 months), standing (14 months), and walking (>24 months). Rotary chair and caloric tests performed at the ages of 19 yrs, 17 yrs, 15 yrs, and 13 yrs for II.1, II.2, II.3, and II.4, respectively, indeed demonstrated vestibular hyporeflexia in all four affected subjects. A computed tomography (CT) scan in subject II.2 revealed no inner ear abnormalities.
Figure 3.
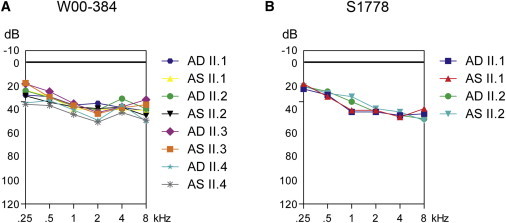
Audiometric Characterization of OTOG Families
(A) Binaural mean air-conduction pure tone threshold values of affected members of family W00-384. These represent mean values of audiograms at different ages for each affected individual; II.1 (7.2–19.6 yrs), II.2 (5.4–17.2 yrs), II.3 (3.5–15.3 yrs), and II.4 (3.9–13.8 yrs).
(B) Mean air-conduction pure tone threshold values of family S1778. These represent mean values of audiograms at different ages for each affected individual; 5–30 years for subject II.1 and 6–24 years for subject II.2. AD (aurio dextra) and AS (aurio sinister) represent the right and left ear, respectively.
The two affected subjects of family S1778 had bilateral hearing impairment, which was sensorineural, moderate, and stable, as supported by serial audiograms at different ages. The shape of the audiograms was slightly down-sloping across all frequencies (Figure 3B). Although the clinical diagnoses were established at ages 4 (subject II.1) and 5 years (subject II.2), speech development was delayed in both subjects, which suggests a prelingual onset. The affected individuals had a normal motor development, and they have not complained of dizziness or walking instability during adulthood. A putative subclinical vestibular dysfunction was investigated. Responses obtained from oculomotor testing (saccade, optokinetics) and rotatory chair testing by videonystagmography were normal in both subjects. However, bithermal caloric testing did reveal a bilateral deficit.
Two mouse models have been described for OTOG, the Otogtm1Prs mouse with targeted disruption of Otog by deletion of the first three exons and the twister (twt) mouse with a spontaneous recessive mutation leading to the absence of Otog expression.14,15 Both mouse models have a very similar phenotype with deafness and severe imbalance.14,15 The hearing loss is bilateral and highly progressive after birth, ranging from mild to profound.15 Otog−/− pups start to show signs of vestibular dysfunction as early as postnatal day (P)4.15
Unfortunately audiometric data before the age of 3.5 yrs was missing for both families W00-384 and S1778. Therefore, it is not clear whether, comparable to the Otog mouse models, an early progressive hearing loss was preceding the later stable phase of the hearing loss in the affected human subjects. For all affected subjects of both families, the hearing loss was stable since the first audiogram. The delayed motor development in two of the affected subjects from family W00-384 suggests early vestibular dysfunction. Vestibular function was affected in all four subjects of W00-384 as confirmed by clear absence of asymmetry of the directional nystagmus preponderance meausured with rotational chair in all four subjects. On the contrary, motor development in the two affected subjects of family S1778 was normal and only bicaloric thermal testing (performed in adulthood) revealed an anomaly suggestive of a subclinical vestibular dysfunction. The differences observed between humans and mice suggest that central compensation may be more efficient in humans, as reported for other audiovestibular conditions. The variability in vestibular dysfunction among the subjects in this study might be related to their different OTOG genotypes (homozygous truncating mutations versus heterozygous truncating/nontruncating mutations) and/or to other hypothetical modifier factors.
