

Abstract
SOX2 is an early developmental transcription factor and marker of stem cells that has recently been implicated in the development of the pituitary gland. Heterozygous SOX2 mutations have been described in patients with hypopituitarism and severe ocular abnormalities. In the majority of published cases, the pituitary gland is either small or normal in size. Here, we report two unrelated patients with SOX2 haploinsufficiency (a heterozygous gene deletion and a novel c.143TC>AA/p.F48X mutation) who developed nonprogressive pituitary tumors of early onset, suggesting a congenital etiology. The truncating mutation resulted in significant loss of function and impaired nuclear localization of the mutant protein, in addition to a failure to repress β-catenin transcriptional activity in vitro. This is the first indication that SOX2 haploinsufficiency is implicated in the generation of pituitary tumors with distinct clinical characteristics, possibly mediated via its effects on the Wnt signaling pathway. 32:1376–1380, 2011. ©2011 Wiley Periodicals, Inc.
Keywords: SOX2, pituitary, tumors, ß-catenin
SOX2 (MIM# 184429), a member of the SOXB1 family of transcription factors, is a widely expressed marker of progenitor and stem cells. The single exon gene encodes a 317 amino acid protein that contains an N-terminal domain, a DNA-binding high-mobility group (HMG) domain and a C-terminal transcriptional activation domain [Collignon et al., 1996]. SOX2 haploinsufficiency in both mouse and human has been associated with variable hypopituitarism associated with anterior pituitary hypoplasia, suggesting that it has a critical role in the development of the anterior pituitary [Kelberman et al., 2006]. Heterozygous de novo mutations in humans are associated with severe ocular phenotypes (bilateral anophthalmia or severe microphthalmia) and hypogonadotropic hypogonadism (HH) with or without associated abnormalities such as esophageal atresia, male genital anomalies, developmental delay, sensorineural deafness, hippocampal malformation, hypoplasia of the corpus callosum, and hypothalamic hamartoma [Kelberman et al., 2006, 2008]. In humans, SOX2 expression is detected within Rathke's pouch and maintained throughout the development of the anterior pituitary, as well as in the presumptive hypothalamus and neural ectoderm [Kelberman et al., 2008]. In neural progenitors, SOX2 downregulation is associated with progression from a proliferating undifferentiated state to a committed phenotype [Graham et al., 2003]. In the murine adult pituitary, SOX2 expression is maintained in a small population of cells lining the pituitary cleft, which show many of the properties of progenitor cells and have the ability to differentiate into all hormone-producing cell types [Fauquier et al., 2008].
The specificity of the action of SOX proteins depends largely on their interaction with partner proteins. Recent data have suggested an interaction with components of another early developmental pathway, the Wnt/β-catenin signaling pathway. Members of the Wnt/β-catenin pathway are expressed in the developing pituitary and are implicated in the maintenance of normal morphology of the gland and the determination of hormone-secreting cell types [Olson et al., 2006; Potok et al., 2008]. Xenopus XSox3 and XSox17 as well as murine SOX2 interact with β-catenin and repress its activity in vitro [Mansukhani et al., 2005; Zorn et al., 1999]. We have shown that human SOX2 is also capable of inhibiting β-catenin-mediated transcriptional activation [Kelberman et al., 2008]. Recent studies have suggested that aberrant activation of the Wnt pathway, resulting from sustained β-catenin activation or downregulation of Wnt-inhibitors is associated with the development of pituitary tumors [Buslei et al., 2005; Elston et al., 2008; Gaston-Massuet et al., 2011]. Here we report, for the first time to our knowledge, the identification of heterozygous SOX2 mutations in two unrelated patients in association with pituitary tumors of likely congenital origin and we provide in vitro evidence that disruption of the SOX2/β-catenin interaction may be the molecular mechanism underlying some human pituitary tumors.
