

Abstract
Background
Chronic disease accelerates endothelial dysfunction in aging, a process associated with cell senescence. However, the mechanisms underlying this process are unclear. We examined whether endothelial cell (EC)-derived microparticles (MPs) facilitate EC senescence and questioned the role of reactive oxygen species in this process.
Methods and Results
Senescence was induced by sequential passaging of primary mouse ECs. Cells retained phenotypic characteristics of ECs from passage 4 through passage 21. Passage 21 ECs exhibited features of senescence, including increased staining of senescence-associated β-galactosidase (SA-βgal), a greater percentage of cells in G1/G0 phase of the cell cycle, and increased phosphorylation of p66Shc (P<0.05). Microparticle formation from passage 21 ECs was increased versus passage 4 ECs (∼2.2-fold increase versus passage 4, P<0.05), and the Rho kinase inhibitor fasudil blocked this increase. Exposure of passage 4 ECs to MPs shifted cells from a proliferating to a nonproliferating phenotype, as indicated by cell cycle analysis and increased senescence-associated β-galactosidase staining. MPs increased EC generation of O2•− (∼2.7-fold) and H2O2 (∼2.6-fold), effects blocked by apocynin (nicotinamide adenine dinucleotide phosphate oxidase inhibitor) and rotenone (mitochondrial oxidase inhibitor) but not by allopurinol (xanthine oxidase inhibitor). MPs increased expression of cell cycle proteins p 21 cip1 and p16ink4a and stimulated phosphorylation of p66Shc in ECs (P<0.05 versus untreated ECs). Pretreatment with the reactive oxygen species scavenger sodium 4,5-dihydroxybenzene-1,3-disulfonate (tiron) abrogated the prosenescent effects of MPs.
Conclusions
MPs promote EC senescence through nicotinamide adenine dinucleotide phosphate oxidase- and mitochondrial-derived reactive oxygen species. Such redox-sensitive processes may be important in vascular dysfunction in aging. (J Am Heart Assoc. 2012;1:e001842 doi: 10.1161/JAHA.112.001842.)
Keywords: cell cycle, endothelium, microparticles, oxidative stress, senescence
Introduction
Aging is a primary risk factor for the development of cardiovascular disease and is associated with impaired vascular function.1 With aging, structural and functional changes in the vasculature occur gradually and include increased arterial thickness, reduced lumen diameter, and a shift to a dysfunctional endothelium, with impaired vasorelaxation, a pro-oxidative, proinflammatory phenotype, and decreased antithrombotic capacity.2,3 In chronic disease states, such as hypertension, these age-associated changes are accelerated in a process termed “early vascular aging.”4,5 Evidence indicates that during both normal and early vascular aging, healthy endothelial cells (ECs) progressively enter into a senescent, nonreplicative state in which they are metabolically active but do not respond to mitogenic stimuli.4,5 Many of the alterations in senescent ECs are consistent with the decline in endothelial function during vascular aging. In this regard, EC senescence is associated with increased reactive oxygen species (ROS) production, decreased nitric oxide production, increased expression of cellular adhesion molecules, increased adhesion of monocytes, and increased sensitivity to proapoptotic stimuli. 6 Accordingly, EC senescence has been implicated in the pathogenesis of endothelial dysfunction in early and normal vascular aging.7
A central component of vascular aging is the increased production of ROS.8,9 In the endothelium, sources of aging-related increases in ROS production include mitochondria, uncoupled endothelial nitric oxide synthase (eNOS), and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.9,10 Regardless of the source, increased ROS has been implicated in the functional alterations of the endothelium as well as in EC inflammation, apoptosis, and senescence.8,10 These impairments appear causally linked to longevity, as mice that lack the gene encoding p66Shc exhibit reduced systemic and intracellular ROS, improved endothelial function, and a 30% increase in lifespan.11,12 However, while ROS-mediated effects on EC senescence and vascular dysfunction are well established, the stimuli responsible for ROS-mediated increases in EC senescence are unclear.
Associated with EC damage and endothelial dysfunction is the formation of endothelial microparticles (MPs).13–15 MPs are small heterogeneous membranous structures (0.1–1 μm), formed from activated or stressed cells as a result of cytoskeletal reorganization, membrane blebbing, and shedding of membrane fragments into extracellular space.13 MPs of endothelial, platelet, and leukocyte origin are consistently detectable in plasma samples, are increased under conditions of vascular stress/dysfunction, and appear to reflect vascular health.13,15 Recently, our laboratory and others have reported that MPs exert direct effects on the endothelium by increasing endothelial oxidative stress and inflammation, decreasing NO production, or stimulating platelet and macrophage adhesion to ECs.16–18 However, the impact of aging on MP formation and effects of MPs on EC senescence are unclear.
In the present study, we tested the hypothesis that MP formation is increased in senescent ECs and that MPs promote premature endothelial senescence through redox-sensitive processes.
Methods
Cell Culture
The study was approved by the Animal Ethics Committee of the University of Ottawa and performed according to the recommendations of the Canadian Council for Animal Care. ECs were isolated from the aortas of C57BL6 mice and cultured as described previously.17 ECs were seeded on attachment factor-coated polystyrene dishes in Dulbecco's modified Eagle's medium (DMEM, Gibco) containing 10% FCS (Gibco), 50 mg/L of EC growth supplement (Sigma), 10 U/mL heparin, 100 U/mL penicillin/streptomycin, and 1× minimal essential amino acids (Gibco) and placed in a humidified incubator at 37°C and 5% CO2.
For studies involving replicative senescence, ECs were cultured in 10% FCS as mentioned earlier. When cells reached approximately 80% confluence, serial passage was performed (1:5 split ratio) by trypsinization with trypsin-EDTA (Gibco). Experiments used cells in either passage 4 (p4; young, little senescence) or passage 21 (p21; old, highly senescent). Cell viability was assessed by Trypan blue dye exclusion.
For studies examining stress-induced premature senescence, ECs were cultured in 10% FCS as above, and cells were stimulated with either 100 μmol/L H2O2 or MPs (105/mL) in the presence and absence of the O2•− scavenger (O2•− semiquinone-forming) sodium 4,5-dihydroxybenzene-1,3-disulfonate (tiron; 10 μmol/L, Sigma-Aldrich). Tiron forms a semiquinone radical through an electron trapping transfer from O2•−. The doses of H2O2 and MPs were chosen from pilot experiments aimed at identifying optimal induction of senescence. All experiments in which premature senescence was induced were performed in cells from p4 or p5.
Immunocytochemistry
ECs were seeded on glass slides, cultured to subconfluence, and fixed in 4% paraformaldehyde for 15 minutes. Cells were then incubated overnight at 4°C with a fluorescein isothiocyanate–conjugated antibody to vascular endothelial cadherin (Santa Cruz Biotechnology, 1:100) or unconjugated rabbit anti-eNOS (Cell Signaling, 1:100). For eNOS labeling, protein was visualized by incubation with a fluorescein isothiocyanate–conjugated secondary antibody (1:500, BD Biosciences) for 1 hour. Nuclei were stained with Hoechst 33342 (1 μg/mL), and slides were mounted with fluorescent mounting medium (Dako Scientific) and visualized with a Zeiss Axioskop 2 MOT.