Otogelin is one of the three main noncollagenous components of the TM besides α-tectorin (TECTA; MIM 602574) and β-tectorin (TECTB; MIM 602653). Also, collagen types II, V, IX, and XI are main components of the TM.16 In addition, a number of less abundant TM proteins have been identified, e.g., CEACAM16. Moderate hearing impairment with a U-shaped to flat audiogram is the most common type of nonsyndromic hearing impairment associated with defects in components of the TM. This is seen for recessive TECTA mutations with one exception known,17 dominant CEACAM16 (MIM 614591) mutations, dominant COL11A2 (MIM 120290) mutations, and now also for recessive OTOG mutations.17–21 Recessive mutations in STRC (MIM 606440), which encodes stereocilin that functions in attaching the outer hair cell stereocilia to the TM, also result in mild-to-moderate hearing impairment.22,23 Exceptions are recessive mutations in COL11A2. However, this gene has a much broader expression pattern in the inner ear than, for example, TECTA or OTOG.20, 24 Besides recessive mutations, also dominant mutations occur in TECTA resulting in moderate mid-frequency or high-frequency hearing impairment depending on the protein domain affected by the mutation.25–29 Because the severity of hearing loss associated with recessive TECTA and OTOG mutations is very similar and the translated proteins are both part of the TM, OTOG might also be a candidate gene for dominant hearing loss. So far, no TECTB (MIM 602653) mutations have been described for humans. Based on the presence of β-tectorin in the TM, TECTB is a good candidate gene for moderate to severe hearing loss with a flat to a shallow U-shaped audiogram; Tectb−/− mice, however, display low-frequency hearing loss.30
The precise function of otogelin in the gelatinous structures overlying the sensory epithelia of the inner ear is still elusive. In mouse, Otog is expressed prenatally both in the cochlea and the vestibular organ and our qPCR data indicate an early expression in humans as well (week 8 of gestation).31 In mouse, expression of otogelin is higher in the vestibular organ as compared to the cochlea.15,31 Otogelin is not required for the formation of the acellular gelatinous membranes of the inner ear because these are all formed in complete absence of otogelin in mice. However, detachment of the otoconial membranes and cupulae from the sensory epithelia in the vestibule demonstrates that otogelin is required for anchoring of these membranes to the epithelia.15 This is not the case for the TM in the cochlea. The precise localization of otogelin in the TM is still unclear. Ultrastructural analysis of the TM of Otog−/− mice demonstrated that all components of the TM are formed including the type-A and type-B fibers. However, because of the presence of some abnormal fibrilar or rod-like structures in the TM of these Otog-deficient mice, otogelin was proposed to be implicated in the interaction or stabilization of the fibers in the TM.15 Multiple roles for the TM have recently been identified using Tecta-deficient or mutant mice.32–36 The TM ensures that outer hair cells can effectively respond to basilar membrane motion and that feedback is delivered with the appropriate gain and timing required for amplification.33,35 A second role for the TM is that it enables the motion of the basilar membrane to optimally drive the inner hair cells at their best frequencies.34 Similar detailed investigations of the properties of the TM in Otog−/− mice could provide more insight into the function of otogelin in the TM.
In conclusion, we report the identification of mutations in OTOG as a cause of moderate nonsyndromic hearing loss. The shapes of the audiograms closely resemble those associated with recessive TECTA mutations and can be a clue to orientate molecular diagnostic testing. Recessive TECTA mutations have not been reported to cause vestibular symptoms, which are present in some subjects with OTOG mutations. This phenotypic difference can be a further clue to directed molecular diagnostic testing.
Acknowledgments
We are grateful to the families for their participation in this study. This work was financially supported by grants from the Heinsius Houbolt Foundation (to H.K.), the INTERREG IV A-program Germany-the Netherlands, The Oticon Foundation (09-3742, to H.K.), ZonMW (40-00812-98-09047, to H.K. and 90700388 to R.J.E.P.), Netherlands Genomics Initiative (40-41009-98-9073, to M.S.), Fondo de Investigaciones Sanitarias (PI 08/0818 and PI 11/00612, to I.d.C.), Fundación Ramón Areces (to I.d.C.), and Spanish Ministerio de Ciencia e Innovación (SAF2008-03216, to F.M.) and Karadeniz Technical University Research Fund (2006.114.001.1 to E.K.). L.R.-P. was a recipient of a predoctoral fellowship from Instituto de Salud Carlos III.
