

Abstract
Background
The expression of calcium/calmodulin-dependent kinase IV (CaMKIV) was hitherto thought to be confined to the nervous system. However, a recent genome-wide analysis indicated an association between hypertension and a single-nucleotide polymorphism (rs10491334) of the human CaMKIV gene (CaMK4), which suggests a role for this kinase in the regulation of vascular tone.
Methods and Results
To directly assess the role of CaMKIV in hypertension, we characterized the cardiovascular phenotype of CaMK4−/− mice. They displayed a typical hypertensive phenotype, including high blood pressure levels, cardiac hypertrophy, vascular and kidney damage, and reduced tolerance to chronic ischemia and myocardial infarction compared with wild-type littermates. Interestingly, in vitro experiments showed the ability of this kinase to activate endothelial nitric oxide synthase. Eventually, in a population study, we found that the rs10491334 variant associates with a reduction in the expression levels of CaMKIV in lymphocytes from hypertensive patients.
Conclusions
Taken together, our results provide evidence that CaMKIV plays a pivotal role in blood pressure regulation through the control of endothelial nitric oxide synthase activity. (J Am Heart Assoc. 2012;1:e001081 doi: 10.1161/JAHA.112.001081.)
Keywords: angiogenesis, arrhythmia, endothelium, hypertension, hypertrophy
Introduction
A growing body of evidence bears out the rising interest in the function of calcium/calmodulin-dependent kinases (CaMKs) in cardiovascular pathophysiology. In particular, although it is now established that CaMKII is an important player in the regulation of cardiac responses, both in terms of electrophysiology and of cardiac myocyte hypertrophy,1–4 less is known about the role of other members of the CaMK family, such as CaMKIV, in the cardiovascular system.5
The recent genome-wide analysis of the Framingham Heart Study 100K Project6 showed an association between elevated diastolic blood pressure (BP) and the rs10491334 T/C single-nucleotide polymorphism (SNP) of the human CaMKIV gene (CaMK4). Such a finding suggests that this kinase, the expression of which once was thought to be confined to the nervous tissue,7–10 has a yet unidentified role in the control of vascular tone.
We therefore hypothesized that CaMKIV could affect endothelial functions, such as the control of vascular resistance, and that changes in its level of expression or activity in endothelial cells (ECs) might alter the fine regulation of vascular responses, causing hypertension. To ascertain whether CaMKIV signaling is involved in endothelial dysfunction, a hallmark of the hypertensive state,11–14 we used a murine model of genetic deletion of CaMK4. Finally, a population study was carried out in normotensive and hypertensive patients to investigate the effects of the CaMK4 rs10491334 SNP in humans.
Methods
In Vivo Studies
Animals
All animal procedures were performed in accordance with the policies and guidelines of the “Position of the American Heart Association on Research Animal Use”15 and were approved by the Ethics Committee of the “Federico II” University. The European Commission Directive 2010/63/EU was followed. We studied male mice with global homozygous deletion of the CaMK4 gene (CaMK4−/−), backcrossed >12 generations onto a C57Bl/6J background. The mice were kindly provided by Anthony Means (Duke University, Durham, NC).9 Age-matched wild-type littermates (CaMK4+/+) were used as controls. The animals were housed in a 22°C room with a 12-hour light/dark cycle and were allowed food and tap water ad libitum. Two groups of mice (CAMK4+/+ and CAMK4−/−) were subjected to a long-term furosemide treatment (0.35 mg/kg per day) from the age of 3 months until 6 or 18 months. The drug was added directly to the drinking water. The development of the typical target-organ damage (vascular, cardiac, and kidney damage)16–18 was evaluated in 18-month-old mice. Longitudinal survival observation on CAMK4+/+ and CAMK4−/− mice was performed over a period of 24 months. Genotypes were determined by polymerase chain reaction amplification of tail DNA. The individual performing all experiments was blinded to the mouse genotype until all data were fully analyzed.
