

Abstract
Right ventricular (RV) function is the major determinant of mortality in pulmonary arterial hypertension and male sex is a strong predictor of mortality in this disease. The effects of testosterone on RV structure and function in load stress are presently unknown. We tested whether testosterone levels affect RV hypertrophic responses, fibrosis, and function. Male C57BL/6 mice underwent castration or sham followed by pulmonary artery banding (PAB) or sham. After recovery, testosterone pellets were placed in a subset of the castrated mice and mice were maintained for at least two weeks, when they underwent hemodynamic measurements and tissues were harvested. Plasma levels of testosterone were reduced by castration and repleted by testosterone administration. In PAB, castration resulted in lower right ventricle/left ventricle + septum (RV/LV+S), and myocyte diameter (P < 0.05). Replacement of testosterone normalized these parameters and increased RV fibrosis (P < 0.05). Two weeks of PAB resulted in increased RV systolic pressure in all groups with decreased markers of RV systolic and diastolic function, specifically reduced ejection fraction and increased time constant, and dPdt minimum (P < 0.05), though there was minimal effect of testosterone on hemodynamic parameters. Survival was improved in mice that underwent castration with PAB compared with PAB alone (P < 0.05). Testosterone affects RV hypertrophic response to load stress through increased myocyte size and increased fibrosis in mice. Castration and testosterone replacement are not accompanied by significant alterations in RV in vivo hemodynamics, but testosterone deprivation appears to improve survival in PAB. Further study of the role of testosterone in RV dysfunction is warranted to better understand these findings in the context of human disease.
Keywords: pulmonary hypertension, right ventricle, sex hormones, testosterone
Right ventricular (RV) dysfunction is a major source of morbidity in many types of pulmonary vascular diseases including pulmonary arterial hypertension (PAH), pulmonary embolism, and left heart failure.[1–4] In PAH, RV dysfunction is the most important prognostic feature.[1] Despite the importance of the RV, little is known about the determinants of RV structure and function or its response to stress.[5]
Emerging data have begun to elucidate the determinants of RV size and function in the healthy human population, including the effects of physical activity, lung function, and sex hormones.[6–8] In the general healthy population, higher levels of the anabolic sex hormones testosterone in men and dihydroepiandrosterone in women have been associated with higher RV mass, volume, and stroke volume.[8] The functional consequences of these finding in the stressed human RV are presently unknown.
In contrast, large-scale registry data of patients with PAH have shown shortened survival in male patients.[9–11] Although propensity to develop PAH is closely linked with female sex, survival in established disease appears to be worse in male patients. Although the influence of testosterone on the pulmonary vasculature is a potential mechanism underlying this finding, the importance of RV function in determining survival in PAH suggests that male sex hormones may be exerting a direct influence on the RV. While androgen receptors have been shown to have expression in both left and right ventricles (LV and RV), most research on the effects of testosterone and its metabolite dihydrotestosterone have focused on the LV. In the LV, testosterone has been shown to induce myocyte hypertrophy through the mammalian target of rapamycin complex 1 pathway and focal adhesion kinases.[12,13] On a physiologic level, testosterone induces LV concentric remodeling. Finally, supraphysiologic testosterone administration in a rodent model of LV hypertrophy increased fibrosis formation.[14] However, there have been no studies to date on the effects of testosterone on the RV and given the unique anatomy, physiology, and molecular milieu, findings in the LV may not be applicable to the RV.[5,15–17]
In order to study the RV-specific effects of testosterone independent of the pulmonary vasculature, we chose to use the pulmonary artery banding (PAB) model. In this model, we hypothesized that testosterone would increase hypertrophy but without improvement in RV function. Here we report on the effects of castration and testosterone replacement on RV structure and function with and without load stress.
MATERIALS AND METHODS
The Institutional Animal Care and Uses Committee at Vanderbilt University approved all animal procedures (protocol number M08-083).
Animal model
Male C57BI/6 mice (in-house breeding) at six to seven weeks of age underwent surgical castration or sham operation after anesthesia with 3% inhaled isoflurane. The mouse was placed prone on surgical table, the scrotum was incised, testes identified and removed, and the skin sutured. Mice in each group later underwent PAB or sham at 10 weeks of age as previously described.[18] Briefly, mice were anesthetized with 3% isoflurane, administered intraperitoneal etomidate (20-40 mL/kg), and orotracheally intubated. Animals were mechanically ventilated with isoflurane general anesthesia. A sternal incision was made using a sterile technique, the pericardium was removed, and a partially occlusive tantalum clip was placed around the pulmonary artery (Weck, Research Triangle Park, N.C.). The animal was then sutured closed and allowed to recover from anesthesia. Mice received post-operative analgesic with a subcutaneous injection of 5 mg/kg of carprofen. Sham-treated animals underwent the same procedure without vascular clip placement.