Supplemental Data
Web resources
The URLs for data presented herein are as follows:
NHLBI Exome Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim
Polyphen, http://genetics.bwh.harvard.edu/pph/
Sorting Intolerant from Tolerant (SIFT), http://sift.jcvi.org/
The 1000 Genomes Browser, http://browser.1000genomes.org/index.html
The ConSeq Server, http://conseq.bioinfo.tau.ac.il/
The Ensembl Project, http://www.ensembl.org/index.html
The Hereditary Hearing loss Homepage, http://hereditaryhearingloss.org/
The National Center for Biotechnology, http://www.ncbi.nlm.nih.gov/
UCSC Human Genome Database Build hg19, February 2009, http://www.genome.ucsc.edu
References
- 1.Lenz D.R., Avraham K.B. Hereditary hearing loss: from human mutation to mechanism. Hear. Res. 2011;281:3–10. doi: 10.1016/j.heares.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 2.Brown S.D., Hardisty-Hughes R.E., Mburu P. Quiet as a mouse: dissecting the molecular and genetic basis of hearing. Nat. Rev. Genet. 2008;9:277–290. doi: 10.1038/nrg2309. [DOI] [PubMed] [Google Scholar]
- 3.Dror A.A., Avraham K.B. Hearing impairment: a panoply of genes and functions. Neuron. 2010;68:293–308. doi: 10.1016/j.neuron.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Goodyear R.J., Legan P.K., Wright M.B., Marcotti W., Oganesian A., Coats S.A., Booth C.J., Kros C.J., Seifert R.A., Bowen-Pope D.F., Richardson G.P. A receptor-like inositol lipid phosphatase is required for the maturation of developing cochlear hair bundles. J. Neurosci. 2003;23:9208–9219. doi: 10.1523/JNEUROSCI.23-27-09208.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grillet N., Schwander M., Hildebrand M.S., Sczaniecka A., Kolatkar A., Velasco J., Webster J.A., Kahrizi K., Najmabadi H., Kimberling W.J. Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am. J. Hum. Genet. 2009;85:328–337. doi: 10.1016/j.ajhg.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schraders M., Oostrik J., Huygen P.L., Strom T.M., van Wijk E., Kunst H.P., Hoefsloot L.H., Cremers C.W., Admiraal R.J., Kremer H. Mutations in PTPRQ are a cause of autosomal-recessive nonsyndromic hearing impairment DFNB84 and associated with vestibular dysfunction. Am. J. Hum. Genet. 2010;86:604–610. doi: 10.1016/j.ajhg.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Charizopoulou N., Lelli A., Schraders M., Ray K., Hildebrand M.S., Ramesh A., Srisailapathy C.R., Oostrik J., Admiraal R.J., Neely H.R. Gipc3 mutations associated with audiogenic seizures and sensorineural hearing loss in mouse and human. Nat. Commun. 2011;2:201. doi: 10.1038/ncomms1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y., Pohl E., Boulouiz R., Schraders M., Nürnberg G., Charif M., Admiraal R.J., von Ameln S., Baessmann I., Kandil M. Mutations in TPRN cause a progressive form of autosomal-recessive nonsyndromic hearing loss. Am. J. Hum. Genet. 2010;86:479–484. doi: 10.1016/j.ajhg.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schraders M., Lee K., Oostrik J., Huygen P.L., Ali G., Hoefsloot L.H., Veltman J.A., Cremers F.P., Basit S., Ansar M. Homozygosity mapping reveals mutations of GRXCR1 as a cause of autosomal-recessive nonsyndromic hearing impairment. Am. J. Hum. Genet. 2010;86:138–147. doi: 10.1016/j.ajhg.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ouyang X.M., Xia X.J., Verpy E., Du L.L., Pandya A., Petit C., Balkany T., Nance W.E., Liu X.Z. Mutations in the alternatively spliced exons of USH1C cause non-syndromic recessive deafness. Hum. Genet. 2002;111:26–30. doi: 10.1007/s00439-002-0736-0. [DOI] [PubMed] [Google Scholar]
- 11.Ahmed Z.M., Smith T.N., Riazuddin S., Makishima T., Ghosh M., Bokhari S., Menon P.S., Deshmukh D., Griffith A.J., Riazuddin S. Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC. Hum. Genet. 2002;110:527–531. doi: 10.1007/s00439-002-0732-4. [DOI] [PubMed] [Google Scholar]
