

Abstract
Multiple-respiratory-chain deficiency represents an important cause of mitochondrial disorders. Hitherto, however, mutations in genes involved in mtDNA maintenance and translation machinery only account for a fraction of cases. Exome sequencing in two siblings, born to consanguineous parents, with severe encephalomyopathy, choreoathetotic movements, and combined respiratory-chain defects allowed us to identify a homozygous PNPT1 missense mutation (c.1160A>G) that encodes the mitochondrial polynucleotide phosphorylase (PNPase). Blue-native polyacrylamide gel electrophoresis showed that no PNPase complex could be detected in subject fibroblasts, confirming that the substitution encoded by c.1160A>G disrupts the trimerization of the protein. PNPase is predominantly localized in the mitochondrial intermembrane space and is implicated in RNA targeting to human mitochondria. Mammalian mitochondria import several small noncoding nuclear RNAs (5S rRNA, MRP RNA, some tRNAs, and miRNAs). By RNA hybridization experiments, we observed a significant decrease in 5S rRNA and MRP-related RNA import into mitochondria in fibroblasts of affected subject 1. Moreover, we found a reproducible decrease in the rate of mitochondrial translation in her fibroblasts. Finally, overexpression of the wild-type PNPT1 cDNA in fibroblasts of subject 1 induced an increase in 5S rRNA import in mitochondria and rescued the mitochondrial-translation deficiency. In conclusion, we report here abnormal RNA import into mitochondria as a cause of respiratory-chain deficiency.
Main Text
Mitochondrial disorders represent a heterogeneous group of genetic diseases characterized by an oxidative-phosphorylation (OXPHOS) deficiency. OXPHOS is carried out by the mitochondrial respiratory chain (RC), a complex pathway linking cellular respiration to the synthesis of adenosine triphosphate. The RC consists of five complexes composed of more than 80 distinct subunits, 13 of which are encoded by mtDNA. Moreover, several hundred nuclear genes are also needed for various functions of the RC. OXPHOS deficiency results from either mitochondrial or nuclear gene mutations.1 Among them, multiple-RC deficiencies are associated with a variety of disease mechanisms that alter mtDNA maintenance, translation of mitochondrially encoded proteins, and cardiolipin synthesis.2 Several point mutations and large deletions of mtDNA have been shown to cause multiple-RC deficiency. However, an increasing number of disease mutations are being identified in this group of mitochondrial diseases. Almost all of them encode mitochondrial proteins that are synthesized within the cytosol and further imported into the mitochondria. Mitochondria import not only proteins but also a few small RNAs that are essential for replication, transcription, and translation of the mitochondrial genome. Almost all species, including humans, import small RNAs. 5S rRNA (MIM 180420) is the most abundant imported RNA in human mitochondria3 and is critical for mitochondrial translation. Two other RNAs are imported, and these are (1) MRP RNA (MIM 157660), a noncoding RNA component of RNase MRP, a site-specific endoribonuclease involved in primer RNA cleavage during replication of mtDNA4 and (2) RNase P RNA component (MIM 608513), thought to participate in the maturation of mitochondrial tRNA (mt-tRNA). In addition, several nuclear tRNAs were reportedly targeted to mitochondria either in vitro5 or in vivo6,7 and were shown to rescue a mtDNA mutation linked to myoclonic epilepsy associated with ragged-red fibers (MERFF) syndrome (MIM 545000).8 Finally, several nuclearly encoded miRNAs are localized in the mitochondria and could represent a new regulatory pathway for nuclear-mitochondrial cross-talk.9 The way in which these small RNAs are imported into the mitochondria is not well understood, and various mechanisms have been considered.3,6,10 Recently, the mitochondrial protein PNPase has been shown to facilitate mitochondrial import of 5S rRNA, MRP RNA, and RNaseP RNA into mammalian cells.11 Here, we report an abnormal mitochondrial RNA import triggered by a PNPase mutation as a cause of mitochondrial RC deficiency.