EC MP Isolation, Quantification, and Electron Microscopy
Endothelial MPs were isolated from media collected from EC cultures as previously described.17 Briefly, samples were centrifuged at 1500g for 20 minutes at 20°C to obtain cell-free media. MPs were then pelleted from cell-free media by centrifugation at 18 000g for 20 minutes at 20°C. Endothelial MPs were confirmed as such and quantified by flow cytometry with an Alexa-647–labeled Annexin V (0.5 μg/mL, Biolegend) to identify events as MPs. As a negative control, a subpopulation of MPs was resuspended in Annexin V binding buffer lacking calcium, which is necessary for Annexin V binding to phosphatidylserine. MPs were centrifuged and resuspended in PBS for treatment. A final concentration of 105 endothelial MPs/mL, previously identified to exert effects on cultured ECs, was used for all experiments.17
In addition, MPs from low- and high-passage ECs were examined by electron microscopy. MP-containing suspensions were centrifuged at 18 000g for 20 minutes at 20°C, the supernatants were aspirated, and pellets were fixed in 2.5% gluteraldehyde in 1× PBS (overnight at 20°C). The pellets were then washed in 0.1 mol/L Na cacodylate buffer, postfixed in 2% OsO4, and dehydrated in graded ethanol. Samples were embedded in Spurrs Resin, and 60-nm sections were prepared on copper grids. Samples were visualized on a Hitachi H7100 electron microscope. MPs were identified as small (0.1–1.0 μm), rounded objects with clear, intact membranes, as described previously.17
Cell Cycle Analysis
Cell cycle analysis was conducted by using a modified propidium iodine–based flow cytometry protocol.20,21 Subconfluent cells were trypsinized and centrifugated at 1000g for 5 minutes. The supernatant was discarded and pelleted cells washed in PBS and centrifuged as before. Cells were resuspended in 300 μL PBS and fixed in 70% ethanol at 4°C overnight. Fixed cells were pelleted at 8000g for 10 minutes and then incubated in KRISHIAN buffer (0.1% sodium citrate, 0.02 mg/mL RNAse A [Sigma-Aldrich], 0.3% NP-40 [Sigma-Aldrich], and 0.05 mg/mL propidium iodide [Invitrogen]) for 1 hour at 4°C in the dark. Cell suspensions were filtered and analyzed for propidium iodide labeling of DNA by flow cytometry.
Measurement of Rho Kinase Activity
Rho kinase (ROCK) activity was assessed in ECs with the ROCK Activity Assay Kit (Cell Biolabs Inc) as described previously.17
Measurement of Superoxide and Hydrogen Peroxide Production
Superoxide (O2•−) and hydrogen peroxide (H2O2) production was measured in ECs by dihydroethidium high-performance liquid chromatography (HPLC) and the Amplex Red hydrogen Peroxide/Peroxidase Assay Kit (Molecular Probes), respectively, as we have previously described.17,22
Western Blot Analysis
Western blotting was used to examine levels of cell cycle/senescence and autophagy-related proteins in ECs and to determine protein levels in endothelial MPs. Membranes were probed with anti-phospho (ser36) p66-Shc (1:1000; Calbiochem), anti–total-Shc (1:1000, Calbiochem), anti–p16- ink4a (1:500, Santa Cruz Biotechnology), anti-p21cip1 (1:2000; Santa Cruz Biotechnology), anti-p27kip1 (1:2000; Santa Cruz Biotechnology), anti-Bax (1:1000; Santa Cruz Biotechnology), anti-Bcl-2 (1:1000; Santa Cruz Biotechnology), anti–microtubule-associated protein 1 Light Chain 3 (LC3, 1:1000, Cell Signaling Technology), anti-eNOS (1:1000, Cell Signaling Technology), anti–vascular endothelial cadherin (1:1000, Santa Cruz Biotechnology), anti–flotillin-2 (1:4000, BD Biosciences), anti–caveolin-1 (1:2000, Santa Cruz Biotechnology), and anti-GAPDH (1:4000, Millipore). Membranes were then washed in Tris buffered saline with tween-20 and incubated with horse radish peroxidase–conjugated secondary antibodies (1:2000; Santa Cruz Biotechnology) in milk for 1 hour. Membranes were probed for immunoreactivity by chemiluminescence. Quantification of blots was performed by densitometry (ImageJ).
β-Galactosidase Cytochemical Staining Assay
Senescence-associated β-galactosidase (SA-βgal) activity at pH 6 was measured with a SA-βgal Staining Kit (Cell Signaling Technology). SA-βgal activity at pH 6 is a well-characterized hallmark of senescence, absent in replicating, quiescent, and immortal cells. 23 Cultured ECs in 35-mm 6-well plates were washed in 1× PBS, and. A total of 1 mL of Fixative Solution (2% formaldehyde, 0.2% glutaraldehyde, 1× PBS) was added to each well for 15 minutes. Plates were washed in PBS, and then β-galactosidase Staining Solution (40 mmol/L citric acid/sodium phosphate pH 6, 0.15 mol/L NaCl, 2 mmol/L MgCl2, 500 nmol/L potassium ferrocyanide, 50 μL 20 mg/mL X-gal in N-N-dimethylformamide) was added to each well and incubated overnight at 37°C. Cells were overlayed with 70% glycerol and stored at 4°C. Slides were imaged via Axiovision 4.6 software with an Axio HRC camera (Zeiss). Twenty-five high-powered fields imaged at 200× magnification (approximately 1000 cells) were analyzed.
Fluorescence-Based Caspase-3 Activity Assay
Caspase-3 activity was measured with the QuantiZyme Assay System (BioMol) as described previously.24 Briefly, cells were lysed in a buffer containing 1 mol/L HEPES (pH 7.4), 0.2% Triton X-100, 140 μg/mL dithiothreitol, and 0.5 mol/L EDTA. Cell lysates (50 μg protein/sample) were then incubated at 37°C for 40 hours in the presence of the BioMol AC-DEVD-AMC Substrate (200 μmol/L) with and without the BioMol AC-DEVD-CHO inhibitor (4 μmol/L) in a buffer containing 1 mol/L HEPES (pH 7.4), 3 mol/L NaCl, 140 μg/mL dithiothreitol, 0.5 mol/L EDTA, 100% glycerol, and 0.2% Triton X-100. Fluorescence was measured on a FLUOstar Galaxy microplate reader (Inter Departmental Equipment) with an excitation wavelength of 360 nm and an emission wavelength of 460 nm. Results are expressed as arbitrary units (AU)/μg protein.
Statistical Analysis
Results are expressed as mean ± SEM and were analyzed by using a Mann-Whitney test and one-way or two-way ANOVA with a Bonferoni's posttest as appropriate. For parametric statistical tests, data were verified to not deviate from Gaussian distributions with a Kolmogorov-Smirnov test. Bonferoni's correction was applied when P values were calculated for multiple comparisons. All statistical analyses were performed with Graphpad Prism 4.0 (GraphPad Software, Inc). P<0.05 was considered significant.
Results
In Vitro Model of Aged ECs
An in vitro model of replicative senescence was used with cultured mouse aortic ECs. Long-term culture of ECs was used as a model of endothelial aging and is associated with decreased proliferation and increased SA-βgal activity.25,26 In our studies, culture of mouse aortic ECs to p21 was associated with an increase in the number of cells staining positive for SA-βgal activity (Figure 1A) and a reduction in the number of proliferating cells as determined by propidium iodide cell cycle analysis (Figure 1B). Senescent cells appeared larger and more granular with occasional vacuolization of the cytoplasm (Figure 1A). Importantly, cells retained expression of EC markers including eNOS and vascular endothelial cadherin (Figure 2). Both short-term (p4) and long-term (p21) cultured ECs were viable as assessed by Trypan Blue exclusion (96±1% and 95±2%, respectively, n=5–6). In addition to displaying a senescent phenotype, p21 ECs were characterized by a reduction in levels of LC3-II (Figure 3A), which is indicative of reductions in autophagy,27 increased ROCK activity (Figure 3B), and increased phosphorylation of the longevity determinant adaptor protein p66Shc (Figure 3C). No differences in the percentage of apoptotic nuclei (Hoechst staining) or Bax/Bcl-2 ratio were observed (Figure 4A and 4B) suggesting that levels of apoptosis were similar between early- and late-passage ECs. A small but significant increase in levels of caspase-3 activity was observed in late-passage ECs (Figure 4C).
Figure 1.