- 12.Jain P.K., Lalwani A.K., Li X.C., Singleton T.L., Smith T.N., Chen A., Deshmukh D., Verma I.C., Smith R.J., Wilcox E.R. A gene for recessive nonsyndromic sensorineural deafness (DFNB18) maps to the chromosomal region 11p14-p15.1 containing the Usher syndrome type 1C gene. Genomics. 1998;50:290–292. doi: 10.1006/geno.1998.5320. [DOI] [PubMed] [Google Scholar]
- 13.Cohen-Salmon M., El-Amraoui A., Leibovici M., Petit C. Otogelin: a glycoprotein specific to the acellular membranes of the inner ear. Proc. Natl. Acad. Sci. USA. 1997;94:14450–14455. doi: 10.1073/pnas.94.26.14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simmler M.C., Zwaenepoel I., Verpy E., Guillaud L., Elbaz C., Petit C., Panthier J.J. Twister mutant mice are defective for otogelin, a component specific to inner ear acellular membranes. Mamm. Genome. 2000;11:961–966. doi: 10.1007/s003350010197. [DOI] [PubMed] [Google Scholar]
- 15.Simmler M.C., Cohen-Salmon M., El-Amraoui A., Guillaud L., Benichou J.C., Petit C., Panthier J.J. Targeted disruption of otog results in deafness and severe imbalance. Nat. Genet. 2000;24:139–143. doi: 10.1038/72793. [DOI] [PubMed] [Google Scholar]
- 16.Richardson G.P., Lukashkin A.N., Russell I.J. The tectorial membrane: one slice of a complex cochlear sandwich. Curr. Opin. Otolaryngol. Head Neck Surg. 2008;16:458–464. doi: 10.1097/MOO.0b013e32830e20c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mustapha M., Weil D., Chardenoux S., Elias S., El-Zir E., Beckmann J.S., Loiselet J., Petit C. An alpha-tectorin gene defect causes a newly identified autosomal recessive form of sensorineural pre-lingual non-syndromic deafness, DFNB21. Hum. Mol. Genet. 1999;8:409–412. doi: 10.1093/hmg/8.3.409. [DOI] [PubMed] [Google Scholar]
- 18.Naz S., Alasti F., Mowjoodi A., Riazuddin S., Sanati M.H., Friedman T.B., Griffith A.J., Wilcox E.R., Riazuddin S. Distinctive audiometric profile associated with DFNB21 alleles of TECTA. J. Med. Genet. 2003;40:360–363. doi: 10.1136/jmg.40.5.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Leenheer E.M., McGuirt W.T., Kunst H.P., Huygen P.L., Smith R.J., Cremers C.W. The phenotype of DFNA13/COL11A2. Adv. Otorhinolaryngol. 2002;61:85–91. doi: 10.1159/000066804. [DOI] [PubMed] [Google Scholar]
- 20.De Leenheer E.M., Bosman A.J., Kunst H.P., Huygen P.L., Cremers C.W. Audiological characteristics of some affected members of a Dutch DFNA13/COL11A2 family. Ann. Otol. Rhinol. Laryngol. 2004;113:922–929. doi: 10.1177/000348940411301112. [DOI] [PubMed] [Google Scholar]
- 21.Zheng J., Miller K.K., Yang T., Hildebrand M.S., Shearer A.E., DeLuca A.P., Scheetz T.E., Drummond J., Scherer S.E., Legan P.K. Carcinoembryonic antigen-related cell adhesion molecule 16 interacts with alpha-tectorin and is mutated in autosomal dominant hearing loss (DFNA4) Proc. Natl. Acad. Sci. USA. 2011;108:4218–4223. doi: 10.1073/pnas.1005842108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Francey L.J., Conlin L.K., Kadesch H.E., Clark D., Berrodin D., Sun Y., Glessner J., Hakonarson H., Jalas C., Landau C. Genome-wide SNP genotyping identifies the Stereocilin (STRC) gene as a major contributor to pediatric bilateral sensorineural hearing impairment. Am. J. Med. Genet. A. 2012;158A:298–308. doi: 10.1002/ajmg.a.34391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verpy E., Leibovici M., Michalski N., Goodyear R.J., Houdon C., Weil D., Richardson G.P., Petit C. Stereocilin connects outer hair cell stereocilia to one another and to the tectorial membrane. J. Comp. Neurol. 2011;519:194–210. doi: 10.1002/cne.22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W., Kahrizi K., Meyer N.C., Riazalhosseini Y., Van Camp G., Najmabadi H., Smith R.J. Mutation of COL11A2 causes autosomal recessive non-syndromic hearing loss at the DFNB53 locus. J. Med. Genet. 2005;42:e61. doi: 10.1136/jmg.2005.032615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alloisio N., Morlé L., Bozon M., Godet J., Verhoeven K., Van Camp G., Plauchu H., Muller P., Collet L., Lina-Granade G. Mutation in the zonadhesin-like domain of alpha-tectorin associated with autosomal dominant non-syndromic hearing loss. Eur. J. Hum. Genet. 1999;7:255–258. doi: 10.1038/sj.ejhg.5200273. [DOI] [PubMed] [Google Scholar]