Subject 1, a girl, was born to first-cousin, healthy Moroccan parents after a full-term pregnancy and normal delivery. Her birth weight was 3,470 g, her height was 49 cm, and her occipitofrontal circumference [OFC] was 35.5 cm. She did apparently well in the first months of life. She could smile and follow with her eyes at the age of 2 months and acquired head control at the age of 4 months. However, she presented with vomiting, poor eating, and swallowing difficulties of unexplained origin, and she could never sit unaided. At 9 months of age, she suddenly developed trunk hypotonia with disorganized, asynchronic, and erratic dystonic and choreoathetotic movements of all four limbs and bucofacial dyskinesias. She lost purposeful hand movements and could no longer hold her bottle. When she was first referred, she was a 10-year-old bed- and wheel-chair-bound, severely hypotrophic girl (weight = 17 kg [−3 standard deviations (SDs)]; OFC = 49 cm [−2.5 SDs]). She could neither stand nor sit unaided. Her voluntary movements were slow and markedly hampered by dystonia, dyskinesia, and choreoathetosis. She had global hypotonia, severe muscle weakness, no head control, and permanent bucofacial dyskinesias. Deep-tendon reflexes of the inferior limbs were barely detected, owing to muscle atrophy and permanent abnormal movements, but her nerve-conduction velocity was reduced (32–35 m/s; controls = 40–45 m/s). She could not speak, but she understood and obeyed simple orders. She was otherwise fully conscious and alert, and she could smile, burst out laughing, and follow with her eyes with a direct gaze and no abnormal eye movements. Major swallowing difficulties required gastrostomy, and major retractions of the right hip and ankles prompted a consideration of multiple tenotomies.
The second child (subject 2), a boy, was also born after a full-term pregnancy and normal delivery. His birth weight was 3,580 g, his height was 50 cm, and his OFC was 36.5 cm. He also did well in the first few months of life and could smile and follow with his eyes, but he never acquired head control. At 6 months of age, he suddenly developed motor regression with trunk hypotonia and choreoathetotic movements. He lost the ability to hold his bottle and grasp. When he was first referred at 3.5 years old, he had major dystonia of the limbs, global hypotonia, and permanent choreoathetotic movements (his weight was 14.6 kg, and his height was 96 cm). All subject samples were acquired according to the Necker Hospital Ethical Committee, and informed consent was given prior to sample collection.
The two affected children therefore had a fixed, severe, but nonprogressive encephalopathy and mildly elevated plasma and cerebrospinal-fluid lactate. Brain magnetic resonance images (MRIs) (axial T2, fast-spin-echo-weighted images) of the two children showed hyperintensities in the bilateral putamen and caudate nuclei (Figure 1A). Neither white-matter involvement nor pontocerebellar anomalies were noted. Electroencephalographic evidence of clinically silent erratic myoclonies was noted.
Figure 1.
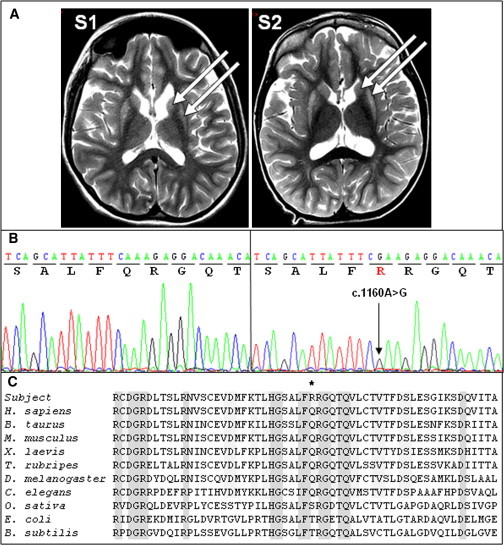
Brain MRIs and Sequence Analyses of the Affected Children
(A) Brain MRIs of subjects 1 (S1) and 2 (S2) at 14 and 3.5 years of age, respectively. The arrows show hyperintensities in the bilateral putamen and caudate nuclei.
(B) Sequence analysis of exon 13 of PNPT1 (RefSeq accession number NM_033109) in a control (left) and subject 1 (right). The arrow indicates the mutation.
(C) Sequence alignment of the PNPase proteins from human and nonhuman sources. The asterisk indicates the altered amino acid.
A severe decrease in the activity of RC complexes III and IV was found in the liver of subject 1, whereas normal enzyme-activity values were found in her skeletal muscle biopsy and in cultured skin fibroblasts of subject 2 (Table 1). Histopathological examination showed predominant type I fibers, mild atrophy of type II fibers, and peripheral accumulation of abnormal mitochondria. Large-scale mtDNA deletions, mtDNA depletion, and common point mutations were excluded by the use of appropriate techniques in liver DNA of the two children. Blue-native gel electrophoresis (BN-PAGE) in cultured skin fibroblasts showed normal RC assembly in subject 2.