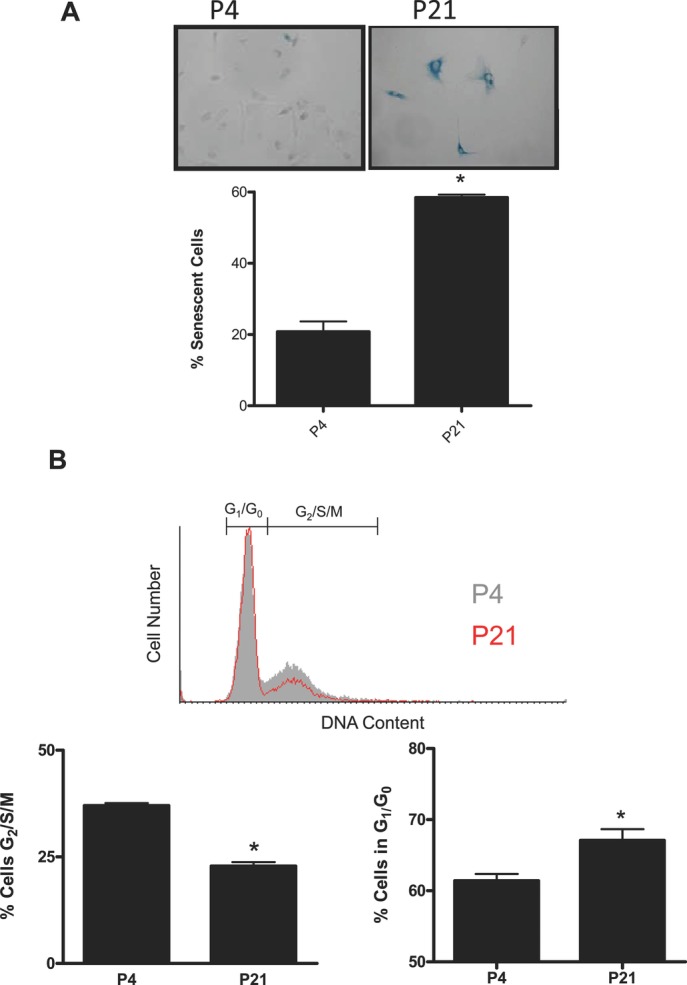
SA-βgal activity and cell cycle analysis in aged ECs. Mouse aortic ECs were serially passaged from p4 to p21. A, SA-βgal staining in p4 and p21 ECs expressed as a percentage of the total EC population. Inset: representative image. B, Distribution of cells in G1 and G2/S/M expressed as a percentage of total cells. Inset: representative histograms of DNA content during cell cycle; p4 populations are visible as a shaded gray histogram, while p21 populations are visible as the red open histogram. Data are expressed as mean±SEM; *P<0.05 vs p4 cells, n=4.
Figure 2.
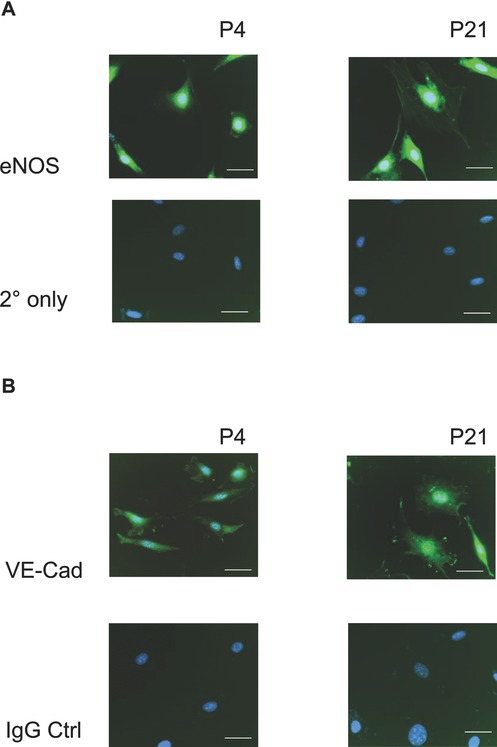
Fluorescent micrographs of p4 and p21 mouse aortic ECs. A, eNOS expression in p4 and p21 ECs. Green fluorescence (FITC) is eNOS; blue fluorescence (4′,6-diamidino-2-phenylindole, DAPI) is Hoechst 33342 nuclear stain. Shown are representative micrographs as well as autofluorescence controls consisting of cells exposed to secondary antibody only. B, VE-cadherin expression in p4 and p21 ECs. Green fluorescence (FITC) is VE-cadherin; blue fluorescence (DAPI) is Hoechst 33342 nuclear stain. Shown are representative micrographs as well as autofluorescence controls consisting of cells exposed to a FITC-conjugated isotype control. Bar represents 10 μm. FITC indicates fluorescein isothiocyanate; VE-cadherin, vascular endothelial cadherin.
Figure 3.
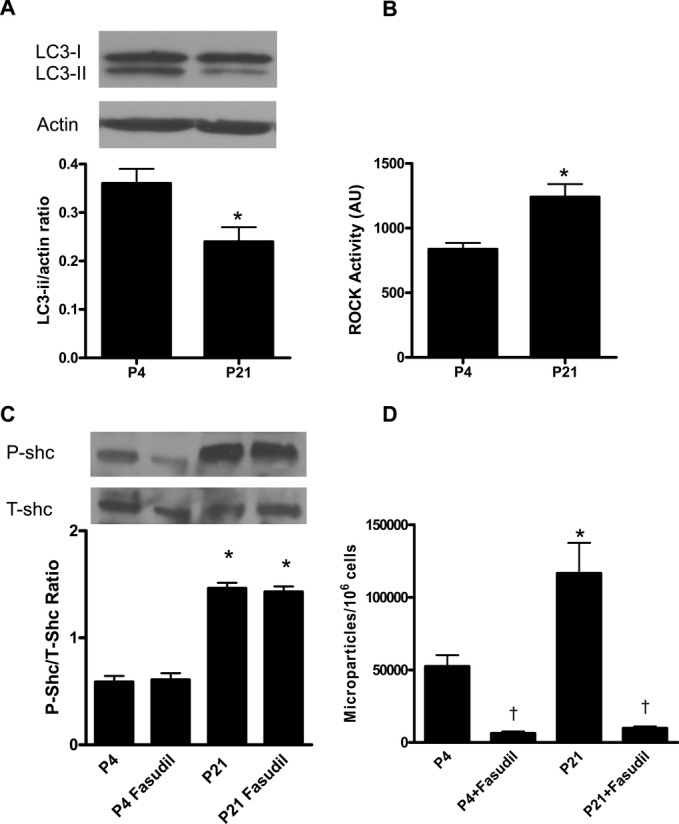
Characterization of p4 and p21 ECs. A, Western analysis of LC3-I and LC3-II levels in p4 and p21 ECs. Levels of LC3-II, the active form, were significantly reduced in p21 cells. Inset: representative blot. Data are expressed as mean±SEM; *P<0.05 vs p4, n=4. B, ROCK activity was significantly increased in p21 vs p4 ECs. Data are expressed as mean±SEM; *P<0.05 vs p4, n=4. C, Phosphorylation of p66Shc was increased in p21 cells compared with p4 cells. Coculture with fasudil (10 μmol/L, 24 hours) had no effect on p66Shc phosphorylation. Data are expressed as mean±SEM; *P<0.05 vs p4, †P<0.05 vs untreated cells, n=6. D, MP formation from p4 and p21 ECs was determined by measuring the amount of Annexin V+ve MPs released into media over 24 hours and expressed as the number of MPs/106 cells. Data are expressed as mean±SEM; *P<0.05 vs p4, †P<0.05 vs untreated cells, n=6 to 7.
Figure 4.
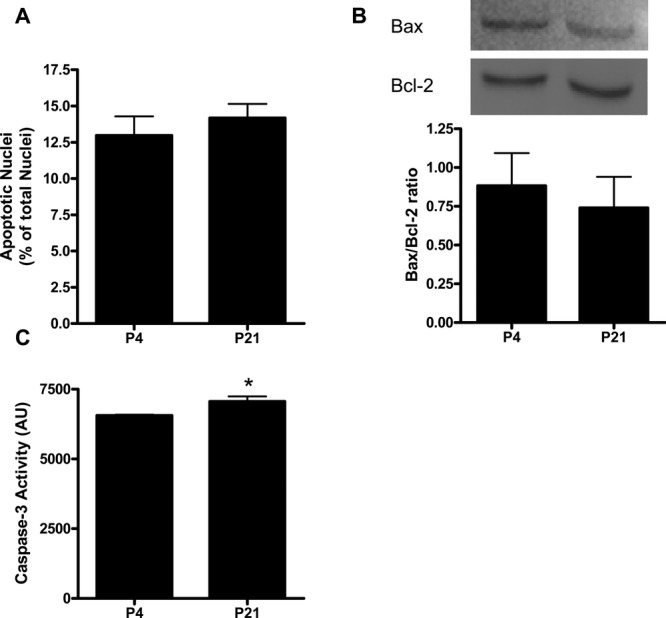
Assessment of apoptosis in p4 and p21 ECs. Mouse aortic ECs were serially passaged from p4 to p21, and apoptosis was assessed by Hoechst nuclear staining (A), Bax/Bcl-2 ratio (B), and caspase-3 activity (C). Inset: representative blot. Data are expressed as mean±SEM; *P<0.05 vs p4 cells, n=5 to 6.