Case 1 is a female patient with bilateral anophthalmia who presented for the first time at the age of 18 years for assessment of pubertal delay. She was the second child of nonconsanguineous parents born at term with a birth weight of −1.0 standard deviation scor (SDS), and had severely impaired language development and delayed motor milestones. At presentation, she was prepubertal (Tanner staging 1) with a height of 144.8 cm (−3.12 SDS). Basal endocrine investigations demonstrated undetectable estradiol with low basal gonadotropins and a flat luteinising hormone (LH) and follicle stimulating hormone (FSH) response to GnRH stimulation confirming a diagnosis of HH (Table 1). Magnetic resonance imaging (MRI) revealed a sellar tumor with a cystic component, extending into the suprasellar area (Fig. 1A), without evidence of compression syndrome. Hormone replacement treatment was declined and at the age of 24 years, she went on to develop spontaneous but incomplete puberty (breast Tanner stage 2). Repeat MRI at that stage as well as sequential MR imaging over a period of 10 years did not show any significant change in the size or morphology of the tumor (Fig. 1B). There was no evidence of development of additional pituitary hormone deficiencies (Table 1) and the patient has been managed conservatively.
Table 1.
Endocrine investigations in patients carrying SOX2 heterozygous mutations
| Patient 1 (Ht SOX2 deletion) | Patient 2 (c.143TC>AA, p.F48X) | ||||
|---|---|---|---|---|---|
| 1a | 1b | normal range | normal range | ||
| Total T4 (µg/dl) | – | 8.98 | 4.5–12.5 | ||
| FT4 (ng/dl) | 1.40 | 0.9–1.9 | 1.24 | 0.7–1.94 | |
| TSH (mU/L) | 1.41 | 1.68 | 0.4–4.5 | – | |
| IGF-1 (µg/L) | 270.7 | 291 | 96–502 | 48 | 51–303 |
| IGFBP3 (mg/L) | 1.42 | 0.80–3.90 | |||
| Random GH (ng/ml) | 6.84 | 0.60 | – | – | – |
| Basal cortisol (µg/dl) | 13.70 | 22.51 | 5–25 | 18.05 | 5–25 |
| Peak cortisol (µg/dl) | – | – | 27.80 | ||
| Basal LH (U/L) | 0.19 | 0.10 | |||
| Basal FSH (U/L) | 0.31 | 0.47 | |||
| Estradiol (pg/ml) | 25.60 | 9.16 | |||
| Basal testosterone (µg/L) | – | – | – | 0.20 | – |
| Testosterone at 3–week HCG test (µg/L) | 1.73 | ||||
Figure 1.
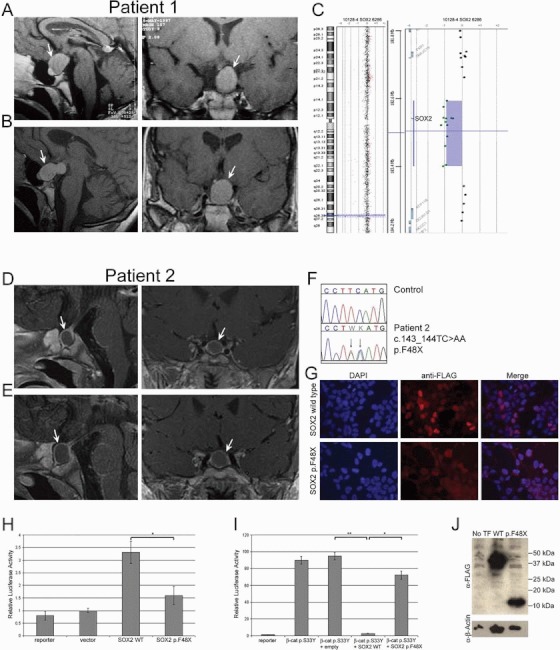
Identification and characterization of novel SOX2 heterozygous mutations in association with slow-growing sellar tumors. (A and B) Midline sagittal (left) and coronal (right) MRI scans of patient 1 at 18 years (A) and 28 years of age (B) revealing a large mass in the location of the pituitary gland (arrows). (C) Array-CGH profile from patient 1 showing the ratio of probes, each represented by a single dot plotted as a function of chromosome position; loss of copy number of a probe shifts the ratio to the left showing deletion of SOX2. (D and E) Midline sagittal (left) and coronal (right) MRI scans of patient 2 at 17 months (D) and at 32 months of age (E) showing a large mass in the location of the pituitary gland (arrows). (F) Sequence electropherogram showing a two-base substitution c.143_144TT>AA (p.F48X) in patient 2. Nucleotide numbering reflects cDNA