Table 1.
Respiratory-Chain Activities in Liver Homogenate, Muscle Mitochondria, and Fibroblasts
|
Liver Homogenate |
Muscle Mitochondria |
Fibroblasts |
||||
|---|---|---|---|---|---|---|
| Subject 1 | Control | Subject 1 | Control | Subject 2 | Control | |
| Absolute Activity (nmol/min/mg of Protein) | ||||||
| CI | 20 | 25 ± 5 | 108 | 73 ± 15 | 14 | 13 ± 3 |
| CII | 140 | 146 ± 20 | 143 | 98 ± 20 | 28 | 21 ± 2 |
| CIII | 185 | 400 ± 60 | 2,343 | 1,458 ± 257 | 247 | 181 ± 20 |
| CIV | 48 | 187 ± 29 | 1,073 | 740 ± 146 | 132 | 103 ± 11 |
| CV | 149 | 112 ± 29 | 539 | 335 ± 68 | 61 | 65 ± 6 |
| CS | 48 | 61 ± 9 | 573 | 399 ± 70 | 75 | 75 ± 7 |
| Activity Ratios | ||||||
| CI/CS | 0.41 | 0.41 ± 0.08 | 0.19 | 0.18 ± 0.02 | 0.19 | 0.17 ± 0.03 |
| CII/CS | 2.93 | 2.23 ± 0.30 | 0.25 | 0.25 ± 0.02 | 0.38 | 0.29 ± 0.02 |
| CIII/CS | 3.86 | 6.43 ± 0.85 | 4.09 | 3.91 ± 0.40 | 3.31 | 2.53 ± 0.20 |
| CIV/CS | 0.99 | 3.03 ± 0.38 | 1.87 | 1.96 ± 0.15 | 1.77 | 1.35 ± 0.11 |
| CV/CS | 3.11 | 1.80 ± 0.34 | 0.94 | 0.80 ± 0.06 | 0.27 | 0.27 ± 0.03 |
Abnormal values are in bold. The following abbreviations are used: CI–CV, complexes I–V; and CS, citrate synthase.
To identify the causative nuclear gene mutation, we sequenced the exomes of subjects 1 and 2. The subjects’ genomic DNA (1 μg) was isolated from blood leukocytes. We captured exons by the in-solution enrichment methodology (SureSelect Human All Exon Kits v.3, Agilent, Massy, France) by using the company’s biotinylated oligonucleotide probe library (Human All Exon v.3 50 Mb, Agilent). Each genomic DNA sample was then sequenced on a sequencer as paired-end 75 bp reads (Illumina HISEQ2000, Illumina, San Diego, USA). Image analyses and base calling were performed with Real Time Analysis Pipeline v.1.9 with default parameters (Illumina). Sequences were aligned to the human genome reference sequence (hg19 assembly), and SNPs were called on the basis of the allele calls and read depth with the use of the CASAVA (Consensus Assessment of Sequence and Variation 1.8 [Illumina]) pipeline. From the 48,068 and 48,069 SNPs and indels identified in subjects 1 and 2, respectively, the pathogenic variant was selected according to the following criteria. (1) The known SNPs reported in dbSNP, 1000 Genomes, and the Exome Variant Server were excluded. (2) Intergenic variants were excluded because most mutations disrupt protein-coding sequences in Mendelian disorders. (3) Given the consanguinity of the parents, only homozygous variations were considered. (4) Finally, variations shared by both subjects 1 and 2 were selected. This filtering resulted in a list of 25 genes, only one of which (PNPT1 [MIM 610316]) encodes a known mitochondrial protein. The homozygous c.1160A>G mutation in exon 13 of PNPT1 (RefSeq accession number NM_033109.3) on chromosome 2 resulted in the transition of a neutral glutamine into a basic arginine (p.Gln387Arg) in the protein. This change is predicted to be probably damaging and deleterious by PolyPhen and SIFT software, respectively. Sanger sequencing allowed us to confirm this mutation in the two affected individuals (Figure 1B) and showed that the parents were heterozygous and that the healthy brother was wild-type (WT) homozygous (not shown). We then sequenced PNPT1 in a series of nine individuals with a similar clinical presentation and/or brain MRI, but we failed to detect any other mutations. Finally, this mutation was not found in 100 controls of north African origin. The p.Gln387 substitution lies in a highly conserved domain of the protein and is present in amino acid sequences from humans to C. elegans (Figure 1C).