MP Formation Is Increased in Aged ECs
To examine the formation of MPs in aged ECs, media was collected from p4 and p21 ECs over 24 hours, and the number of Annexin V+ve MPs released into the media was quantified by flow cytometry. Long-term culture of ECs was associated with a significant increase in the rate of spontaneous MP formation from ECs (Figure 3D). On the basis of the observation that ROCK activity was increased in late-passage ECs, we examined whether ROCK plays a mechanistic role in MP formation in senescent ECs. Inhibition of ROCK with fasudil (10 μmol/L) significantly reduced MP formation in both p4 and p21 cells (Figure 3D). Conversely, inhibition of ROCK had no effect on phosphorylation of p66Shc (Figure 3C), suggesting that p66Shc does not play a downstream role in regulating MP formation.
Characterization of MPs by electron microscopy demonstrated no obvious phenotypic difference between MPs derived from senescent (p21) ECs and MPs from young (p4) ECs (Figure 5A). Similarly, the levels of EC markers vascular endothelial cadherin and eNOS were comparable between p4- and p21-derived MPs (Figure 5B). Levels of caveolin-1 and flotillin-2, markers of lipid rafts/caveolae, which we previously identified as highly expressed in endothelial MPs, were comparable in p4 and p21 cells (Figure 5B).
Figure 5.
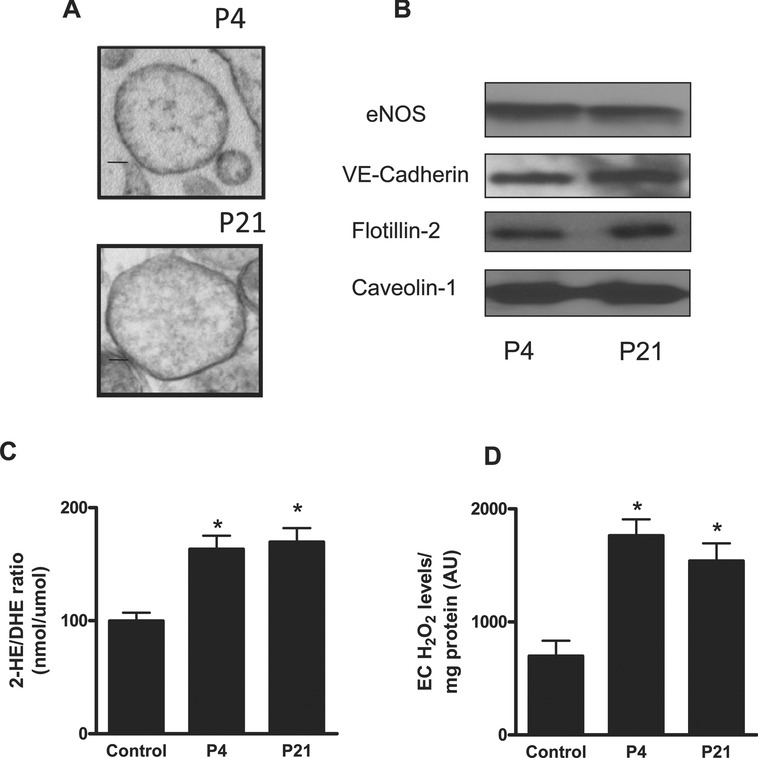
Characterization of EC-derived MPs from p4 and p21 ECs. A, Transmission electron micrographs of MPs from p4 and p21 ECs. MPs were isolated from media, and samples were fixed and examined by electron microscopy at 50 000× magnification. Bar=0.1 μm. B, MPs were isolated from the medium of p4 and p21 ECs, and expression of eNOS, VE-cadherin, flotillin-2, and caveolin-1 was measured by Western blot analysis. C and D, Pro-oxidative effects of endothelial MPs. ECs were cultured and treated with endothelial MPs (105 MPs/mL) for 4 hours, and superoxide (O2•−) production was measured by DHE/HPLC (C) and H2O2 was measured by amplex red (D). Treatment with both p4 and p21 MPs was associated with significant increases in O2•− and H2O2 production in ECs. Data are expressed as mean±SEM; *P<0.05 vs control untreated cells, n=5 to 7. FITC indicates fluorescein isothiocyanate.
MPs Derived From Senescent and Young ECs Promote Oxidative Stress
Our laboratory and others have reported that MPs promote oxidative stress in cultured ECs.16,17 To determine whether functional differences exist between MPs formed from healthy and senescent ECs, low-passage ECs were treated with MPs derived from young (p4) and aged (p21) ECs. Treatment of ECs with MPs was associated with a significant increase in O2•− (Figure 5C) and H2O2 (Figure 5D) formation as measured by dihydroethidium (DHE)-HPLC and Amplex Red, respectively. However, no differences were observed between ECs treated with p4 and p21 MPs (Figure 5C and 5D).
MPs Induce Premature Senescence in Cultured ECs
To examine whether endothelial MPs contribute to premature EC senescence, mouse aortic ECs (p4 to p5) were treated with MPs obtained from low-passage ECs (105 MPs/mL). As a positive control, ECs were treated with 100 μmol/L H2O2, which has been previously shown to induce premature senescence in ECs.8,28 Neither H2O2 nor MPs had any effect on apoptosis as determined by caspase-3 activity or Bax/Bcl-2 ratio (data not shown). However, treatment with MPs or H2O2 was associated with a shift from a proliferating to a nonproliferating phenotype as determined by propidium iodide DNA staining (Figure 6A and 6B). In addition, the expression of the cyclin-dependent kinase (Cdk) inhibitors p21cip and p16ink4a was significantly increased after treatment with H2O2 or MPs at 8 hours (Figure 7A and 7B). Similarly, phosphorylation of the longevity determinant adaptor protein p66Shc, which is both activated by ROS and a stimulus for ROS production, was increased after treatment with H2O2 or MPs (Figure 7C). Expression of p27kip1, a separate Cdk inhibitor, was not changed after treatment (Figure 7D). Finally, treatment with endothelial MPs was associated with induction of premature EC senescence, typified by an increase in the proportion of cells staining positive (bright blue) for β-galactosidase activity at pH 6 for 48 hours (P<0.05, Figure 6C).
Figure 6.
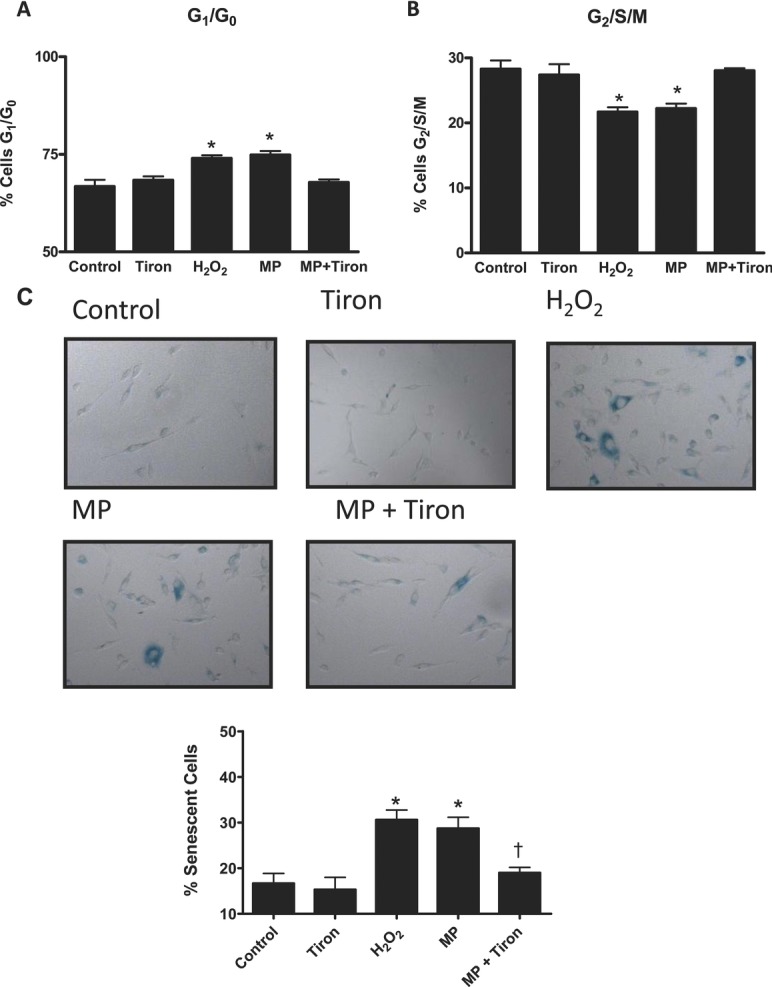
Cell cycle progression and SA-βgal activity in ECs treated with H2O2 (100 μmol/L), or MPs (105/mL). A, Treatment with MPs or H2O2 for 8 hours increased the proportion of nonproliferative cells (G1/G0). B, Treatment with MPs or H2O2 for 8 hours significantly decreased the proportion of proliferating cells (G2/M). C, SA-βgal staining of ECs after 48 hours. Upper panels: Representative images show SA-βgal staining in ECs exposed to 4,5-dihydroxybenzene-1,3-disulfonate (tiron), H2O2 (positive control), and MPs in the absence and presence of tiron pretreatment. Lower panel is a bar graph of SA-βgal staining expressed as a percentage of the total EC population and presented as means±SEM of 4 to 5 experiments. Treatment with MPs or H2O2 increased the number of cells exhibiting SA-βgal activity. Treatment with the superoxide scavenger sodium tiron (10 μmol/L) alone had no effect on cell cycle progression (A, B) or SA-βgal activity (C), but cotreatment with MPs blocked MP-mediated reductions in proliferation and increases in SA-βgal activity. Data are expressed as mean±SEM; *P<0.05 vs control, *P<0.05 vs MP treatment, n=5.
