

Abstract
Primary ciliary dyskinesia (PCD) is a group of autosomal-recessive disorders resulting from cilia and sperm-flagella defects, which lead to respiratory infections and male infertility. Most implicated genes encode structural proteins that participate in the composition of axonemal components, such as dynein arms (DAs), that are essential for ciliary and flagellar movements; they explain the pathology in fewer than half of the affected individuals. We undertook this study to further understand the pathogenesis of PCD due to the absence of both DAs. We identified, via homozygosity mapping, an early frameshift in LRRC6, a gene that encodes a leucine-rich-repeat (LRR)-containing protein. Subsequent analyses of this gene mainly expressed in testis and respiratory cells identified biallelic mutations in several independent individuals. The situs inversus observed in two of them supports a key role for LRRC6 in embryonic nodal cilia. Study of native LRRC6 in airway epithelial cells revealed that it localizes to the cytoplasm and within cilia, whereas it is absent from cells with loss-of-function mutations, in which DA protein markers are also missing. These results are consistent with the transmission-electron-microscopy data showing the absence of both DAs in cilia or flagella from individuals with LRRC6 mutations. In spite of structural and functional similarities between LRRC6 and DNAAF1, another LRR-containing protein involved in the same PCD phenotype, the two proteins are not redundant. The evolutionarily conserved LRRC6, therefore, emerges as an additional player in DA assembly, a process that is essential for proper axoneme building and that appears to be much more complex than was previously thought.
Main Text
Primary ciliary dyskinesia (PCD [MIM 244400]) is a heterogeneous group of genetic disorders affecting 1 in 15,000–30,000 individuals and is usually transmitted as an autosomal-recessive trait.1 This disease results from structural and functional defects of cilia and leads to impaired mucociliary transport responsible for early recurrent respiratory-tract infections. Approximately 50% of individuals with PCD display situs inversus, which defines Kartagener syndrome (MIM 244400).2 Moreover, most male individuals are infertile as a result of functional and ultrastructural abnormalities in sperm flagella, whose axonemal structure is similar to that of motile cilia.3
The axoneme, the core of motile cilia and flagella, consists of nine peripheral microtubule doublets and a central complex composed of two single microtubules. Attached to the peripheral doublets, the inner and outer dynein arms (IDAs and ODAs, respectively) are multiprotein ATPase complexes that are essential for normal ciliary and flagellar movements.4 Various axonemal ultrastructural defects have been reported in PCD, and most of them concern ODAs and/or IDAs.5 A second level of heterogeneity in PCD is attested to by the fact that the same ultrastructural defect can result from mutations in distinct genes.6 Most of the genes implicated in PCD encode structural proteins that participate in the composition of diverse axonemal components.6,7 The identification of other mutations has revealed the importance of some genes in the proper assembly of ciliary axonemes: CCDC398 (MIM 613807) and CCDC409 (MIM 613808) mutations are implicated in individuals with axonemal disorganization and an absence of IDAs. Mutations in RPGR (MIM 300455), a gene that was first involved in nonsyndromic retinitis pigmentosa and that encodes a protein implicated in intraflagellar transport, have been found in a PCD phenotype characterized by a complex ultrastructural respiratory-cilia defect associated with retinitis pigmentosa.6 As for PCD cases due to the absence of both dynein arms (DAs), mutations in genes involved in cytoplasmic preassembly of DAs, such as DNAAF2 (previously KTU [MIM 612518]),10 DNAAF1 (previously LRRC50 [MIM 613193]),11,12 and DNAAF3 (MIM 606763), have been identified.13 Just recently, a fourth gene called CCDC103 (MIM 614679) and believed to play a role in anchoring DAs to microtubules has been associated with this phenotype.14 However, although the absence of both DAs represents a frequent ultrastructural defect present in at least one-third of PCD cases,5 mutations in those genes account for only a small proportion of cases: DNAAF1 mutations explain 17% of PCD cases with an absence of both DAs;11 DNAAF2 mutations have been reported in only two unrelated families;10 and DNAAF3 and CCDC103 have been found to be mutated in three and six families, respectively.13,14