with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence. (G) Overexpression of FLAG-tagged SOX2 p.F48X in HEK293T cells results in impaired localization of the protein whereas FLAG-tagged wild type SOX2 localizes in the nucleus. Nuclei are counterstained with DAPI. (H) Luciferase assay using the Hesx1 minimal promoter demonstrates impaired activation of the promoter through expression of SOX2 p.F48X (t-test, P = 0.0006). (I) Luciferase assay using the TOPFLASH reporter reveals that expression of the SOX2 p.F48X mutant protein does not result in repression of Wnt signaling, activated by expression of a stable mutant form of β-catenin (p.S33Y) (t-test, P = 0.0002), whereas expression of wild-type SOX2 represses this activation (t-test, P < 0.0001). (J) Western blot analysis showing similar expression levels of both wild-type (WT) and p.F48X proteins. Although the intensity of the signal obtained with the α-FLAG antibody is not comparable, this is due to unequal loading of protein extracts in the gel, as shown by the loading control (α-β-Actin).
Case 2 is a male infant, diagnosed at birth with bilateral anophthalmia, who was referred to the pediatric endocrine department at the age of 17 months for investigation of micropenis. He was the first child of nonconsanguineous parents, born at term with a birth weight of 2.83 kg (−1.7 SDS). At the time of presentation, his weight was 8.6 kg (−2.59 SDS) with a length of 74.8 cm (−2.24 SDS). He had a stretched penile length of 2.5 cm and a hypoplastic scrotum with testes of 0.5–1 ml palpable high in the scrotal sacs. Endocrine investigations revealed a normal FT4 of 1.24 ng/dl (16 pmol/L, normal range 9–25 pmol/L), peak cortisol to synacthen of 27.8 µg/dl (769 nmol/L) with a low IGF1 48 µg/L (normal range 51–303 µg/L). A 3-week hCG test demonstrated a testosterone response that was consistent with HH (1.73 µg/L, 6.0 nmol/L). MRI at that point revealed a pituitary mass with a cystic component extending to the suprasellar area (Fig. 1D). Review of an MRI scan performed in the neonatal period, which was thought to have been normal apart from absent prechiasmatic optic nerves, revealed that the mass had been present at that stage. Subsequent imaging demonstrated a modest increase in size between the age of 17 and 32 months (Fig. 1E). Based on the auxological data and the low IGF-1, he commenced treatment with rhGH (0.025 mg/kg/day) and he is under regular clinical and neuroradiological follow-up. In both cases, there was no family history of eye abnormalities, pubertal delay, or infertility.
DNA analysis using array CGH identified a heterozygous 731-kb deletion on Chr3q26 encompassing SOX2 in patient 1 (Fig. 1C); this deletion was not identified in the unaffected mother, but paternal DNA was unavailable. Patient 2 was heterozygous for a mutation (c.143TC>AA) resulting in the substitution of phenylalanine at position 48 by a stop codon, predicted to generate a truncated protein lacking most of the HMG domain and the C-terminal domain of SOX2 (Fig. 1F). DNA from either parent was unavailable.