Figure 7.
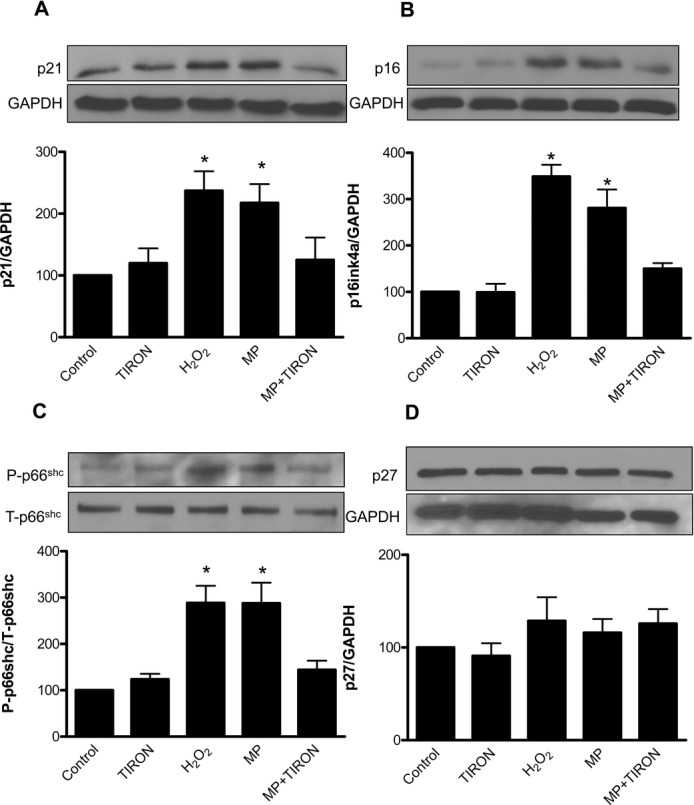
Effects of MPs on cell cycle and markers of cell senescence in ECs. Effect of MPs (105/mL, 8 hours), H2O2 (100 μmol/L, 8 hours), and sodium 4,5-dihydroxybenzene-1,3-disulfonate (tiron, 10 μmol/L, 8 hours) on the expression of p21cip1 (p21), p16ink4a (p16), and p27kip1 (p27) and phosphorylation of p66Shc. Treatment with MPs or H2O2 increased expression of p21 (A) and p16 (B), increased phosphorylation of p66Shc (C), but had no effect on p27 expression (D). Treatment with the superoxide scavenger sodium 4,5-dihydroxybenzene-1,3-disulfonate (tiron, 10 μmol/L) had no significant effect on SA-βgal staining, but cotreatment with MPs blocked MP-mediated effects on p21, p16, and p66Shc. Data are expressed as mean±SEM; *P<0.05 vs control, n=6 to 8.
MPs Induce EC Senescence via a ROS-Dependent Pathway
As MPs are known to induce ROS production, and MP treatment was associated with phosphorylation of the ROS-sensitive protein p66Shc, implicated in senescence, we hypothesized that MP-mediated induction of EC senescence was dependent on ROS production. Using tiron, a O2•− scavenger, we examined whether MP-induced premature EC senescence in p4 ECs is due to ROS production. Tiron treatment (8 hours) alone caused no changes in cell cycle progression but attenuated MP-mediated increases in nonproliferative G0/G1 populations (Figure 6A and 6B). Similarly, tiron blocked MP-induced increases in p21cip1 and p16ink4a expression and p66Shc phosphorylation (Figure 7A through 7C). Finally, tiron treatment (48 hours) alone had no effect on SA-βgal activity but attenuated MP-induced increases in SA-βgal activity (Figure 6C).
Mechanisms of MP-Mediated Increases in Oxidative Stress
Finally, to determine the mechanisms by which MPs promote oxidative stress in ECs, mouse aortic ECs (p4 to p5) were treated with MPs obtained from low-passage ECs (105 MPs/mL) in the presence of inhibitors of NADPH oxidase (apocynin, 10 μmol/L), xanthine oxidase (allopurinol, 100 μmol/L), and mitochondrial respiration (rotenone, 5 μmol/L). O2•−(Figure 8A) and H2O2 (Figure 8B) formation was measured by DHE-HPLC and Amplex Red, respectively. Treatment with MPs was associated with a significant increase in EC ROS production. Cotreatment with allopurinol had no effect on MP-mediated increases in ROS production (Figure 8). Conversely, cotreatment with both apocynin and rotenone significantly reduced EC O2•− and H2O2 production, suggesting that both NADPH oxidase and mitochondria, but not xanthine oxidase, play a role in MP-mediated increase in oxidative stress.
Figure 8.
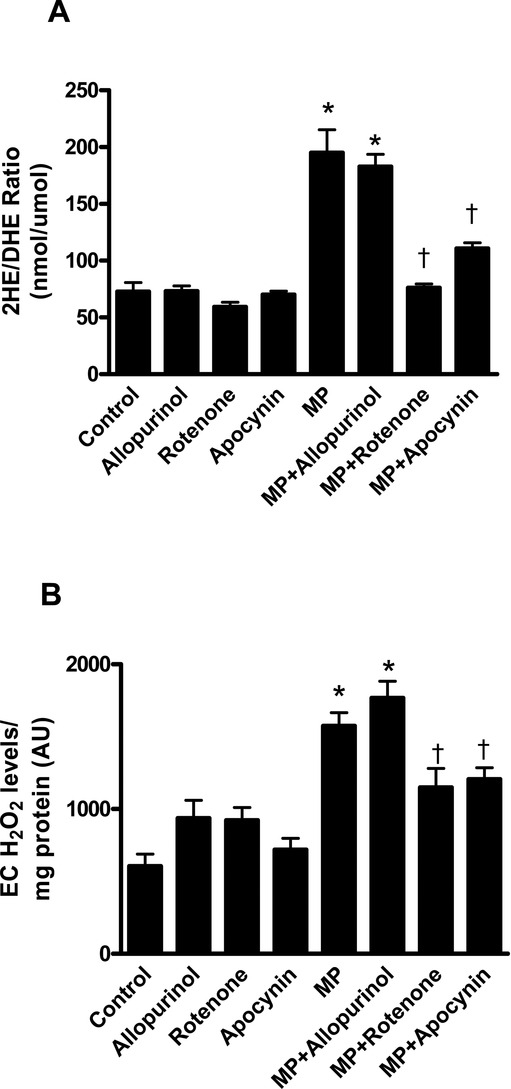
Role of prooxidant systems in MP-mediated oxidative stress. ECs were cultured and treated with endothelial MPs (10 5/mL) for 4 hours in the presence and absence of apocynin (10 μM), rotenone (5 μM), or allopurinol (100 μM). Treatment with MPs significantly increased EC superoxide (O2•−, A) and hydrogen peroxide (H2O2, B) production as measured by DHE/HPLC (A) and Amplex Red (B), respectively. Cotreatment with apocynin and rotenone diminished MP-mediated increases in O2•− and H2O2 production. Data are expressed as mean±SEM; *P<0.05 vs untreated control, *P<0.05 vs MP treatment, n=6.