We undertook this study in order to identify additional genes involved in the PCD phenotype characterized by the absence of both DAs. Forty-seven unrelated families (containing 50 affected individuals) of our cohort have PCD and/or male sterility with an absence of both DAs. In all of them, the axonemal defect was confirmed by transmission electron microscopy (TEM) showing a lack of ODAs and IDAs on all microtubule doublets of all examined cilia and/or spermatozoa. By screening the genes already known to be involved in this phenotype, we identified mutations in only seven families: DNAAF1 mutations in six families (Duquesnoy et al.11 and unpublished data) and DNAAF2 mutations in one family (unpublished data). DNAAF3 has so far not been found to be involved in our cohort, whereas the very recently identified CCDC103 remains to be analyzed. Among the 42 individuals (31 males and 11 females) belonging to the remaining 40 families, 37 had a PCD diagnosis with typical clinical features (sinopulmonary syndrome beginning in early childhood; this was associated with situs inversus in 17 of them). The remaining five persons (from five unrelated families) were included because their male sterility with immotile sperm flagella was related to an absence of both DAs (documented by TEM); three of them also presented with minor respiratory problems (bronchorrhea and nasal obstruction). Infertility was noted in a total of 15 adults (13 males and 2 females). Seven individuals were born to a consanguineous union; one of these families (DC28) had two affected siblings (DCP16 and DCP17). The current study was approved by the Ile-de-France ethics committee (CPP07729), and written informed consent was obtained from all individuals and/or their parents.
Genomic DNAs from the seven individuals of the six consanguineous families were first analyzed by means of homozygosity mapping. A large homozygous region identified in chromosomal region 8q24.22, spanning 4,852,130 bp between rs6997983 and rs7825610, was found to be shared by the two affected siblings of family DC28 (Figure S1A, available online). This region contains 20 genes (Figure S1B), including leucine-rich-repeat (LRR)-containing 6 (LRRC6), which several arguments prompted us to consider as an excellent candidate for PCD. As reported in a previous proteomic study,15 it is expressed in the flagella of Chlamydomonas reinhardtii and in human cilia. In mice, Lrrc6 transcripts have been found to be expressed in tissues that have flagella or motile cilia.16,17 In addition, a putative PCD locus containing LRRC6 has been identified in a genome-wide linkage study performed in familial cases of PCD.18 Most importantly, the phenotypic features of several animal models with mutations in LRRC6 orthologs are consistent with ciliary defects. In zebrafish, seahorse mutants display a curved body with pronephric cysts,19 associated with left-right abnormalities in half cases.20 In Drosophila, a null allele of the LRRC6 ortholog, called tilB, is responsible for ciliary dysfunction of sensory neurons of the auditory organ and male sterility.21
In humans, LRRC6 consists of 12 exons; the only predicted transcript (RefSeq accession number NM_012472.3) encodes a 466 residue protein with five N-terminal LRR motifs (Figure 1A). The LRR repeats of LRRC6 (Figure 1B) contain the consensus sequence LxxLxLxxNxIxxIxxLxzxLxx (“z” indicates frequent deletions), which defines the SDS22-like subfamily of LRR-containing proteins.22 Each LRR repeat is a β-strand-turn-α-helix structure, and all together, these motifs are known to form a solenoid (repeated structural units that form a continuous superhelix).23 LRRC6 also contains an LRRcap that shields the solenoid, a coiled-coil (CC) domain, a polylysine motif, and a C-terminal α-crystallin-p23-like domain.
Figure 1.