To characterize the functional consequences of the p.F48X truncated protein, we first analyzed the cellular localization of this mutant protein in HEK293T cells (Fig. 1G). Anti-FLAG immunostaining of cells transfected with wild-type FLAG-SOX2 revealed normal nuclear localization of the protein. However, FLAG-p.F48X protein was detected mainly in the cytoplasm, suggesting a failure of the mutant protein to concentrate in the nucleus. This is not unexpected as this mutation introduces a stop codon prior to the HMG DNA-binding domain, which contains the nuclear localization signals.
Next, we assessed the transcriptional activity of the p.F48X mutant protein on the murine Hesx1 promoter, which contains SOX-binding sites and has been previously shown to be regulated by SOX2 [Eroshkin et al., 2002] (Fig. 1H). Transfection of plasmids expressing wild-type SOX2 led to a 3.31-fold (±0.44 SD) increase in luciferase expression levels relative to cells transfected with empty vector. In contrast, expression of the p.F48X mutant protein resulted in only a 1.59-fold (±0.36 SD) activation of the basal reporter activity (t-test, P = 0.0006). Similar effects have been reported for other SOX2 mutant proteins previously identified in patients with hypopituitarism and eye defects [Kelberman et al., 2006, 2008].
Finally, we investigated the ability of the p.F48X mutant protein to antagonize the β-catenin-mediated activation of the TOPFLASH reporter [Korinek et al., 1997] (Fig. 1I). Transfection of the constitutively active p.S33Y mutant β-catenin resulted in an 89.7-fold (±4.6 SD) activation of the basal TOPFLASH reporter activity. This transactivation of the reporter was significantly reduced when cells were cotransfected with plasmids expressing wild-type SOX2 (2.8-fold, ±0.34 SD, t-test, P < 0.0001). In contrast, cotransfection of a construct expressing the p.F48X mutant SOX2 failed to suppress the β-catenin-mediated activation of TOPFLASH compared with the wild-type SOX2 (72.3-fold, ±4.91 SD, t-test, P = 0.0002). These differences were not due to impaired synthesis of SOX2 protein, as confirmed by western blot using whole-cell extracts following transfection with wild-type and p.F48X FLAG-SOX2 (Fig 1J). Together, these experiments demonstrate that the p.F48X mutant protein is severely impaired in its transactivation activity as well as its ability to antagonise β-catenin-mediated transcriptional activation.
The possible oncogenic potential of SOX2 has been demonstrated in breast cancer [Chen et al., 2008] and its expression is upregulated in up to 23% of squamous cell lung cancers [Bass et al., 2009] as well as in small cell lung cancer [Maddison et al., 2010], squamous head and neck carcinomas [Freier et al., 2010], meningiomas [Comtesse et al., 2005], glioblastomas [Schmitz et al., 2007], pancreatic [Sanada et al., 2006], hepatocellular and bladder carcinomas, prostate cancer, and in seminomas [Schoenhals et al., 2009]. These findings are in contrast to our report whereby SOX2 haploinsufficiency, rather than its upregulation, resulted in the development of a pituitary mass, which would suggest that SOX2 can also act as a tumor suppressor. Indeed, the oncogenic/tumor suppressor potential of SOX2 seems to be cell-type dependent. SOX2 expression is downregulated in gastric carcinomas [Li et al., 2004; Otsubo et al., 2008] and in regions of intestinal metaplasia in patients with Barrett's esophagus. In addition, the esophagus of hypomorphic mutant mice (Sox2EGFP/COND), expressing only 17% of wild-type SOX2 levels, has an appearance resembling mucus metaplasia [Que et al., 2007]. In these cell types, as in the developing pituitary, SOX2 may play a role in the control of the cell cycle through its interaction with the canonical Wnt signaling pathway. Wnt signaling plays an essential role during pituitary development in the control of proliferation of Rathke's pouch precursors and in the differentiation of the Pouf1-lineage (also known as Pit1) [Olson et al., 2006]. Overactivation of the Wnt pathway in the mouse leads to hyperplasia of the embryonic pituitary due to a significant increase in proliferation [Gaston-Massuet et al., 2008]. Moreover, overactivating mutations in β-catenin have been detected in up to 90% of adamantinomatous craniopharyngiomas (ACP) in humans [Buslei et al., 2005; Kato et al., 2004], a benign and slow-dividing rare hypothalamo-pituitary tumor, while Wnt inhibitors are downregulated in other pituitary tumors [Elston et al., 2008]. We have recently shown that mice expressing a degradation-resistant mutant β-catenin in Rathke's pouch develop pituitary tumors that closely resemble human ACP [Gaston-Massuet et al., 2011]. Our luciferase data clearly show that the p.F48X mutant protein cannot repress β-catenin-mediated transcriptional activation, which is compatible with a model whereby the two identified patients may have developed a pituitary mass during development due to increased proliferation. The nonprogressive nature of these otherwise impressive pituitary lesions supports the suggestion that they have an embryological origin. It could be postulated that in these cases failure of SOX2 to repress β-catenin activity may not be sufficient to sustain the growth of the pituitary mass and other factors (including other members of the SOX family) may be compensating for the impaired SOX2 function.