Testosterone replacement
After anesthesia with 3% inhalational isoflurane, a 25 mg testosterone pellet (A-151, Innovative Research of America, Sarasota, Fla.) was placed subcutaneously between the scapulae in a portion of the castrated animals on the day following PAB or sham. Animals were sacrificed 14 days after pellet placement. Pellets were engineered for up to 21 days of continuous release of testosterone.
Echocardiography
Two-dimensional echocardiography was performed using Vivo 770 High-Resolution Image System (VisualSonics Toronto, Canada). Echocardiograms were obtained the day prior to sacrifice under isoflurane anesthetic. In the short axis, LV and RV end-systolic and -diastolic diameters and heart rate were recorded in M-mode.[19,20]
In vivo hemodynamics
Two weeks after PAB or sham, mice were anesthetized with 3% isoflurane, administered intraperitoneal etomidate, and orotracheally intubated. Animals were mechanically ventilated with vaporized isoflurane general anesthesia. Mice were positioned supine, ventral side up on a heated operating table. A vertical incision over the abdomen was made and cautery was used to cut the diaphragm and expose the heart. A 1.4F Mikro-tip catheter was introduced into the surgically exposed heart. Open-chested catheterization was chosen since this method provides better quality pressure-volume loops than the close-chested technique in our hands[18] and has been validated by other groups.[21] Hemodynamics were continuously recorded with a Millar MPVS-300 unit coupled to a Powerlab 8-SP analog-to-digital converter acquired at 1000 Hz and captured to a Macintosh G4 (Millar Instruments Houston, Tex.). Mice were sacrificed using a cervical dislocation. The right and left heart were weighed separately and snap-frozen in liquid nitrogen or placed in formalin for histology.
Testosterone assay
A commercially available ELISA kit was used to assess serum concentrations of testosterone at the time of sacrifice in male mice (RandD systems, Minneapolis, Minn.).
Histology
Hearts were fixed in 10% formalin overnight, then embedded in paraffin, sectioned at a thickness of 5 mm, and stained with Masson's trichrome stain and hematoxylin and eosin. The percentage of collagen per cardiac tissue area was obtained using Nikon NIS-Elements Advanced Research software (Melville, N.Y.) with observer blinded as to tissue source. At least three separate fields per heart were examined each for fibrosis and myocyte diameter. The ratio of area affected by fibrosis (blue color) to total cardiac area in each section was calculated and expressed as percent fibrotic area.[22,23] Myocyte diameter was measured at the maximum cross-sectional area in at least 25 cells per field at 20× magnification for four fields per sample.
PCR
RNA was obtained using Qiagen RNeasy Mini Kit (Qiagen, Valencia, Calif.). First-strand cDNA was made using QuantiTect Reverse Transcription Kit (Qiagen) from 1 mg total RNA. Quantitative real-time PCR was performed using a total reaction volume of 25 μl, containing 5 μl of diluted cDNA, 12.5 μl SYBR Green Supermix (Applied Biosystems Foster City, Calif.), and 0.03 μl of each oligonucleotide primer (250 mM). PCR was carried out in a StepOnePlus Real-Time PCR System (Applied Biosystems) using 40 cycles of 95°C for 15 sec followed by 60°C for one minute with a 10-min 95°C initial soak. Each measurement was made in triplicate and expressed relative to the detection of the standard HPRT. PCR was preformed for the primer sets HPRT, atrial natriuretic peptide (ANP), and brain natriuretic peptide (BNP; Integrated DNA Technologies IDTH Coralville, Iowa).
Statistics
All data are presented as mean ± SEM except as otherwise noted. Statistical tests were two-way ANOVA with post-hoc Bonferroni test performed using GraphPad Prism Plus version 5.0 Software (San Diego, Calif.) or Kaplan-Meier survival curve as appropriate. A P value < 0.05 was considered significant.