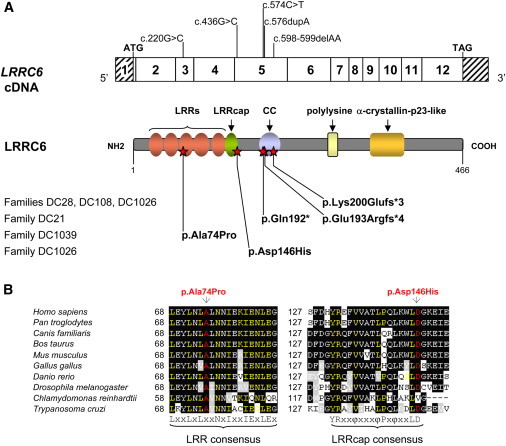
LRRC6 Mutations and Their Impact at the Protein Level in Individuals with PCD
(A) Exonic organization of the human LRRC6 cDNA, in which are shown the mutations (top) and domain-organization model of the corresponding protein (middle). The mutations’ impact at the protein level is shown for the five families described in this study (bottom). The twelve exons are indicated by empty or hashed boxes, depicting translated or untranslated sequences, respectively. According to the LRRC6 domain-organization model, derived from predictions by NCBI and UniProt/Swiss-Prot, the protein contains four LRR domains (amino acids 23–42, 46–65, 68–87, and 90–112), one modified LRR domain (amino acids 115–130) and a subsequent LRRcap (amino acids 131–146), a coiled-coil domain (amino acids 178–204), a polylysine motif (amino acids 272–286), and an α-crystallin-p23-like domain (amino acids 332–381).
(B) A partial protein alignment of LRRC6 shows the evolutionary conservation of the third LRR motif and the LRRcap domain, which contains the two amino acid substitutions identified in this study.
With the aim of testing the implication of LRRC6 in PCD, we first determined its expression by quantitative RT-PCR. The analysis revealed that LRRC6 is mainly expressed in the testis, as well as in respiratory epithelial cells obtained from nasal brushing; this expression pattern is typical of cilia-associated genes (Figure S2A). Consistent with this, LRRC6 expression levels in those tissues were found to be similar to those of DNAAF1, DNAAF2, DNAAF3, and CCDC103 (Figure S2B). We subsequently screened LRRC6 for mutations in individual DCP16 from family DC28. Sequence-variant databases, such as dbSNP, Ensembl, and the National Heart, Lung, and Blood Institute (NHLBI) Exome Variant Server (EVS), were used for filtering out previously identified SNPs. This analysis identified homozygous frameshift mutation c.598_599delAA (in exon 5), leading to a premature stop codon at position 202 (p.Lys200Glufs∗3). The same homozygous mutation was also present in her affected brother (DCP17). Their asymptomatic mother was found to harbor the mutation in the heterozygous state (Figure S3), and paternal DNA was not available. We subsequently screened for LRRC6 mutations in the remaining 39 independent families (40 individuals) affected by the absence of both DAs. Four of them (DC21, DC108, DC1039, and DC1026) had biallelic LRRC6 mutations, consistent with a loss of function of the corresponding protein (Figure 1A and Figure S3). Individual DCP18 (family DC21) had two premature stop codons in exon 5: the c.574C>T (p.Gln192∗) nonsense mutation on one allele and the frameshift c.576dupA (p.Glu193Argfs∗4) mutation on the other, as shown by the sequencing of cloned PCR products. Individual DCP152 (family DC108) was also shown to be homozygous for the c.598_599delAA (p.Lys200Glufs∗3) mutation identified in family DC28. His asymptomatic mother had the mutation in the heterozygous state, and paternal DNA was not available. Individual DCP193 (family DC1039) had a homozygous transversion in exon 3: c.220G>C (p.Ala74Pro). Homozygosity and consanguinity were confirmed in DCP152 and DCP193 by whole-genome genotyping with the Human CytoSNP-12 chip from Illumina (data not shown). Individual DCP339 (family DC1026) was a compound heterozygote and had the frameshift c.598_599delAA (p.Lys200Glufs∗3, exon 5) mutation on one allele and a transversion (c.436G>C in exon 5) introducing a missense variation (p.Asp146His) on the other allele, as demonstrated by the sequencing of cloned PCR products. If translated, the p.Gln192∗, p.Glu193Argfs∗4, and p.Lys200Glufs∗3 molecular defects are predicted to result in severely truncated proteins lacking more than half of the protein, including the polylysine motif and the α-crystallin-p23-like domain (Figure 1A). The p.Ala74Pro amino acid substitution, which replaces an amino acid of low steric hindrance with a cyclic amino acid, involves a residue that is invariant throughout evolution and that belongs to the third LRR motif of LRRC6. Proline is a breaker of α helices and β strands, so the p.Ala74Pro substitution is expected to disrupt this motif. The p.Asp146His (c.436G>C) amino acid substitution lies in the LRRcap, an LRR-associated motif commonly found in members of the SDS22-like subfamily of LRR-containing proteins. This motif is defined by the consensus sequence YRxxφxxxφPxφxxLD (Figure 1B), in which “φ” represents any hydrophobic residue. The first and last amino acids (tyrosine and aspartic acid, respectively) of the LRRcap are hydrogen bonded24 and shield the LRR superhelix. The p.Asp146His amino acid substitution, which involves a residue that is highly conserved throughout evolution (Figure 1B), most likely has a detrimental effect on LRRC6 conformation.