Discussion
The present study examined the role of endothelial MPs in EC senescence. Major findings demonstrate that (1) long-term culture of mouse aortic ECs leads to a senescent phenotype associated with increased ROCK activity and formation of MPs, (2) MPs stimulate endothelial ROS production via NADPH oxidase and mitochondrial respiratory enzymes, and (3) MPs induce premature EC senescence in a ROS-dependent manner. Our results suggest that MPs contribute to the progression of vascular aging via a feed-forward mechanism wherein increased MP formation from senescent ECs promotes further EC senescence through increased ROS production. These processes are associated with upregulation of cell cycle inhibitor proteins (Figure 9).
Figure 9.
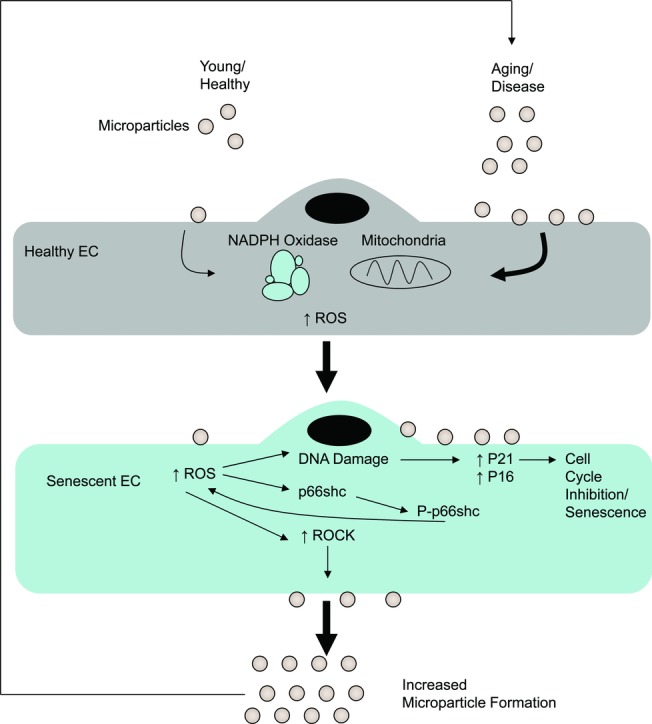
Putative mechanisms whereby MPs influence vascular cell aging and senescence. MP formation is increased in aging or under stress (disease) conditions, MPs, in turn, promote oxidative stress in ECs via NADPH oxidase and mitochondria. Increased oxidative stress ultimately results in EC senescence through DNA damage, leading to expression of cell cycle inhibitors p16ink4a (p16) and p21cip1 (p21) and increased activation of p66Shc, a determinant of cell longevity. The resultant EC senescence leads to increased MP formation via ROCK, which further promotes oxidative stress in a feed-forward fashion.
MPs are increasingly recognized as cellular biomarkers of vascular injury/dysfunction in many pathologies (eg, hypertension, diabetes, end-stage renal disease).14 Despite this, little is known about the relationship between MPs and vascular senescence, and there are conflicting reports with regard to plasma levels of MPs during aging. On the one hand, P-selectin–expressing platelet MPs increase with older age in otherwise healthy individuals, 29 endothelial MPs are increased with age in diabetic rats, 30 and leukocyte-derived MPs are increased in thrombosed aged mice.31 Conversely, Forest et al reported that endothelial MPs decrease in individuals aged 75 years and older.32 Each of these studies involved in vivo measurement of plasma MP levels, where MP clearance may be a significant determinant of concentration. As aging is known to impair clearance of apoptotic cells, 33 we examined MP formation in vitro using a model of endothelial senescence through long-term culture as previously detailed. 25,26 Long-term culture of mouse aortic ECs led to phenotypic characteristics of aging/senescence, including decreased cell proliferation and increased SA-βgal staining. In addition to these phenotypic changes, MP release into media was significantly increased in high-passage ECs, suggesting that MP formation increases in senescent cells. This increase appears to be mediated through ROCK, as treatment with fasudil blocked MP formation in these cells, consistent with our previous work.17 Levels of the autophagy-associated protein LC3-II were decreased in high-passage ECs. This observation is consistent with age-related declines in autophagy, which have been reported elsewhere. 34,35 It has been speculated that MP formation represents an adaptation that allows healthy cells to rid themselves of deleterious/toxic components.36 On the basis of our observations, it is conceivable that the increase in MP formation in late-passage ECs represents an adaptive response to compensate for reduced autophagic degradation of undesirable cell components.
Consistent with previous reports, we observed that MPs derived from low-passage and high-passage ECs promote the production of ROS (O2•− and H2O2) in cultured ECs.16,17,18,37 Despite the established pro-oxidative effects of MPs, the mechanisms responsible for ROS production after MP exposure have been elusive, and reports are contradictory. Lymphocytic MPs have been reported to promote EC ROS production via xanthine oxidase 38 or through NADPH oxidase.39 Monocyte MPs stimulate EC ROS production via NADPH oxidase, mitochondria, xanthine oxidase, cyclooxygenase, and uncoupled nitric oxide synthase.40 Recently, Terrisse et al implicated xanthine oxidase and possibly NADPH oxidase in endothelial MP-mediated EC ROS production.18 Our results suggest that these increases in EC ROS production are mediated via NADPH oxidase and mitochondria but not by xanthine oxidase. Reasons for the different results are unclear, but they may relate to differences in MP isolation/handling by various laboratories or to differences in the EC populations being treated.
Surprisingly, despite clear phenotypic differences between low- and high-passage ECs, MPs derived from both origins had a similar capacity for inducing ROS production in ECs. While others reported divergent biological activity of distinct MP populations,41 we found that endothelial MPs obtained from high-passage senescent ECs appear to promote EC oxidative stress to a similar extent to those obtained from low-passage ECs. In support of this, electron microscopy and Western blot analysis revealed that MPs derived from high-passage ECs were similar in composition to MPs derived from low-passage ECs. Thus, MP production appears to increase in senescent ECs while retaining similar biological activity, the product of which would be an increase in MP-mediated pro-oxidative effects during aging.
Under conditions of stress/damage, such as those that occur during oxidative stress or angiotensin II exposure, ECs can become prematurely senescent, which, in turn, may contribute to the early vascular aging seen in chronic disease.42 To determine whether endothelial MPs promote premature EC senescence, we exposed cultured ECs to MPs and examined effects on proliferation, the expression of cell cycle inhibitory proteins, and SA-βgal activity. H2O2, previously reported to rapidly induce a senescent phenotype in ECs, was used as a positive control for stress-induced premature EC senescence.8,28 Consistent with previous reports, H2O2 rapidly induced a reduction in EC proliferation as determined by propidium iodide cell cycle analysis and a significant increase in the number of ECs staining positive for SA-βgal activity. This stress-induced senescent phenotype was recapitulated in ECs treated with MPs. In addition, MP treatment promoted phosphorylation and activation of the mitochondrial adaptor protein p66Shc, a determinant of mitochondrial ROS production and longevity.12,43,44 On the basis of these observations, we hypothesized that MPs promote premature EC senescence through the stimulation of EC ROS production. To address this question, we treated ECs with MPs in the presence of the O2•− scavenger tiron. Cotreatment with tiron blocked MP-induced reductions in proliferation and MP-induced increases in SA-βgal activity. Thus, MPs promote premature EC senescence through ROS-dependent processes.