PNPT1 encodes the mitochondrial polynucleotide phosphorylase, a 3′–5′ exoribonuclease and poly-A polymerase (PNPase) predominantly located in the mitochondrial intermembrane space.12 This protein assembles into a homo-oligomeric complex consisting of a trimer or a dimer of trimers.11 We therefore studied the effect of the PNPT1 mutation on the amount and multimerization of the PNPase. SDS-PAGE analysis of total-protein extracts from cultured skin fibroblasts showed a marked decrease in mutant protein because only 50%–60% of normal PNPase could be detected in subject 1 compared to the control (Figure 2A). However, the amounts of PNPT1 transcripts were similar between the fibroblasts of subject 1 and control fibroblasts (not shown), suggesting that the p.Gln387Arg substitution destabilizes the mutant protein. Proteins (30 μg) solubilized from subject 1 mitoplasts were loaded on a 4%–16% acrylamide nondenaturing gradient gel (Invitrogen).13 After electrophoresis, gels were transferred onto a membrane (GE Healthcare), processed for immunoblotting, and immunoblotted for PNPase. A ∼260 kDa complex containing PNPase was detected in control fibroblasts and might represent PNPase trimers. This complex was totally absent from the subjects’ cultured cells, whereas similar amounts of RC complexes II and III, used as loading tests, were detected in subjects and controls. No assembly intermediate could be detected in either subject or control mitoplasts (Figure 2B). The absence of PNPase complexes in the subjects’ fibroblasts, despite a residual amount of mutant PNPase, suggests that the p.Gln387Arg substitution alters multimerization of the protein, although one cannot exclude that the mutation affects association with proteins from the mitochondrial inner membrane.
Figure 2.
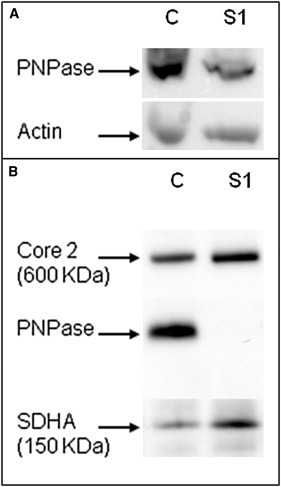
PNPase Analysis
(A) SDS-PAGE and immunoblot analysis of PNPase and actin in subject 1 (S1) and a control (C).
(B) BN-PAGE analysis of mitochondria from cultured skin fibroblasts of subject 1 (S1) and a control (C) with PNPase antibodies. SDHA (complex II) and core 2 (complex III) were used as loading controls.
So far, the way in which small RNA is imported into mitochondria has remained poorly understood. However, it has been recently proposed that PNPase could be involved in RNA targeting to human mitochondria.11 We therefore studied the possible impact of the PNPase substitution on mitochondrial RNA import. We isolated total and mitochondrial RNA (mtRNA) from cultured skin fibroblasts of subjects and controls8,14 and analyzed 5S rRNA import by Northern hybridization by using the following 32P-labeled probes: 5′-CATCCAAGTACTACCAGGCCC-3′ for 5S rRNA, 5′-GGCCGCAAGTGCGTTCGAAG-3′ for 5.8S rRNA (control for the absence of cytosolic contamination), and 5′-GTTGAAATCTCCTAAGTG-3′ for mt-tRNAVal (control for the absence of mtRNA degradation during the procedure). We observed a significant decrease in 5S rRNA import (70% ± 10%) into isolated and cytosol noncontaminated mitochondria from subject 1 compared to the control (Figure 3). To model the effect of PNPase decrease in an independent cell line, we analyzed mitochondrial import of 5S rRNA in a human immortalized HepG2 cell line in the presence of PNPT1 siRNA. HepG2 cells were lipofectamine transfected with a mixture of three dsRNA duplexes (AAGAGUUACAUCUGAAGUCCU, AAAACCUCGAGCAUCUAGAAA, and AAAACAGGUGUAACUAUUA)15 and analyzed 48 hr after a single transfection. Immunoblot analyses confirmed efficient knockdown of PNPT1 given that the amount of PNPase was reduced by at least three to four times (Figure 3). This reduction was accompanied by a 50%–60% decrease in 5S rRNA in mitochondria. Why a three-to-four-times reduction of PNPase levels only reduced the 5S rRNA amount by half is questionable, but these results clearly confirm the importance of PNPase for 5S rRNA import.