With the aim of assessing the effect of this mutation on LRRC6 conformation, we used the resolution structure of the closest crystallized protein, the Chlamydomonas ODA LC1 dynein light chain (structure ID 1DS9). Asp180 (the equivalent of Asp146 in human LRRC6) and Tyr165 (the equivalent of Tyr131 in human LRRC6) form a hydrogen bond. The p.Asp180His variation replaces an acidic amino acid with a noncharged residue containing an imidazole functional group; the formation of a hydrogen bond with Tyr165 would considerably increase the free energy of this molecule (from −2,843 kJ/mol to +1,126 kJ/mol, Figure S4), a result incompatible with the existence of such a structure.
Overall, we identified LRRC6 mutations in six individuals from five families. LRRC6 was therefore found to be mutated in 10.6% (5/47) of the families affected by a PCD phenotype characterized by an absence of both DAs. The nonsense and frameshift mutations, as well as the c.220G>C missense mutation, are not reported in databases such as dbSNP, Ensembl, or EVS. As for the c.436G>C missense variation, it is not described in Ensembl; however, it has just recently been reported in dbSNP (rs200321595; no allele frequency is available) and in EVS at an extremely low frequency (3 of 13,004 alleles; allele frequency = 0.00023), so the homozygous genotype is expected to be extremely rare. Given that the incidence of PCD ranges from 1 in 15,000 to 1 in 30,000 affected individuals, the expected frequency of a PCD allele ranges from 0.006 to 0.008. Therefore, such a low allele frequency for the c.436G>C mutation, together with the fact that this missense mutation was identified in trans to a frameshift, is consistent with the implication of the c.436G>C allele in the disease. The c.598_599delAA mutation is shared by three families (DC28, DC108, and DC1026) not known to be related (Table 1). Genotyping of microsatellites markers that flank LRRC6 (D8S1765, D8S558, and D8S1740) and span ∼1.9 Mb of the disease locus supports a founder effect (Figure S5).
Table 1.
Phenotypic Features of Individuals with Identified LRRC6 Mutations
| Family (Origin) | Individual | Known Consanguinity | Gender | Situs Inversus | Airway Disease | Fertility | CBF (Hz) | Allele 1 | Allele 2 |
|---|---|---|---|---|---|---|---|---|---|
| DC28 (European) |
DCP16 | yes | female | no | NRD, bronchitis, bronchiectasis, lobectomy, rhinosinusitis, otitis | hypofertility | 0 | c.598_599delAA (p.Lys200Glufs∗3) | c.598_599delAA (p.Lys200Glufs∗3) |
| DCP17 | yes | male | yes | bronchitis, rhinosinusitis | asthenospermia | 0 | c.598_599delAA (p.Lys200Glufs∗3) | c.598_599delAA (p.Lys200Glufs∗3) | |
| DC21 (European) |
DCP18a | no | male | no | bronchitis, bronchiectasis, rhinosinusitis, nasal polyposis | asthenospermia | 0 | c.574C>T (p.Gln192∗) | c.576dupA (p.Glu193Argfs∗4) |
| DC108 (European) |
DCP152 | no | male | no | bronchitis, bronchiectasis, rhinosinusitis, otitis | NR | 0 | c.598_599delAA (p.Lys200Glufs∗3) | c.598_599delAA (p.Lys200Glufs∗3) |
| DC1039 (European) |
DCP193 | no | male | no | bronchorrhea | asthenospermia | 0 | c.220G>C (p.Ala74Pro) | c.220G>C (p.Ala74Pro) |
| DC1026 (European) |
DCP339 | no | male | yes | bronchitis, bronchiectasis, rhinosinusitis, otitis | asthenospermia | ND | c.598_599delAA (p.Lys200Glufs∗3) | c.436G>C (p.Asp146His) |
The following abbreviations are used: CBF, ciliary beat frequency; NRD, neonatal respiratory distress; NR, not relevant; and ND, not determined.