Attenuation of cell cycle progression represents a central mechanism in the development of growth arrest and cell senescence. We therefore examined the effect of MP administration on the expression of the Cdk inhibitors p16(ink4a), p21(cip1), and p27kip1. p16ink4a inhibits Cdk 4, thereby reducing phosphorylation of retinoblastoma protein (Rb) and leading to a reduction in pro-proliferative transcription factor E2F-activity.45 p21cip1, a Cdk inhibitor activated by DNA damage and p53, is capable of binding and preventing the activation of cyclin E-Cdk2 and cyclin D-Cdk4, therein promoting growth arrest and senescence.45 p27kip1 functions very similarly to p21cip1, but its regulation is generally not due to endogenous DNA damage but rather through various cell surface signals, kinase activity, and microRNAs (miRNA).46 In the present study, H2O2 and MP-mediated stress-induced premature senescence was also associated with upregulation of cell cycle checkpoint pathways of p21cip1 and p16ink4a but not p27kip1. Cotreatment of MPs with tiron attenuated MP-mediated increases in p21cip1 and p16ink4a. Interestingly, p27kip1 expression was not upregulated in either H2O2- or MP-treated cells. While one might expect ROS-dependent upregulation of p27kip1, Deshpande et al have reported induction of p21cip1, but not p27kip1 after H2O2 administration in vascular smooth muscle cells, suggesting that ROS-dependent induction of cell cycle arrest in the vasculature may not involve changes in p27kip1 expression.21
Mechanisms whereby MP–EC interactions are transduced into intracellular changes and functional responses remain unclear. However, emerging evidence has identified integrin activation, phosphatidylserine-dependent internalization of MPs, interleukin-1 receptor activation, sonic hedgehog signaling, and transfer of miRNAs as possible mediators of MP-induced biological effects.18,47–50 In addition, we recently reported a surface interaction involving epidermal growth factor receptor activation, which contributes to pro-oxidative and proinflammatory effects of MPs.17 While the mechanisms of MP-induced oxidative stress and senescence were not explicitly explored, it is plausible that a similar MP–EC interaction via epidermal growth factor receptor contributes to the ROS-dependent pro-senescent effects seen here.
In summary, we report that endothelial MP formation is increased in senescent ECs and that endothelial MPs induce premature senescence in cultured mouse aortic ECs in a ROS-dependent manner. These novel findings demonstrate a link between endothelial MPs and EC senescence. This phenomenon sheds new light on the role of MPs in vascular pathobiology and suggests that MPs may themselves be key mediators of early vascular aging by promoting cellular senescence.
Sources of Funding
This study was funded by grants from the Canadian Institute of Health Research (CIHR). Dr Touyz was supported through a Canada Research Chair/Canadian Foundation for Innovation award. Dr Burger is supported by a fellowship from the Kidney Research Scientist Core Education and National Training (KRESCENT) program, D.G. Kwart by a studentship from the National Sciences and Engineering Research Council of Canada (NSERC), and Dr Montezano by a fellowship from the CIHR.
Disclosures
None.
References
- 1.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139-146 [DOI] [PubMed] [Google Scholar]
- 2.Herrera MD, Mingorance C, Rodriguez-Rodriguez R, Alvarez de Sotomayor M. Endothelial dysfunction and aging: an update. Ageing Res Rev. 2009;9:142-152 [DOI] [PubMed] [Google Scholar]
- 3.Safar ME. Arterial aging—hemodynamic changes and therapeutic options. Nat Rev Cardiol. 2010;7:442-449 [DOI] [PubMed] [Google Scholar]
- 4.Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100:15-26 [DOI] [PubMed] [Google Scholar]
- 5.Nilsson PM. Early vascular aging (EVA): consequences and prevention. Vasc Health Risk Manag. 2008;4:547-552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol. 2009;106:326-332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci. 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oeseburg H, de Boer RA, Buikema H, van der Harst P, van Gilst WH, Sillje HH. Glucagon-like peptide 1 prevents reactive oxygen species-induced endothelial cell senescence through the activation of protein kinase A. Arterioscler Thromb Vasc Biol. 2010;30:1407-1414 [DOI] [PubMed] [Google Scholar]
- 9.Seals DR, Jablonski KL, Donato AJ. Aging and vascular endothelial function in humans. Clin Sci (Lond). 2011;120:357-375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Touyz RM, Briones AM. Reactive oxygen species and vascular biology: implications in human hypertension. Hypertens Res. 2011;34:5-14 [DOI] [PubMed] [Google Scholar]
- 11.Camici GG, Cosentino F, Tanner FC, Luscher TF. The role of p66Shc deletion in age-associated arterial dysfunction and disease states. J Appl Physiol. 2008;105:1628-1631 [DOI] [PubMed] [Google Scholar]
- 12.Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309-313 [DOI] [PubMed] [Google Scholar]
- 13.Boulanger CM. Microparticles, vascular function and hypertension. Curr Opin Nephrol Hypertens. 2010;19:177-180 [DOI] [PubMed] [Google Scholar]
- 14.Boulanger CM, Amabile N, Tedgui A. Circulating microparticles: a potential prognostic marker for atherosclerotic vascular disease. Hypertension. 2006;48:180-186 [DOI] [PubMed] [Google Scholar]
- 15.Esposito K, Ciotola M, Schisano B, Gualdiero R, Sardelli L, Misso L, Giannetti G, Giugliano D. Endothelial microparticles correlate with endothelial dysfunction in obese women. J Clin Endocrinol Metab. 2006;91:3676-3679 [DOI] [PubMed] [Google Scholar]
- 16.Brodsky SV, Zhang F, Nasjletti A, Goligorsky MS. Endothelium-derived microparticles impair endothelial function in vitro. Am J Physiol Heart Circ Physiol. 2004;286:H1910-H1915 [DOI] [PubMed] [Google Scholar]
- 17.Burger D, Montezano AC, Nishigaki N, He Y, Carter A, Touyz RM. Endothelial microparticle formation by angiotensin II is mediated via AT1R/NADPH oxidase/rho kinase pathways targeted to lipid rafts. Arterioscler Thromb Vasc Biol. 2011 [DOI] [PubMed] [Google Scholar]
- 18.Terrisse AD, Puech N, Allart S, Gourdy P, Xuereb JM, Payrastre B, Sie P. Internalization of microparticles by endothelial cells promotes platelet/endothelial cell interaction under flow. J Thromb Haemost. 2010;8:2810-2819 [DOI] [PubMed] [Google Scholar]
- 19.Taiwo F. Mechanism of tiron as scavenger of superoxide ions and free electrons. Spectroscopy. 2008;22:491 [Google Scholar]
- 20.Darzynkiewicz Z, Huang X. Analysis of cellular DNA content by flow cytometry. Curr Protoc Immunol. 2004Chapter 5:Unit 5 7 [DOI] [PubMed] [Google Scholar]
- 21.Deshpande NN, Sorescu D, Seshiah P, Ushio-Fukai M, Akers M, Yin Q, Griendling KK. Mechanism of hydrogen peroxide-induced cell cycle arrest in vascular smooth muscle. Antioxid Redox Signal. 2002;4:845-854 [DOI] [PubMed] [Google Scholar]
- 22.Xu S, He Y, Vokurkova M, Touyz RM. Endothelial cells negatively modulate reactive oxygen species generation in vascular smooth muscle cells. Hypertension. 2009;54:427-433 [DOI] [PubMed] [Google Scholar]
- 23.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363-9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burger D, Lei M, Geoghegan-Morphet N, Lu X, Xenocostas A, Feng Q. Erythropoietin protects cardiomyocytes from apoptosis via up-regulation of endothelial nitric oxide synthase. Cardiovasc Res. 2006;72:51-59 [DOI] [PubMed] [Google Scholar]