RESULTS
Testosterone affects RV hypertrophy at baseline and in the context of load stress
Plasma testosterone levels were decreased with castration and increased with subcutaneous testosterone pellet placement (P < 0.05 control vs. castrated and vs. testosterone replacement; data not shown). RV hypertrophy, as measured by RV/LV+S, was affected by both PAB and testosterone status (P < 0.05, Fig. 1A). Although testosterone had minimal effects at baseline on RV fractional weight, in the context of PAB, castration prevented RV hypertrophy, and testosterone replacement returned RV/LV+S ratio to normal. As expected with pulmonary artery clip placement, PAB resulted in RV dilation as measured by m-mode echocardiography, but there was no effect of testosterone on RV diastolic diameter (Fig. 1B).
Figure 1.

(A) RV/LV+S ratio in control and PAB mice. (B) Echocardiography of diastolic RV internal diameter (RVIDd) in control and PAB mice. *P < 0.05 by two-way ANOVA for the effect of testosterone status, #P < 0.05 by two-way ANOVA for the effect of PAB n = 4-12 per group.
Testosterone increases RV fibrosis and myocyte hypertrophy in response to load stress
At baseline testosterone had no significant effect on RV fibrosis as measured by Masson Trichrome stain (Fig. 2). After two weeks of PAB with intact testes, there was a marked increase in fibrotic area within the RV. Although castration was not associated with an attenuation of fibrosis in the context of PAB, testosterone replacement affected a nearly fivefold increase in fibrotic area. Myocyte size was measured in HandE-stained histologic specimens both in sham-treated and 2-week PAB animals with intact testes, castration, and testosterone replacement (Fig. 2). At baseline, there was a small effect of both castration and testosterone replacement on RV myocyte size. After two weeks of PAB, there was a 38% increase in RV myocyte size in mice with intact testes. Load-induced myocyte hypertrophy was completely prevented by castration. With testosterone replacement, myocyte hypertrophy was further augmented. We also performed rtPCR for molecular markers of cardiac hypertrophy, ANP and BNP (Fig. 3), and found a significant interaction by two-way ANOVA of both PAB and castration on ANP expression level. In contrast, BNP was increased over 50-fold with PAB, but was not affected by castration.
Figure 2.

(A) Representative images of Masson-Trichrome stain in RV from Control and PAB in intact testes (CON), castration (CAS), or castration + testosterone (CAS+TEST); left column, sham operation; right column, PAB. (B) Percent fibrotic area. (C) Myocyte size. *P < 0.05 by two-way ANOVA for the effect of testosterone status, #P < 0.05 by two-way ANOVA for the effect of PAB, n = 3 per group.
Figure 3.

Molecular expression levels in the RV. (A) Atrial natriuretic peptide (ANP) and (B) brain natriuretic peptide (BNP). Long bar indicates a significant difference associated with PAB by two-way ANOVA. *P < 0.05 by two-way ANOVA for interaction of PAB with castration status, n = 3 per group.
Testosterone has minimal effects on RV function as measured by in vivo hemodynamics
The effects of 14 days of PAB or sham in the context of intact testes, castration, or castration and testosterone replacement were measured in the open chest, mechanically ventilated, and anesthetized mouse (Fig. 4). The heart rate was not altered by any of the experimental conditions (data not shown). Without load stress, there were no significant effects of testosterone status on RV systolic pressure, diastolic pressure, systolic, or diastolic function. PAB resulted in a significant increase in RV systolic pressure. As expected with a fixed vascular clip providing load stress, RV systolic pressure was unchanged in response to testosterone deprivation or administration compared with animals with intact testes. PAB worsened RV systolic and diastolic function as evidenced by increased RV end-diastolic pressure, reduced ejection fraction and contractility index, prolonged time constant (Tau), and decreased dPdt minimum.
Figure 4.

In vivo hemodynamic response in sham controls (Control) and with PAB with intact testes (Control), castration or castration + testosterone. Normalized data are shown for maximum power, ejection fraction, and cardiac output. Long bars indicate P < 0.05 for the effect of PAB by two-way ANOVA, n = 4-6 per group.
Without load stress, castration resulted in a nonsignificant trend to decreased RV maximum power, maximum dPdt, and contractility index. Testosterone did not significantly affect these values. In the context of PAB, castration resulted in a trend toward improved markers of RV function including decreased end-diastolic pressure and time constant with increased ejection fraction, cardiac output, and contractility index.
Castration improves survival in RV load stress
In order to study the long-term consequences of testosterone deprivation on RV load stress responses, mice underwent castration or sham without PAB and were studied for 10 months. Castration resulted in improved survival after PAB in male mice with a mortality of 60% in the PAB group with intact testes and 0% in the group with concomitant castration (Fig. 5). Echocardiography was performed in a portion of this cohort six weeks after PAB and there was no difference in cardiac output between PAB with intact testes from PAB with castration (Fig. 6).