Very low nasal nitric-oxide levels (13 nl/min) were documented in this individual.
Several conclusions can be drawn from the analysis of the PCD phenotype of individuals with LRRC6 mutations (Table 1). Four individuals (DCP16, DCP17, DCP18, and DCP152) who have biallelic nonsense or frameshift mutations display a typical disease phenotype: their cilia are immotile (Movies S1 and S2). Another individual (DCP339), who also has a typical disease phenotype, harbors the c.598_599delAA frameshift mutation in combination with the c.436G>C missense mutation that, at the protein level, is expected to severely impair protein function by conformational change. The only individual with moderate respiratory symptoms is DCP193, who is also the only one with partial absence of both DAs in airway cilia (Figure 2). Noteworthy is that this individual is also the only one who has a homozygous missense mutation (c.220G>C). This suggests that the p.Ala74Pro amino acid substitution would be compatible with the production of a partially functional protein and would thereby lead to a milder phenotype. The situs inversus observed in two individuals (DCP17 and DCP339) supports a key role for LRRC6 in the proper functioning of embryonic nodal cilia. These phenotype-genotype correlations also show that, like in individuals with DNAAF1 or DNAAF2 mutations (Duquesnoy et al.11 and our unpublished data), all adult individuals (four males and one female) with mutations in LRRC6 have fertility problems. In males, as shown in Figure 2, infertility is readily explained by the absence of both DAs in sperm flagella.
Figure 2.
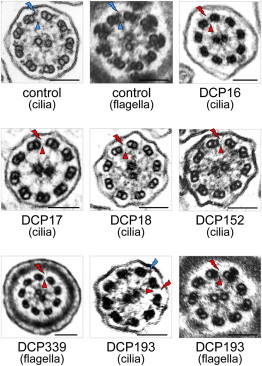
Absence of Both DAs in Respiratory Cilia and/or Spermatozoa Flagella of Individuals with LRRC6 Mutations
The electron micrographs of cross-sections of cilia and/or spermatozoa flagella from a control and individuals with identified LRRC6 mutations are shown. The blue flashes and triangles show the presence of ODAs and IDAs, respectively, and red flashes and triangles show their absence in affected individuals’ axonemes. Note the partial absence of DAs in the airway cilia of DCP193 (blue flash), whereas at the sperm level, the absence of DAs seems complete. Black scale bars represent 0.1 μm.
To gain further insight into the role of LRRC6, we determined its subcellular localization in human ciliated cells by means of high-resolution immunofluorescence microscopy. This was performed in ciliated cells collected by nasal brushing25 from both a control individual and individual DCP18 with the LRRC6 c.[574C>T; 576dupA] genotype. In ciliated cells from a control individual, strong LRRC6 labeling was observed in the cytoplasm and within the cilia (Figures 3A–3C). By contrast, LRRC6 was not detected in airway epithelial cells from individual DCP18 (Figures 3D–3F). This latter result, which confirms the specificity of the labeling obtained with the LRRC6 antibody used in these experiments, is consistent with the loss-of-function mutations identified in this individual. Immunostaining of the ODA intermediate chain DNAI2 (Figures 3H and 3K) and of the IDA component DNALI1 (Figures 3N and 3Q) indicates that the loss of LRRC6 in ciliated cells affects the assembly of DAs; these results confirm the TEM data clearly showing the absence of ODAs and IDAs in cilia or flagella from individuals with LRRC6 mutations (Figure 2).