- 25.Vasa M, Breitschopf K, Zeiher AM, Dimmeler S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ Res. 2000;87:540-542 [DOI] [PubMed] [Google Scholar]
- 26.Khaidakov M, Wang X, Mehta JL. Potential involvement of LOX-1 in functional consequences of endothelial senescence. PLoS One. 2011;6:e20964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di Sano F, Dice JF, Difiglia M, Dinesh-Kumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, Dunn WA, Jr, Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA, Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, Komatsu M, Kominami E, Kondo S, Kovacs AL, Kroemer G, Kuan CY, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei HY, Lenardo MJ, Levine B, Lieberman A, Lim KL, Lin FC, Liou W, Liu LF, Lopez-Berestein G, Lopez-Otin C, Lu B, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mautner J, Meijer AJ, Melendez A, Michels P, Miotto G, Mistiaen WP, Mizushima N, Mograbi B, Monastyrska I, Moore MN, Moreira PI, Moriyasu Y, Motyl T, Munz C, Murphy LO, Naqvi NI, Neufeld TP, Nishino I, Nixon RA, Noda T, Nurnberg B, Ogawa M, Oleinick NL, Olsen LJ, Ozpolat B, Paglin S, Palmer GE, Papassideri I, Parkes M, Perlmutter DH, Perry G, Piacentini M, Pinkas-Kramarski R, Prescott M, Proikas-Cezanne T, Raben N, Rami A, Reggiori F, Rohrer B, Rubinsztein DC, Ryan KM, Sadoshima J, Sakagami H, Sakai Y, Sandri M, Sasakawa C, Sass M, Schneider C, Seglen PO, Seleverstov O, Settleman J, Shacka JJ, Shapiro IM, Sibirny A, Silva-Zacarin EC, Simon HU, Simone C, Simonsen A, Smith MA, Spanel-Borowski K, Srinivas V, Steeves M, Stenmark H, Stromhaug PE, Subauste CS, Sugimoto S, Sulzer D, Suzuki T, Swanson MS, Tabas I, Takeshita F, Talbot NJ, Talloczy Z, Tanaka K, Tanida I, Taylor GS, Taylor JP, Terman A, Tettamanti G, Thompson CB, Thumm M, Tolkovsky AM, Tooze SA, Truant R, Tumanovska LV, Uchiyama Y, Ueno T, Uzcategui NL, van der Klei I, Vaquero EC, Vellai T, Vogel MW, Wang HG, Webster P, Wiley JW, Xi Z, Xiao G, Yahalom J, Yang JM, Yap G, Yin XM, Yoshimori T, Yu L, Yue Z, Yuzaki M, Zabirnyk O, Zheng X, Zhu X, Deter RL. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151-175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oeseburg H, Iusuf D, van der Harst P, van Gilst WH, Henning RH, Roks AJ. Bradykinin protects against oxidative stress-induced endothelial cell senescence. Hypertension. 2009;53:417-422 [DOI] [PubMed] [Google Scholar]
- 29.van der Zee PM, Biro E, Ko Y, de Winter RJ, Hack CE, Sturk A, Nieuwland R. P-selectin- and CD63-exposing platelet microparticles reflect platelet activation in peripheral arterial disease and myocardial infarction. Clin Chem. 2006;52:657-664 [DOI] [PubMed] [Google Scholar]
- 30.Brodsky SV, Gealekman O, Chen J, Zhang F, Togashi N, Crabtree M, Gross SS, Nasjletti A, Goligorsky MS. Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Circ Res. 2004;94:377-384 [DOI] [PubMed] [Google Scholar]
- 31.McDonald AP, Meier TR, Hawley AE, Thibert JN, Farris DM, Wrobleski SK, Henke PK, Wakefield TW, Myers DD., Jr Aging is associated with impaired thrombus resolution in a mouse model of stasis induced thrombosis. Thromb Res. 2010;125:72-78 [DOI] [PubMed] [Google Scholar]
- 32.Forest A, Pautas E, Ray P, Bonnet D, Verny M, Amabile N, Boulanger C, Riou B, Tedgui A, Mallat Z, Boddaert J. Circulating microparticles and procoagulant activity in elderly patients. J Gerontol A Biol Sci Med Sci. 2010;65:414-420 [DOI] [PubMed] [Google Scholar]
- 33.Aprahamian T, Takemura Y, Goukassian D, Walsh K. Ageing is associated with diminished apoptotic cell clearance in vivo. Clin Exp Immunol. 2008;152:448-455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000;275:31505-31513 [DOI] [PubMed] [Google Scholar]
- 35.Ward WF. Protein degradation in the aging organism. Prog Mol Subcell Biol. 2002;29:35-42 [DOI] [PubMed] [Google Scholar]
- 36.Willekens FL, Werre JM, Groenen-Dopp YA, Roerdinkholder-Stoelwinder B, de Pauw B, Bosman GJ. Erythrocyte vesiculation: a self-protective mechanism?. Br J Haematol. 2008;141:549-556 [DOI] [PubMed] [Google Scholar]
- 37.Mezentsev A, Merks RM, O'Riordan E, Chen J, Mendelev N, Goligorsky MS, Brodsky SV. Endothelial microparticles affect angiogenesis in vitro: role of oxidative stress. Am J Physiol Heart Circ Physiol. 2005;289:H1106-H1114 [DOI] [PubMed] [Google Scholar]
- 38.Mostefai HA, Agouni A, Carusio N, Mastronardi ML, Heymes C, Henrion D, Andriantsitohaina R, Martinez MC. Phosphatidylinositol 3-kinase and xanthine oxidase regulate nitric oxide and reactive oxygen species productions by apoptotic lymphocyte microparticles in endothelial cells. J Immunol. 2008;180:5028-5035 [DOI] [PubMed] [Google Scholar]
- 39.Yang C, Mwaikambo BR, Zhu T, Gagnon C, Lafleur J, Seshadri S, Lachapelle P, Lavoie JC, Chemtob S, Hardy P. Lymphocytic microparticles inhibit angiogenesis by stimulating oxidative stress and negatively regulating VEGF-induced pathways. Am J Physiol Regul Integr Comp Physiol. 2008;294:R467-R476 [DOI] [PubMed] [Google Scholar]
- 40.Essayagh S, Xuereb JM, Terrisse AD, Tellier-Cirioni L, Pipy B, Sie P. Microparticles from apoptotic monocytes induce transient platelet recruitment and tissue factor expression by cultured human vascular endothelial cells via a redox-sensitive mechanism. Thromb Haemost. 2007;98:831-837 [PubMed] [Google Scholar]
- 41.Agouni A, Lagrue-Lak-Hal AH, Ducluzeau PH, Mostefai HA, Draunet-Busson C, Leftheriotis G, Heymes C, Martinez MC, Andriantsitohaina R. Endothelial dysfunction caused by circulating microparticles from patients with metabolic syndrome. Am J Pathol. 2008;173:1210-1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Erusalimsky JD, Skene C. Mechanisms of endothelial senescence. Exp Physiol. 2009;94:299-304 [DOI] [PubMed] [Google Scholar]
- 43.Francia P, delli Gatti C, Bachschmid M, Martin-Padura I, Savoia C, Migliaccio E, Pelicci PG, Schiavoni M, Luscher TF, Volpe M, Cosentino F. Deletion of p66shc gene protects against age-related endothelial dysfunction. Circulation. 2004;110:2889-2895 [DOI] [PubMed] [Google Scholar]
- 44.Napoli C, Martin-Padura I, de Nigris F, Giorgio M, Mansueto G, Somma P, Condorelli M, Sica G, De Rosa G, Pelicci P. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc Natl Acad Sci USA. 2003;100:2112-2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bringold F, Serrano M. Tumor suppressors and oncogenes in cellular senescence. Exp Gerontol. 2000;35:317-329 [DOI] [PubMed] [Google Scholar]
- 46.le Sage C, Nagel R, Agami R. Diverse ways to control p27Kip1 function: miRNAs come into play. Cell Cycle. 2007;6:2742-2749 [DOI] [PubMed] [Google Scholar]
- 47.Camussi G, Deregibus MC, Tetta C. Paracrine/endocrine mechanism of stem cells on kidney repair: role of microvesicle-mediated transfer of genetic information. Curr Opin Nephrol Hypertens. 2010;19:7-12 [DOI] [PubMed] [Google Scholar]
- 48.Soleti R, Benameur T, Porro C, Panaro MA, Andriantsitohaina R, MartÃnez MC. Microparticles harboring sonic hedgehog promote angiogenesis through the upregulation of adhesion proteins and proangiogenic factors. Carcinogenesis. 2009;30:580-588 [DOI] [PubMed] [Google Scholar]
- 49.Wang JG, Williams JC, Davis BK, Jacobson K, Doerschuk CM, Ting JP, Mackman N. Monocytic microparticles activate endothelial cells in an IL-1beta-dependent manner. Blood. 2011;118:2366-2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuan A, Farber EL, Rapoport AL, Tejada D, Deniskin R, Akhmedov NB, Farber DB. Transfer of microRNAs by embryonic stem cell microvesicles. PLoS One. 2009;4:e4722. [DOI] [PMC free article] [PubMed] [Google Scholar]