It is intriguing that only the two patients described in this study, out of many known patients with SOX2 haploinsufficiency, developed pituitary tumors. Suprasellar lesions have so far been reported in six patients with SOX2 mutations, including three cases of hypothalamic hamartomas [Kelberman et al., 2006; Scheider et al., 2009] and three cases of suprasellar arachnoid cysts [Kelberman et al., 2008; Scheider et al., 2009; Wang et al., 2008]. SOX2 mutations are associated with considerable variability in phenotypes and apart from the severe ocular defects, patients rarely exhibit the full spectrum of clinical manifestations. The sensitivity of different tissues to altered dosage of SOX2 may in part explain this observation. As illustrated in the two cases reported here, it is possible that for some congenital pituitary tumors careful follow-up, rather than surgical excision, may be the treatment of choice.
Acknowledgments
We thank Owen Clark for imaging assistance.
Supplementary material
References
- Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, Ramos AH, Woo MS, Weir BA, Getz G, Beroukhim R, O'Kelly M, Dutt A, Rozenblatt-Rosen O, Dziunycz P, Komisarof J, Chirieac LR, Lafargue CJ, Scheble V, Wilbertz T, Ma C, Rao S, Nakagawa H, Stairs DB, Lin L, Giordano TJ, Wagner P, Minna JD, Gazdar AF, Zhu CQ, Brose MS, Cecconello I, Jr UR, Marie SK, Dahl O, Shivdasani RA, Tsao MS, Rubin MA, Wong KK, Regev A, Hahn WC, Beer DG, Rustgi AK, Meyerson M. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41:1238–1242. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buslei R, Nolde M, Hofmann B, Meissner S, Eyupoglu IY, Siebzehnrubl F, Hahnen E, Kreutzer J, Fahlbusch R. Common mutations of beta-catenin in adamantinomatous craniopharyngiomas but not in other tumours originating from the sellar region. Acta Neuropathol. 2005;109:589–597. doi: 10.1007/s00401-005-1004-x. [DOI] [PubMed] [Google Scholar]
- Chen Y, Shi L, Zhang L, Li R, Liang J, Yu W, Sun L, Yang X, Wang Y, Zhang Y, Shang Y. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J Biol Chem. 2008;283:17969–17978. doi: 10.1074/jbc.M802917200. [DOI] [PubMed] [Google Scholar]
- Collignon J, Sockanathan S, Hacker A, Cohen-Tannoudji M, Norris D, Rastan S, Stevanovic M, Goodfellow PN, Lovell-Badge R. A comparison of the properties of Sox-3 with Sry and two related genes, Sox-1 and Sox-2. Development. 1996;122:509–520. doi: 10.1242/dev.122.2.509. [DOI] [PubMed] [Google Scholar]
- Comtesse N, Zippel A, Walle S, Monz D, Backes C, Fischer U, Mayer J, Ludwig N, Hildebrandt A, Keller A, Steudel WI, Lenhof HP, Meese E. Complex humoral immune response against a benign tumor: frequent antibody response against specific antigens as diagnostic targets. Proc Natl Acad Sci USA. 2005;102:9601–9606. doi: 10.1073/pnas.0500404102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston MS, Gill AJ, Conaglen JV, Clarkson A, Shaw JM, Law AJ, Cook RJ, Little NS, Clifton-Bligh RJ, Robinson BG, McDonald KL. Wnt pathway inhibitors are strongly down-regulated in pituitary tumors. Endocrinology. 2008;149:1235–1242. doi: 10.1210/en.2007-0542. [DOI] [PubMed] [Google Scholar]