Figure 3.
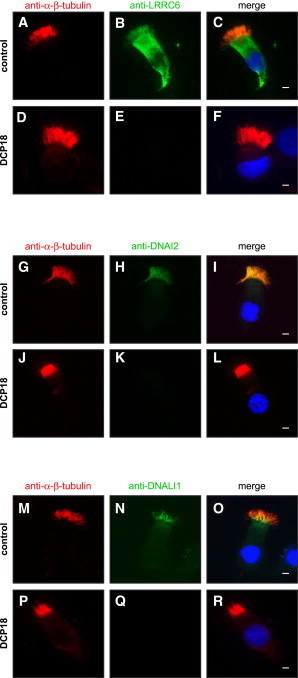
LRRC6 Localizes to the Cytoplasm and Cilia and Is Absent in DCP18
(A–F) In control cells, LRRC6 (green) localizes to the cytoplasm and within cilia, whereas it is absent from the individual’s cells. Axoneme-specific antibodies directed against α-β-tubulin were used as a control. Nuclei were stained with DAPI (blue).
Similar experiments were performed with antibodies directed against the ODA component DNAI2 (G–L) and the IDA component DNALI1 (M–R). In control cells, DNAI2 and DNALI1 localize within the cilia, whereas labeling is absent from the individual’s cells. Airway epithelial cells were examined after labeling with a mouse LRRC6 antibody (Abnova) and a secondary goat anti-mouse Alexa Fluor-488 (green) antibody (Invitrogen). For controls, we used antibodies directed against various axonemal components: α-β-tubulin (Cell Signaling Technology), DNAI2 (Abnova), and DNALI1 (Abnova) for the visualization of microtubules, ODAs, and IDAs, respectively; primary antibodies were revealed with a secondary Alexa Fluor-594 (red) or Alexa Fluor-488 (green) antibody (Invitrogen). White scale bars represent 5 μm.
All together, these data raise the larger question of the mechanism by which the loss of function of LRRC6 results in the absence of DAs in cilia and flagella. One possible answer relies on the striking similarities between LRRC6 and DNAAF1, a gene that has been shown to play a key role in cytoplasmic preassembly of DAs.11 Mutations in LRRC6 and DNAAF1 result in molecular defects that have the same impact on ciliary ultrastructure. These two genes have a similar expression pattern—high expression levels in ciliated tissues—especially in nasal brushings (Figure S2B). The corresponding proteins are both mainly localized within the cytoplasm of respiratory epithelial cells, and strikingly, they share similar domains: an LRR solenoid shielded by an LRRcap followed by a coiled-coil domain. The data suggest that LRRC6 is an additional player in cytoplasmic preassembly of DAs but that, in spite of its structure and functional similarities with DNAAF1, the two proteins are not redundant. Additional possible clues in the physiological role of LRRC6 come from the fact that LRRC6 and its Drosophila ortholog, TilB, share an α-crystallin-p23-like domain. Proteins with an α-crystallin domain belong to the family of small heat-shock proteins, HSPB, and it has been suggested that HSPB members can act as cytoskeleton-specific chaperones.26 These data are of particular interest in light of a recently proposed model for preassembly of DAs.13 This model hypothesizes that oda7, pf13, and pf22 (the orthologs of DNAAF1, DNAAF2, and DNAAF3, respectively), which are proteins required for the recognition and proper folding of the dynein-heavy chains present in Chlamydomonas reinhardtii ODAs, work within a chaperone complex.13 It has been shown that ODA7 interacts with both ODAs and IDAs27 and that loss of function in one of those three proteins results in the absence of both DAs in humans.10,11,13 Although the precise process whereby LRRC6 is involved in preassembly and/or transport of DAs remains to be clarified, it is therefore tempting to speculate that LRRC6 is a part of this chaperone complex by acting via its α-crystallin-p23-like domain in this elaborated mechanism of DA preassembly.