Figure 5.

Kaplan-Meier survival analysis in long-term PAB with intact testes (CON.PAB) and castration (CAS.PAB), P < 0.05, n = 10 each group.
Figure 6.
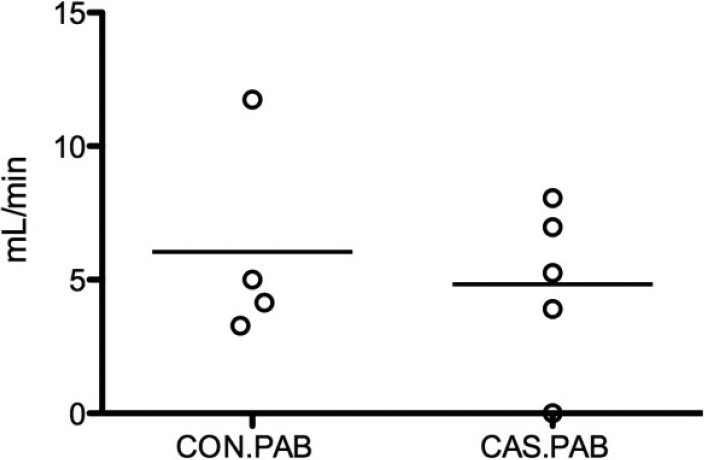
Echocardiographic measurement of cardiac output after 6 weeks of PAB in male mice with intact testes (CON.PAB) and with castration (CAS. PAB). P = non-signficant. Bars indicate the mean value.
DISCUSSION
In this study, we show that the hypertrophic response in the RV to load stress is diminished by castration and that replacement of testosterone is associated with return of RV hypertrophy. Fibrosis is enhanced with testosterone replacement. Although there were no significant in vivo hemodynamic alterations in response to testosterone manipulation in mice that have undergone PAB, long-term survival was markedly worse in mice with PAB and intact testes compared to castrated mice. Taken together, these findings demonstrate a potentially negative effect of testosterone in the stressed RV mediated through dysfunctional RV hypertrophy.
Recent studies have demonstrated that survival rate is substantially worse in males with PAH and, as survival is closely linked to RV function, worse mortality in males may be due to a negative effect of testosterone on the RV.[1,9–11] Recent human data have shown that serum testosterone levels correlate with RV mass, volume, and stroke volume in a control population free from cardiovascular disease, suggesting a role for this sex hormone in regulation of RV size and function.[8] The effects of testosterone on the RV have not been extensively studied; however, there is evidence that androgen receptors are present in rodent ventricular tissue, where they are more abundant than atrial tissue and predominantly located in the nucleus and cytosol.[24] In the LV, gonadectomy in spontaneously hypertensive rats results in diminished heart weight and is accompanied by a shift in the myosin isoenzyme pattern.[25] Similarly, testosterone infusion in the guanylyl cyclase-A knockout mouse resulted in increased cardiac mass and fibrosis, whereas androgen deprivation through flutamide attenuated these findings.[14] Isolated cardiomyocytes increase size and leucine incorporation with testosterone exposure through the mammalian target of rapamycin complex 1 pathway.[12] Additionally, dihydrotestosterone, a metabolite of testosterone, also increases cardiomyocyte hypertrophic response and gross organ hypertrophy.[13,26] In summary, testosterone appears to support LV cardiomyocyte hypertrophy, but long-term and functional effects of this hormone in the myocardium are little studied. Moreover, the RV has a unique embryology, structure, and function, and thus may have different responses to testosterone from what has been found in the LV.[5]
Our data are congruent with other LV data showing increased cardiomyocyte size in mice with intact testes or testosterone replacement. Again, similar to others, we found that fibrosis is accentuated by testosterone in the unstressed RV and this augmentation with testosterone is markedly enhanced in the context of load stress.
Because of the important functional significance of the RV in human disease, we chose to examine markers of RV stress response using in vivo hemodynamics and survival analysis. Despite changes in morphometry of the RV in relation to testosterone levels and PAB, we found minimal change in RV function as assessed by in vivo hemodynamics. As expected in this mechanical model, PAB resulted in constant load stress as assessed by RV systolic pressure in all three hormone exposure groups. PAB decreased ejection fraction, and worsened diastolic function as measured by the end-diastolic pressure, Tau (time constant), and minimum dPdt. Although there was a trend to improved end-diastolic pressure with castration and improved ejection fraction and diastolic function markers, these were not statistically significant. It is possible that longer exposure to either castration or testosterone administration may be required to assess the hemodynamic effects of this sex hormone on the RV. In sum, the effects of testosterone levels on RV in vivo hemodynamics in PAB are less significant than the effects on RV hypertrophy and fibrosis.