- Eroshkin F, Kazanskaya O, Martynova N, Zaraisky A. Characterization of cis-regulatory elements of the homeobox gene Xanf-1. Gene. 2002;285:279–286. doi: 10.1016/s0378-1119(02)00393-1. [DOI] [PubMed] [Google Scholar]
- Fauquier T, Rizzoti K, Dattani M, Lovell-Badge R, Robinson IC. SOX2-expressing progenitor cells generate all of the major cell types in the adult mouse pituitary gland. Proc Natl Acad Sci USA. 2008;105:2907–2912. doi: 10.1073/pnas.0707886105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freier K, Knoepfle K, Flechtenmacher C, Pungs S, Devens F, Toedt G, Hofele C, Joos S, Lichter P, Radlwimmer B. Recurrent copy number gain of transcription factor SOX2 and corresponding high protein expression in oral squamous cell carcinoma. Genes Chrom Canc. 2010;49:9–16. doi: 10.1002/gcc.20714. [DOI] [PubMed] [Google Scholar]
- Gaston-Massuet C, Andoniadou CL, Signore M, Jayakody SA, Charolidi N, Kyeyune R, Vernay B, Jacques TS, Taketo MM, Le Tissier P, Dattani MT, Martinez-Barbera JP. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc Natl Acad Sci USA. 2011;12:11482–11487. doi: 10.1073/pnas.1101553108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston-Massuet C, Andoniadou CL, Signore M, Sajedi E, Bird S, Turner JM, Martinez-Barbera JP. Genetic interaction between the homeobox transcription factors HESX1 and SIX3 is required for normal pituitary development. Dev Biol. 2008;324:322–333. doi: 10.1016/j.ydbio.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham V, Khudyakov J, Ellis P, Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39:749–765. doi: 10.1016/s0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- Kato K, Nakatani Y, Kanno H, Inayama Y, Ijiri R, Nagahara N, Miyake T, Tanaka M, Ito Y, Aida N, Tachibana K, Sekido K, Tanaka Y. Possible linkage between specific histological structures and aberrant reactivation of the Wnt pathway in adamantinomatous craniopharyngioma. J Pathol. 2004;203:814–821. doi: 10.1002/path.1562. [DOI] [PubMed] [Google Scholar]
- Kelberman D, de Castro SC, Huang S, Crolla JA, Palmer R, Gregory JW, Taylor D, Cavallo L, Faienza MF, Fischetto R, Achermann JC, Martinez-Barbera JP, Rizzoti K, Lovell-Badge R, Robinson IC, Gerrelli D, Dattani MT. SOX2 plays a critical role in the pituitary, forebrain, and eye during human embryonic development. J Clin Endocrinol Metab. 2008;93:1865–1873. doi: 10.1210/jc.2007-2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JM, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson IC, Dattani MT. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest. 2006;116:2442–2455. doi: 10.1172/JCI28658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van WD, de WR, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Li XL, Eishi Y, Bai YQ, Sakai H, Akiyama Y, Tani M, Takizawa T, Koike M, Yuasa Y. Expression of the SRY-related HMG box protein SOX2 in human gastric carcinoma. Int J Oncol. 2004;24:257–263. [PubMed] [Google Scholar]
- Maddison P, Thorpe A, Silcocks P, Robertson JF, Chapman CJ. Autoimmunity to SOX2, clinical phenotype and survival in patients with small-cell lung cancer. Lung Canc. 2010;70:335–339. doi: 10.1016/j.lungcan.2010.03.002. [DOI] [PubMed] [Google Scholar]