In summary, this study shows that, similarly to mutations in DNAAF1, DNAAF2, DNAAF3, and CCDC103, mutations in LRRC6 result in the most frequent type of PCD phenotype (i.e., absence of both DAs). Overall, among the 47 families affected by an absence of both DAs in our cohort, 12 have mutations in one of those genes. The current model for cytoplasmic preassembly of DAs involves four actors: DNAAF1, DNAAF2, DNAAF3, and a chaperone complex in which LRRC6, on the basis of its predicted domain organization, is likely to participate. Given that no molecular defect has yet been identified for several individuals with an absence of both DAs, it is likely that other genes contribute to the disease. Identification of those genes should help in better defining the molecular mechanisms involved in DA assembly, a process that is essential for proper axoneme building and that appears to be much more complex than was previously thought.
Acknowledgments
We are grateful to the affected persons and their families, whose cooperation made this study possible, and we thank all referring physicians. This work was supported by grants from the Legs Poix from the Chancellerie des Universités, the Milena Carvajal ProKartagener Foundation, the Assistance Publique-Hôpitaux de Paris (PHRC AOM06053 and P060245), and the Fondation pour la Recherche Médicale (DEQ20120323689).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ciliome, http://www.ciliome.com
Ensembl, http://www.ensembl.org
NHLBI Exome Sequencing Project Exome Variant Server, http://evs.gs.washington.edu/EVS/
GenBank, http://www.ncbi.nlm.nih.gov/Genbank
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Protein Data Bank, http://www.rcsb.org
UniProt, http://www.uniprot.org
References
- 1.Afzelius B.A. A human syndrome caused by immotile cilia. Science. 1976;193:317–319. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- 2.Kartagener M. Zur Pathogenese der Bronchiektasien: Bronchiektasien bei Situs viscerum inversus. Beiträge Zur Klinik Der Tuberkulose. 1933;83:489–501. [Google Scholar]
- 3.Afzelius B.A. The immotile-cilia syndrome: A microtubule-associated defect. CRC Crit. Rev. Biochem. 1985;19:63–87. doi: 10.3109/10409238509086788. [DOI] [PubMed] [Google Scholar]
- 4.Satir P., Christensen S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 5.Papon J.F., Coste A., Roudot-Thoraval F., Boucherat M., Roger G., Tamalet A., Vojtek A.M., Amselem S., Escudier E. A 20-year experience of electron microscopy in the diagnosis of primary ciliary dyskinesia. Eur. Respir. J. 2010;35:1057–1063. doi: 10.1183/09031936.00046209. [DOI] [PubMed] [Google Scholar]
- 6.Escudier E., Duquesnoy P., Papon J.F., Amselem S. Ciliary defects and genetics of primary ciliary dyskinesia. Paediatr. Respir. Rev. 2009;10:51–54. doi: 10.1016/j.prrv.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Mazor M., Alkrinawi S., Chalifa-Caspi V., Manor E., Sheffield V.C., Aviram M., Parvari R. Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am. J. Hum. Genet. 2011;88:599–607. doi: 10.1016/j.ajhg.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merveille A.-C., Davis E.E., Becker-Heck A., Legendre M., Amirav I., Bataille G., Belmont J., Beydon N., Billen F., Clément A. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011;43:72–78. doi: 10.1038/ng.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Becker-Heck A., Zohn I.E., Okabe N., Pollock A., Lenhart K.B., Sullivan-Brown J., McSheene J., Loges N.T., Olbrich H., Haeffner K. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011;43:79–84. doi: 10.1038/ng.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Omran H., Kobayashi D., Olbrich H., Tsukahara T., Loges N.T., Hagiwara H., Zhang Q., Leblond G., O’Toole E., Hara C. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duquesnoy P., Escudier E., Vincensini L., Freshour J., Bridoux A.-M., Coste A., Deschildre A., de Blic J., Legendre M., Montantin G. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2009;85:890–896. doi: 10.1016/j.ajhg.