Figure 3.
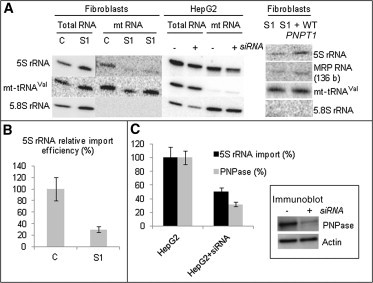
Quantification of Mitochondrial Import of 5S rRNA and MRP RNA by RNA Hybridization
(A) On the left, total and mtRNA from cultured skin fibroblasts of a control (C) and subject 1 (S1) were separated in denaturing 10% PAGE, transferred on Hybond-N filters, and hybridized with oligonucleotide-labeled probes for 5S rRNA, mt-tRNAVal, and 5.8S rRNA. Two deposits (1 and 3 μg) of subject mtRNA were analyzed for 5S-rRNA-import analysis. In the middle is Northern blot hybridization with 5S-rRNA-specific, mt-tRNAVal-specific, and 5.8S-rRNA-specific radio-labeled probes of total and mtRNA from HepG2 cells transfected or not with PNPT1 siRNAs. On the right, Northern blot analysis of mtRNA after transient expression of WT PNPT1 cDNA restored both 5S rRNA and 136 bp MRP RNA amounts in the mitochondria of subject 1 (S1) fibroblasts.
(B) Relative values of 5S rRNA import in subject 1 (S1) fibroblasts normalized to the control (C). Error bars correspond to two independent experiments.
(C) Relative 5S rRNA import and PNPase amount in control HepG2 and treated HepG2 + siRNA cells. The error bars correspond to two independent experiments. Immunoblot analysis of PNPase downregulation is shown in the inset. The actin-specific antibody was used as a loading control.
MRP RNA is the RNA component of RNase MRP, a site-specific endoribonuclease involved in primer-RNA cleavage during the replication of mtDNA.4 MRP RNA is 265 nt long in size. After its PNPase-dependent import into mitochondria, MRP RNA is believed to be at least partially processed into a 136 nt molecule containing a sequence capable of base pairing with a region of its mtRNA substrate.11,16 Indeed, we detected the processed form of MRP RNA in RNase-treated mitochondria of human embryonic kidney (HEK) 293 cells by Northern blot hybridization by using the 32P-labeled probe 5′-GTGGGAAGCGGGGAATGTCTACG-3′ (Figure 4A). Analyzing control and subject fibroblasts, we found similar amounts of full-sized MRP RNA in total-RNA preparations of both subjects and controls. Importantly, however, the 130–140 bp RNA species resulting from MRP-RNA maturation4 was detected in purified mitochondria from control, but not subject, fibroblasts (Figure 4B). The same amount of mt-tRNAVal in purified mitochondria from controls and subjects ruled out RNA degradation. Our results suggest that mitochondrial import of at least two independent RNA species is strongly affected by the PNPT1 mutation in the subjects’ cultured fibroblasts. A decreased MRP RNA amount has apparently little impact on the level of mtDNA given that the mtDNA content in the subjects’ fibroblasts was 42% of control values; this is usually considered normal given the large variability of control ranges. One can hypothesize that the mitochondrial import and processing of MRP RNA mostly impact the switch between asymmetrical and strand-coupled modes of mtDNA synthesis.17
Figure 4.
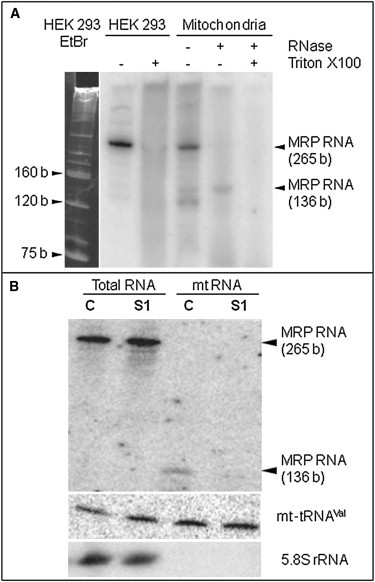
Analysis of MRP RNA
(A) Analysis of MRP RNA isolated from HEK 293 cells. The left panel shows ethidium-bromide (EtBr)-colored 10% denaturating polyacrylamide gel. The sizing of tRNA, 5S rRNA, and 5.8S rRNA, which were used as size markers, was confirmed by hybridization with corresponding oligonucleotide probes. The right panel shows a hybridization autoradiograph with MRP-RNA oligonucleotide-labeled probes of total RNA, total RNA after lysis of cells with Triton X-100 (1%), crude mtRNA, and mtRNA after mitochondria were treated with an RNase mixture.