- Mansukhani A, Ambrosetti D, Holmes G, Cornivelli L, Basilico C. Sox2 induction by FGF and FGFR2 activating mutations inhibits Wnt signaling and osteoblast differentiation. J Cell Biol. 2005;168:1065–1076. doi: 10.1083/jcb.200409182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson LE, Tollkuhn J, Scafoglio C, Krones A, Zhang J, Ohgi KA, Wu W, Taketo MM, Kemler R, Grosschedl R, Rose D, Li X, Rosenfeld MG. Homeodomain-mediated beta-catenin-dependent switching events dictate cell-lineage determination. Cell. 2006;125:593–605. doi: 10.1016/j.cell.2006.02.046. [DOI] [PubMed] [Google Scholar]
- Otsubo T, Akiyama Y, Yanagihara K, Yuasa Y. SOX2 is frequently downregulated in gastric cancers and inhibits cell growth through cell-cycle arrest and apoptosis. Br J Cancer. 2008;98:824–831. doi: 10.1038/sj.bjc.6604193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potok MA, Cha KB, Hunt A, Brinkmeier ML, Leitges M, Kispert A, Camper SA. WNT signaling affects gene expression in the ventral diencephalon and pituitary gland growth. Dev Dyn. 2008;237:1006–1020. doi: 10.1002/dvdy.21511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que J, Okubo T, Goldenring JR, Nam KT, Kurotani R, Morrisey EE, Taranova O, Pevny LH, Hogan BL. Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development. 2007;134:2521–2531. doi: 10.1242/dev.003855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanada Y, Yoshida K, Ohara M, Oeda M, Konishi K, Tsutani Y. Histopathologic evaluation of stepwise progression of pancreatic carcinoma with immunohistochemical analysis of gastric epithelial transcription factor SOX2: comparison of expression patterns between invasive components and cancerous or nonneoplastic intraductal components. Pancreas. 2006;32:164–170. doi: 10.1097/01.mpa.0000202947.80117.a0. [DOI] [PubMed] [Google Scholar]
- Schmitz M, Temme A, Senner V, Ebner R, Schwind S, Stevanovic S, Wehner R, Schackert G, Schackert HK, Fussel M, Bachmann M, Rieber EP, Weigle B. Identification of SOX2 as a novel glioma-associated antigen and potential target for T cell-based immunotherapy. Br J Cancer. 2007;96:1293–1301. doi: 10.1038/sj.bjc.6603696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheider A, Bardakjian T, Reis LM, Tyler RC, Semina EV. Novel SOX2 mutations and genotype-phenotype correlation in anophthalmia and microphthalmia. Am J Med Genet. 2009;149:2706–2715. doi: 10.1002/ajmg.a.33098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenhals M, Kassambara A, De VJ, Hose D, Moreaux J, Klein B. Embryonic stem cell markers expression in cancers. Biochem Biophys Res Commun. 2009;383:157–162. doi: 10.1016/j.bbrc.2009.02.156. [DOI] [PubMed] [Google Scholar]
- Wang P, Liang X, Yi J, Zhang Q. Novel SOX2 mutation associated with ocular coloboma in a Chinese family. Arch Ophthalmol. 2008;126:709–713. doi: 10.1001/archopht.126.5.709. [DOI] [PubMed] [Google Scholar]
- Zorn AM, Barish GD, Williams BO, Lavender P, Klymkowsky MW, Varmus HE. Regulation of Wnt signaling by Sox proteins: XSox17 alpha/beta and XSox3 physically interact with beta-catenin. Mol Cell. 1999;4:487–498. doi: 10.1016/s1097-2765(00)80200-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.