Because of disparate effects of testosterone levels on RV structure and short term in vivo hemodynamics, we undertook a study of prolonged exposure to PAB with and without castration. We found that castration resulted in substantially improved survival in the context of PAB, suggesting that lower testosterone levels may be protective in mice with RV load stress and that hypertrophy of the RV in the PAB model is likely not a form of physiologic hypertrophy, but rather represents pathologic hypertrophy. Because of the short-term nature of testosterone supplementation by subcutaneous pellets, we were not able to administer testosterone for an extended period of time in order to determine if improved survival with castration was normalized with testosterone administration.
These data suggest that testosterone may play a detrimental role in the RV in load stress and these findings may underlie some of the differences found in survival between males and females with established PAH. However, it is possible that a beneficial effect of estrogens on the RV underlies this human observation. Alternatively, there may be a benefit to different doses of testosterone, and future studies on lower doses of testosterone or replication of these study results in a different mouse strain with higher levels of testosterone at baseline (e.g., CD-1) may elicit a subtle beneficial effect. Additionally, these findings are in rodents and further studies of sex hormones in humans with RV failure are needed to confirm the clinical significance of these data. The downstream molecular mechanisms underlying the influence of testosterone on the RV will be important future directions of study in order to define how this hormone may negatively influence RV stress responses.
In conclusion, we found that testosterone affects RV hypertrophic response to load stress through increased myocyte size and increased fibrosis in mice. Castration and testosterone replacement are not accompanied by significant alterations in RV in vivo hemodynamics in the short term, but testosterone deprivation appears to improve survival in PAB. Future study of the influence of sex hormones on RV structure and function in health and disease is warranted.
Footnotes
Source of Support: NIH K08 HL093363.
Conflict of Interest: None declared.
REFERENCES
- 1.D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115:343–9. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 2.Tapson VF. Acute pulmonary embolism. N Engl J Med. 2008;358:1037–52. doi: 10.1056/NEJMra072753. [DOI] [PubMed] [Google Scholar]
- 3.Karatasakis GT, Karagounis LA, Kalyvas PA, Manginas A, Athanassopoulos GD, Aggelakas SA, et al. Prognostic significance of echocardiographically estimated right ventricular shortening in advanced heart failure. Am J Cardiol. 1998;82:329–34. doi: 10.1016/s0002-9149(98)00344-0. [DOI] [PubMed] [Google Scholar]
- 4.Thompson WP. Commonest cause of hypertrophy of the right ventricle-left ventricular strain and failure. Am Heart J. 1936;12:641–9. [Google Scholar]
- 5.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, et al. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation. 2006;114:1883–91. doi: 10.1161/CIRCULATIONAHA.106.632208. [DOI] [PubMed] [Google Scholar]
- 6.Aaron CP, Tandri H, Barr RG, Johnson WC, Bagiella E, Chahal H, et al. Physical activity and right ventricular structure and function: the MESA-right ventricle study. Am J Respir Crit Care Med. 2011;183:396–404. doi: 10.1164/rccm.201003-0469OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barr RG, Bluemke DA, Ahmed FS, Carr JJ, Enright PL, Hoffman EA, et al. Percent emphysema, airflow obstruction, and impaired left ventricular filling. N Engl J Med. 2010;362:217–27. doi: 10.1056/NEJMoa0808836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ventetuolo CE, Ouyang P, Bluemke DA, Tandri H, Barr RG, Bagiella E, et al. Sex hormones are associated with right ventricular structure and function: The MESA-Right ventricle study. Am J Respir Crit Care Med. 2011;183:659–67. doi: 10.1164/rccm.201007-1027OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension. Insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL) Circulation. 2010;122:164–72. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 10.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–63. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 11.Humbert M, Sitbon O, Yaïci A, Montani D, O’Callaghan DS, Jaïs X, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010;36:549–55. doi: 10.1183/09031936.00057010. [DOI] [PubMed] [Google Scholar]