(B) Analysis of mitochondrial import of MRP RNA by RNA hybridization. In the Northern blot analysis, total and mtRNA from cultured skin fibroblasts of a control (C) and subject 1 (S1) were hybridized with MRP-RNA, mt-tRNAVal, and 5.8S rRNA oligonucleotide-labeled probes. Full-sized (265 bp) and processed forms of the MRP RNA (136 bp) are indicated.
Finally, considering that import of 5S rRNA has been shown to play an essential role in mitochondrial translation,3 we questioned whether mitochondrial protein synthesis was affected in the subjects’ cells. Pulse-chase incorporation of 35S-methionine into mitochondrially-synthesized polypeptides was tested in the presence of 0.5 mg/ml emetine for the inhibition of cytoplasmic translation.8 To assure correct quantification, we simultaneously performed immunoblotting by using an actin antibody. We found a reproducible mitochondrial-translation decrease mainly affecting COX subunits in subject, rather than control, fibroblasts (Figure 5A and C). The decrease was much less pronounced than that of the 5S rRNA import (58% ± 4% versus 30% ± 5%), suggesting that the residual 5S rRNA level was sufficient to ensure significant mitochondrial translation of RNA. Such a limited effect was previously observed when 5S rRNA import was only partially abolished.3 Similarly, knocking down PNPase in control fibroblasts with the use of PNPT1-specific siRNAs (as with HepG2 cells, see above) resulted in a translation defect as well (Figure 5A, right panel). In order to confirm that the translational defect was due to the PNPase substitution, we transiently transfected subject fibroblasts with WT PNPT1 cDNA expressed under the control of the CMV promoter (cloned in pCMV6-XL4 vector, purchased in OriGene). Transient cDNA expression induced a 160% increase in the PNPase band intensity 48 hr after transfection (Figure 5). Moreover, an almost full restoration of protein-synthesis rate was observed (Figure 5). Finally, overexpression of WT PNPT1 cDNA in subject fibroblasts restored both mitochondrial 5S rRNA and 136 bp MRP RNA amounts (Figure 3A, right panel). These results clearly demonstrate the deleterious effect of the PNPT1 mutation on mitochondrial translation and RNA import. It is worth remembering that although PNPT1 silencing in HeLa cells has failed to alter the rate of mitochondrial translation,19 a clear defect in mitochondrial protein synthesis was observed in a liver-specific knockout of a Pnpt1 mouse model and RNAi in HEK 293 cells,11 where all mitochondrially-encoded polypeptides appeared to be uniformly affected. This apparent contradiction might be accounted for by HeLa cells’ low glycolytic metabolism compared to active mitochondrial respiration in the liver. Also, because PNPase is involved in various functions such as noncoding RNA import11 and mRNA polyadenylation,15 it should be hypothesized that different cells or tissues might require different functions of PNPase.
Figure 5.
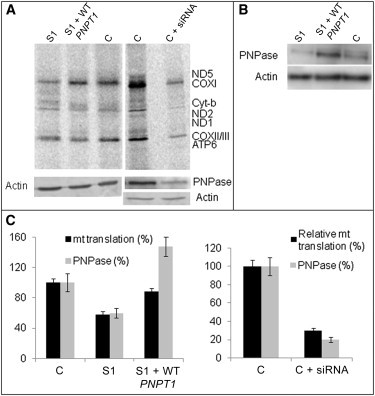
Analysis of Mitochondrial Translation in the Subjects Fibroblasts
(A) In vivo pulse-chase 35S-methionin incorporation in mitochondrially synthesized polypeptides in fibroblasts of subject 1 (S1), S1 fibroblasts transiently transfected with WT PNPT1 cDNA (S1 + WT PNPT1), control (C) fibroblasts, and control fibroblasts transfected with PNPT1 siRNAs (C + siRNA). The mitochondrial-translation products on 12% SDS-PAGE are indicated according to the standard pattern.18 The bottom parts (loading control) show the immunoblot assay with antibodies against actin and PNPase.