- 26.Balciuniene J., Dahl N., Jalonen P., Verhoeven K., Van Camp G., Borg E., Pettersson U., Jazin E.E. Alpha-tectorin involvement in hearing disabilities: one gene—two phenotypes. Hum. Genet. 1999;105:211–216. doi: 10.1007/s004390051091. [DOI] [PubMed] [Google Scholar]
- 27.Govaerts P.J., De Ceulaer G., Daemers K., Verhoeven K., Van Camp G., Schatteman I., Verstreken M., Willems P.J., Somers T., Offeciers F.E. A new autosomal-dominant locus (DFNA12) is responsible for a nonsyndromic, midfrequency, prelingual and nonprogressive sensorineural hearing loss. Am. J. Otol. 1998;19:718–723. [PubMed] [Google Scholar]
- 28.Hildebrand M.S., Morín M., Meyer N.C., Mayo F., Modamio-Hoybjor S., Mencía A., Olavarrieta L., Morales-Angulo C., Nishimura C.J., Workman H. DFNA8/12 caused by TECTA mutations is the most identified subtype of nonsyndromic autosomal dominant hearing loss. Hum. Mutat. 2011;32:825–834. doi: 10.1002/humu.21512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plantinga R.F., de Brouwer A.P., Huygen P.L., Kunst H.P., Kremer H., Cremers C.W. A novel TECTA mutation in a Dutch DFNA8/12 family confirms genotype-phenotype correlation. J. Assoc. Res. Otolaryngol. 2006;7:173–181. doi: 10.1007/s10162-006-0033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russell I.J., Legan P.K., Lukashkina V.A., Lukashkin A.N., Goodyear R.J., Richardson G.P. Sharpened cochlear tuning in a mouse with a genetically modified tectorial membrane. Nat. Neurosci. 2007;10:215–223. doi: 10.1038/nn1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El-Amraoui A., Cohen-Salmon M., Petit C., Simmler M.C. Spatiotemporal expression of otogelin in the developing and adult mouse inner ear. Hear. Res. 2001;158:151–159. doi: 10.1016/s0378-5955(01)00312-4. [DOI] [PubMed] [Google Scholar]
- 32.Moreno-Pelayo M.A., Goodyear R.J., Mencía A., Modamio-Høybjør S., Legan P.K., Olavarrieta L., Moreno F., Richardson G.P. Characterization of a spontaneous, recessive, missense mutation arising in the Tecta gene. J. Assoc. Res. Otolaryngol. 2008;9:202–214. doi: 10.1007/s10162-008-0116-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Legan P.K., Lukashkina V.A., Goodyear R.J., Kössi M., Russell I.J., Richardson G.P. A targeted deletion in alpha-tectorin reveals that the tectorial membrane is required for the gain and timing of cochlear feedback. Neuron. 2000;28:273–285. doi: 10.1016/s0896-6273(00)00102-1. [DOI] [PubMed] [Google Scholar]
- 34.Legan P.K., Lukashkina V.A., Goodyear R.J., Lukashkin A.N., Verhoeven K., Van Camp G., Russell I.J., Richardson G.P. A deafness mutation isolates a second role for the tectorial membrane in hearing. Nat. Neurosci. 2005;8:1035–1042. doi: 10.1038/nn1496. [DOI] [PubMed] [Google Scholar]
- 35.Lukashkin A.N., Lukashkina V.A., Legan P.K., Richardson G.P., Russell I.J. Role of the tectorial membrane revealed by otoacoustic emissions recorded from wild-type and transgenic Tecta(deltaENT/deltaENT) mice. J. Neurophysiol. 2004;91:163–171. doi: 10.1152/jn.00680.2003. [DOI] [PubMed] [Google Scholar]
- 36.Gueta R., Levitt J., Xia A., Katz O., Oghalai J.S., Rousso I. Structural and mechanical analysis of tectorial membrane Tecta mutants. Biophys. J. 2011;100:2530–2538. doi: 10.1016/j.bpj.2011.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.