- 12.Altamirano F, Oyarce C, Silva P, Toyos M, Wilson C, Lavandero S, et al. Testosterone induces cardiomyocyte hypertrophy through mammalian target of rapamycin complex 1 pathway. J Endocrinol. 2009;202:299–307. doi: 10.1677/JOE-09-0044. [DOI] [PubMed] [Google Scholar]
- 13.Koshman YE, Piano MR, Russell B, Schwertz DW. Signaling responses after exposure to 5 alpha-dihydrotestosterone or 17 beta-estradiol in norepinephrine-induced hypertrophy of neonatal rat ventricular myocytes. J Appl Physiol. 2010;108:686–96. doi: 10.1152/japplphysiol.00994.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Kishimoto I, Saito Y, Harada M, Kuwahara K, Izumi T, et al. Androgen contributes to gender-related cardiac hypertrophy and fibrosis in mice lacking the gene encoding guanylyl cyclase-A. Endocrinology. 2004;145:951–8. doi: 10.1210/en.2003-0816. [DOI] [PubMed] [Google Scholar]
- 15.Dai YS, Cserjesi P, Markham BE, Molkentin JD. The transcription factors GATA4 and dHAND physically interact to synergistically activate cardiac gene expression through a p300-dependent mechanism. J Biol Chem. 2002;277:24390–8. doi: 10.1074/jbc.M202490200. [DOI] [PubMed] [Google Scholar]
- 16.Thomas T, Yamagishi H, Overbeek PA, Olson EN, Srivastava D. The bHLH factors, dHAND and eHAND, specify pulmonary and systemic cardiac ventricles independent of left-right sidedness. Dev Biol. 1998;196:228–36. doi: 10.1006/dbio.1998.8849. [DOI] [PubMed] [Google Scholar]
- 17.Hemnes AR, Forfia PR, Champion HC. Assessment of pulmonary vasculature and right heart by invasive haemodynamics and echocardiography. Int J Clin Pract Suppl. 2009;162:4–19. doi: 10.1111/j.1742-1241.2009.02110.x. [DOI] [PubMed] [Google Scholar]
- 18.Johnson JA, West J, Maynard KB, Hemnes AR. ACE2 improves right ventricular function in a pressure overload model. PLoS One. 2011;6:e20828. doi: 10.1371/journal.pone.0020828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Syed F, Diwan A, Hahn HS. Murine echocardiography: A practical approach for phenotyping genetically manipulated and surgically modeled mice. J Am Soc Echocardiogr. 2005;18:982–90. doi: 10.1016/j.echo.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Collins KA, Korcarz CE, Lang RM. Use of echocardiography for the phenotypic assessment of genetically altered mice. Physiol Genomics. 2003;13:227–39. doi: 10.1152/physiolgenomics.00005.2003. [DOI] [PubMed] [Google Scholar]
- 21.Tabima DM, Hacker TA, Chesler NC. Measuring right ventricular function in the normal and hypertensive mouse hearts using admittance-derived pressure-volume loops. Am J Physiol Heart Circ Physiol. 2010;299:H2069–75. doi: 10.1152/ajpheart.00805.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Gao E, Song J, Zhang XQ, Li J, Koch WJ, et al. Phospholemman and beta-adrenergic stimulation in the heart. Am J Physiol Heart Circ Physiol. 2010;298:H807–15. doi: 10.1152/ajpheart.00877.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heineke J, Wollert KC, Osinska H, Sargent MA, York AJ, Robbins J, et al. Calcineurin protects the heart in a murine model of dilated cardiomyopathy. J Mol Cell Cardiol. 2010;48:1080–7. doi: 10.1016/j.yjmcc.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lizotte E, Grandy SA, Tremblay A, Allen BG, Fiset C. Expression, distribution and regulation of sex steroid hormone receptors in mouse heart. Cell Physiol Biochem. 2009;23:75–86. doi: 10.1159/000204096. [DOI] [PubMed] [Google Scholar]
- 25.Lengsfeld M, Morano I, Ganten U, Ganten D, Ruegg JC. Gonadectomy and hormonal replacement changes systolic blood pressure and ventricular myosin isoenzyme pattern of spontaneously hypertensive rats. Circ Res. 1988;63:1090–4. doi: 10.1161/01.res.63.6.1090. [DOI] [PubMed] [Google Scholar]
- 26.Tivesten A, Bollano E, Nystrom HC, Alexanderson C, Bergstrom G, Holmang A. Cardiac concentric remodelling induced by non-aromatizable (dihydro-)testosterone is antagonized by oestradiol in ovariectomized rats. J Endocrinol. 2006;189:485–91. doi: 10.1677/joe.1.06722. [DOI] [PubMed] [Google Scholar]