(B) Quantification of the PNPase expression by immunoblot analysis was performed in parallel with unlabelled aliquots of the same cell lines.
(C) On the left are relative mitochondrial-translation products and PNPase levels in subject 1 (S1) fibroblasts, S1 fibroblasts transiently transfected with WT PNPT1 cDNA (S1 + WT PNPT1), and control (C) fibroblasts. On the right are relative mitochondrial-translation products and PNPase levels in control fibroblasts transfected (C + siRNA) or not (C) with PNPT1 siRNAs. For both diagrams, error bars correspond to two independent experiments.
The exact function(s) of the human PNPase is not completely understood. It was found to be active in phosphorolysis of RNA, but its localization in the mitochondrial intermembrane space (where no RNA is constantly present) suggests that it is not directly involved in the metabolism of mtRNA.20 This protein has also been identified as a T cell leukemia-1 (TCL1) binding partner, whereas the biological implications of TCL1-PNPase complexes remain elusive.21 PNPase is also involved in the processing and adenylation of mitochondrial mRNA,19 but the most recent data support its crucial role in mitochondrial import of small RNAs needed for replication, transcription, and translation of the mitochondrial genome.11
In conclusion, we report here a pathogenic PNPT1 mutation in humans. This mutation destabilizes the protein and abrogates its multimerization. Moreover, we also show that this mutation alters mitochondrial RNA import and mitochondrial translation. These results strongly support the role of PNPase in the import of RNA into mitochondria and provide evidence for its crucial importance in maintaining a functional RC. Impaired RNA import into mitochondria therefore represents a disease mechanism in multiple-RC deficiency. Based on this single family, our study supports the importance of exome sequencing for the identification of rare disease-causing gene mutations.
Acknowledgments
This research was supported in part by the Association Française contre les Myopathies, the French Agence Nationale pour la Recherche, the GIS-Institut des maladies rares, the Institut National de la Santé et de la Recherche Médicale, the Centre National de la Recherche Scientifique, the Fondation pour la Recherche Médicale, and the National Program “Investissement d'Avenir” (Labex MitoCross). A.G. was supported by the Higher Education Comission, Pakistan (SFERE doctoral fellowship).
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in man (OMIM), http://www.omim.org
PolyPhen, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
References
- 1.Chinnery P.F., Turnbull D.M. Epidemiology and treatment of mitochondrial disorders. Am. J. Med. Genet. 2001;106:94–101. doi: 10.1002/ajmg.1426. [DOI] [PubMed] [Google Scholar]
- 2.Rötig A. Genetic bases of mitochondrial respiratory chain disorders. Diabetes Metab. 2010;36:97–107. doi: 10.1016/j.diabet.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Smirnov A., Entelis N., Martin R.P., Tarassov I. Biological significance of 5S rRNA import into human mitochondria: Role of ribosomal protein MRP-L18. Genes Dev. 2011;25:1289–1305. doi: 10.1101/gad.624711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang D.D., Clayton D.A. A mammalian mitochondrial RNA processing activity contains nucleus-encoded RNA. Science. 1987;235:1178–1184. doi: 10.1126/science.2434997. [DOI] [PubMed] [Google Scholar]
- 5.Entelis N.S., Kolesnikova O.A., Dogan S., Martin R.P., Tarassov I.A. 5 S rRNA and tRNA import into human mitochondria. Comparison of in vitro requirements. J. Biol. Chem. 2001;276:45642–45653. doi: 10.1074/jbc.M103906200. [DOI] [PubMed] [Google Scholar]