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loges N.T., Olbrich H., Becker-Heck A., Häffner K., Heer A., Reinhard C., Schmidts M., Kispert A., Zariwala M.A., Leigh M.W. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009;85:883–889. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchison H.M., Schmidts M., Loges N.T., Freshour J., Dritsoula A., Hirst R.A., O’Callaghan C., Blau H., Al Dabbagh M., Olbrich H. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012;44:381–389. doi: 10.1038/ng.1106. S1–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panizzi J.R., Becker-Heck A., Castleman V.H., Al-Mutairi D.A., Liu Y., Loges N.T., Pathak N., Austin-Tse C., Sheridan E., Schmidts M. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat. Genet. 2012;44:714–719. doi: 10.1038/ng.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J.B., Gerdes J.M., Haycraft C.J., Fan Y., Teslovich T.M., May-Simera H., Li H., Blacque O.E., Li L., Leitch C.C. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117:541–552. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- 16.Xue J.C., Goldberg E. Identification of a novel testis-specific leucine-rich protein in humans and mice. Biol. Reprod. 2000;62:1278–1284. doi: 10.1095/biolreprod62.5.1278. [DOI] [PubMed] [Google Scholar]
- 17.McClintock T.S., Glasser C.E., Bose S.C., Bergman D.A. Tissue expression patterns identify mouse cilia genes. Physiol. Genomics. 2008;32:198–206. doi: 10.1152/physiolgenomics.00128.2007. [DOI] [PubMed] [Google Scholar]
- 18.Blouin J.L., Meeks M., Radhakrishna U., Sainsbury A., Gehring C., Saïl G.D., Bartoloni L., Dombi V., O’Rawe A., Walne A. Primary ciliary dyskinesia: A genome-wide linkage analysis reveals extensive locus heterogeneity. Eur. J. Hum. Genet. 2000;8:109–118. doi: 10.1038/sj.ejhg.5200429. [DOI] [PubMed] [Google Scholar]
- 19.Sun Z., Amsterdam A., Pazour G.J., Cole D.G., Miller M.S., Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004;131:4085–4093. doi: 10.1242/dev.01240. [DOI] [PubMed] [Google Scholar]
- 20.Kishimoto N., Cao Y., Park A., Sun Z. Cystic kidney gene seahorse regulates cilia-mediated processes and Wnt pathways. Dev. Cell. 2008;14:954–961. doi: 10.1016/j.devcel.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 21.Kavlie R.G., Kernan M.J., Eberl D.F. Hearing in Drosophila requires TilB, a conserved protein associated with ciliary motility. Genetics. 2010;185:177–188. doi: 10.1534/genetics.110.114009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsushima N., Tachi N., Kuroki Y., Enkhbayar P., Osaki M., Kamiya M., Kretsinger R.H. Structural analysis of leucine-rich-repeat variants in proteins associated with human diseases. Cell. Mol. Life Sci. 2005;62:2771–2791. doi: 10.1007/s00018-005-5187-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bella J., Hindle K.L., McEwan P.A., Lovell S.C. The leucine-rich repeat structure. Cell. Mol. Life Sci. 2008;65:2307–2333. doi: 10.1007/s00018-008-8019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ceulemans H., De Maeyer M., Stalmans W., Bollen M. A capping domain for LRR protein interaction modules. FEBS Lett. 1999;456:349–351. doi: 10.1016/s0014-5793(99)00965-5. [DOI] [PubMed] [Google Scholar]
- 25.Tamalet A., Clement A., Roudot-Thoraval F., Desmarquest P., Roger G., Boulé M., Millepied M.C., Baculard T.A., Escudier E. Abnormal central complex is a marker of severity in the presence of partial ciliary defect. Pediatrics. 2001;108:E86. doi: 10.1542/peds.108.5.e86. [DOI] [PubMed] [Google Scholar]
- 26.Vos M.J., Hageman J., Carra S., Kampinga H.H. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry. 2008;47:7001–7011. doi: 10.1021/bi800639z. [DOI] [PubMed] [Google Scholar]
- 27.Freshour J., Yokoyama R., Mitchell D.R. Chlamydomonas flagellar outer row dynein assembly protein ODA7 interacts with both outer row and I1 inner row dyneins. J. Biol. Chem. 2007;282:5404–5412. doi: 10.1074/jbc.M607509200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.