- 6.Rubio M.A., Hopper A.K. Transfer RNA travels from the cytoplasm to organelles. Wiley Interdiscip Rev RNA. 2011;2:802–817. doi: 10.1002/wrna.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rubio M.A., Rinehart J.J., Krett B., Duvezin-Caubet S., Reichert A.S., Söll D., Alfonzo J.D. Mammalian mitochondria have the innate ability to import tRNAs by a mechanism distinct from protein import. Proc. Natl. Acad. Sci. USA. 2008;105:9186–9191. doi: 10.1073/pnas.0804283105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolesnikova O.A., Entelis N.S., Jacquin-Becker C., Goltzene F., Chrzanowska-Lightowlers Z.M., Lightowlers R.N., Martin R.P., Tarassov I. Nuclear DNA-encoded tRNAs targeted into mitochondria can rescue a mitochondrial DNA mutation associated with the MERRF syndrome in cultured human cells. Hum. Mol. Genet. 2004;13:2519–2534. doi: 10.1093/hmg/ddh267. [DOI] [PubMed] [Google Scholar]
- 9.Bandiera S., Rüberg S., Girard M., Cagnard N., Hanein S., Chrétien D., Munnich A., Lyonnet S., Henrion-Caude A. Nuclear outsourcing of RNA interference components to human mitochondria. PLoS ONE. 2011;6:e20746. doi: 10.1371/journal.pone.0020746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salinas T., Duchêne A.M., Maréchal-Drouard L. Recent advances in tRNA mitochondrial import. Trends Biochem. Sci. 2008;33:320–329. doi: 10.1016/j.tibs.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 11.Wang G., Chen H.W., Oktay Y., Zhang J., Allen E.L., Smith G.M., Fan K.C., Hong J.S., French S.W., McCaffery J.M. PNPASE regulates RNA import into mitochondria. Cell. 2010;142:456–467. doi: 10.1016/j.cell.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen H.W., Rainey R.N., Balatoni C.E., Dawson D.W., Troke J.J., Wasiak S., Hong J.S., McBride H.M., Koehler C.M., Teitell M.A., French S.W. Mammalian polynucleotide phosphorylase is an intermembrane space RNase that maintains mitochondrial homeostasis. Mol. Cell. Biol. 2006;26:8475–8487. doi: 10.1128/MCB.01002-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nijtmans L.G., Henderson N.S., Holt I.J. Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods. 2002;26:327–334. doi: 10.1016/S1046-2023(02)00038-5. [DOI] [PubMed] [Google Scholar]
- 14.Zeharia A., Shaag A., Pappo O., Mager-Heckel A.M., Saada A., Beinat M., Karicheva O., Mandel H., Ofek N., Segel R. Acute infantile liver failure due to mutations in the TRMU gene. Am. J. Hum. Genet. 2009;85:401–407. doi: 10.1016/j.ajhg.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagaike T., Suzuki T., Katoh T., Ueda T. Human mitochondrial mRNAs are stabilized with polyadenylation regulated by mitochondria-specific poly(A) polymerase and polynucleotide phosphorylase. J. Biol. Chem. 2005;280:19721–19727. doi: 10.1074/jbc.M500804200. [DOI] [PubMed] [Google Scholar]
- 16.Topper J.N., Clayton D.A. Characterization of human MRP/Th RNA and its nuclear gene: Full length MRP/Th RNA is an active endoribonuclease when assembled as an RNP. Nucleic Acids Res. 1990;18:793–799. doi: 10.1093/nar/18.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holt I.J. Zen and the art of mitochondrial DNA maintenance. Trends Genet. 2010;26:103–109. doi: 10.1016/j.tig.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 18.Enríquez J.A., Cabezas-Herrera J., Bayona-Bafaluy M.P., Attardi G. Very rare complementation between mitochondria carrying different mitochondrial DNA mutations points to intrinsic genetic autonomy of the organelles in cultured human cells. J. Biol. Chem. 2000;275:11207–11215. doi: 10.1074/jbc.275.15.11207. [DOI] [PubMed] [Google Scholar]
- 19.Slomovic S., Schuster G. Stable PNPase RNAi silencing: Its effect on the processing and adenylation of human mitochondrial RNA. RNA. 2008;14:310–323. doi: 10.1261/rna.697308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Portnoy V., Palnizky G., Yehudai-Resheff S., Glaser F., Schuster G. Analysis of the human polynucleotide phosphorylase (PNPase) reveals differences in RNA binding and response to phosphate compared to its bacterial and chloroplast counterparts. RNA. 2008;14:297–309. doi: 10.1261/rna.698108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.French S.W., Dawson D.W., Chen H.W., Rainey R.N., Sievers S.A., Balatoni C.E., Wong L., Troke J.J., Nguyen M.T., Koehler C.M., Teitell M.A. The TCL1 oncoprotein binds the RNase PH domains of the PNPase exoribonuclease without affecting its RNA degrading activity. Cancer Lett. 2007;248:198–210. doi: 10.1016/j.canlet.2006.07.006. [DOI] [PubMed] [Google Scholar]