Invasive Arterial BP Measurement
Mice were anesthetized by isoflurane (4%) inhalation and maintained by mask ventilation (isoflurane 1.8%). Direct BP and heart rate measurements were performed with the use of a 1.0F Mikro-Tip catheter (SPR1000, Millar Instruments, Houston, TX), which was advanced through the right external carotid artery and placed in the descending aorta. After implantation, the catheter was connected to a transducer (Gould Instruments Systems, Cleveland, OH) to record BP and heart rate for 15 minutes. The pressure catheter then was advanced through the aortic valve into the left ventricle (LV). Subsequent offline evaluation provided the first derivative of the LV pressure curve (maximum and minimum dP/dt). All data were analyzed with dedicated software (PowerLab-Chart 7.1, ADInstruments, Sydney, Australia).
Echocardiography
Transthoracic echocardiography was performed with a small-animal high-resolution imaging system (VeVo770, VisualSonics, Inc, Toronto, Canada) equipped with a 30-MHz transducer (Real-Time Micro Visualization, RMV-707B). The mice, anesthetized as described previously for BP measurement, were placed in a shallow left lateral decubitus position, with strict thermoregulation (37±1°C) to optimize physiological conditions and reduce hemodynamic variability. Fur was removed from the chest by application of a cosmetic cream (Veet, Reckitt Benckiser, Milan, Italy) to gain a clear image. LV end-diastolic and LV end-systolic diameters were measured at the level of the papillary muscles from the parasternal short-axis view.19–20 Intraventricular septal and LV posterior wall thickness were estimated at end diastole. LV fractional shortening was calculated as follows: LVFS = [(LVEDD − LVESD)/ LVEDD] × 100, where LVFS indicates LV fractional shortening; LVEDD, LV end-diastolic diameter; and LVESD, LV end-systolic diameter. LV ejection fraction was calculated automatically by the echocardiography system. All measurements were averaged on 10 consecutive cardiac cycles per experiment and were analyzed by one experienced investigator.
Electrocardiography
Electrocardiography (ECG) was performed under isoflurane (1.5%) anesthesia. Mice were placed on a thermocontrolled plate (37±1°C) and were given 10 minutes to acclimate before ECG recording. Signal-averaged ECG tracings were obtained by subcutaneous placement of 27-gauge steel needle electrodes in each limb, secured with tape. ECG was recorded for 60 minutes with the PowerLab Chart 7.1 system (ADInstruments, Sydney, Australia) and then was analyzed offline.
Angiogenic Response After Peripheral Chronic Ischemia
Peripheral chronic ischemia was induced in 6-month-old mice by means of surgical ligation and excision of the right common femoral artery. We have published a detailed description of the procedure.21–23 The angiogenic response was assessed on postoperative days 3, 7, 14, and 21 by laser Doppler (Perimed Instruments, Järfälla, Sweden).22 Furthermore, 3 weeks after surgery, we performed (1) ultrasound Doppler analysis of the posterior tibial artery with a VeVo770 imaging system equipped with a 20- to 60-MHz scanhead (VisualSonics, Inc, Toronto, Canada), (2) dyed-microbead assay on the gastrocnemius muscle, and (3) histological analysis of the anterior tibial muscle.
Myocardial Infarction
Reproducible infarcts of the anterior LV wall were imposed on 6-month-old mice by cryogeny with a 6-mm2 cryoprobe. Briefly, after isofluorane (2%) anesthesia, a thoracotomy was performed through the fourth left intercostal space, the pericardium was opened, and the heart was exposed. Cryoinfarction was produced by applying the cryoprobe to the anterior LV free wall, followed by freezing for 10 seconds. The exact position of the probe was set carefully by using the left atrium and pulmonary artery as anatomic landmarks. Rinsing with room-temperature saline was performed to allow nontraumatic detachment of the probe from the LV wall after freezing. Cardiac ultrasound analysis was performed 8 weeks after the lesion was created.
Urinary Protein Excretion
As a marker of renal damage,16 we assessed urinary protein excretion nephelometry (bicinchoninic acid method; Pierce, Rockford, IL) by placing the mice (n=12 in each group) in metabolic cages (Tecniplast, Buguggiate, Italy) for 24 hours.
Ex Vivo Studies
Vascular Reactivity
Aortic rings (6 to 9 mm) from 6-month-old mice were suspended in isolated tissue baths (Radnoti Glass Technology, Monrovia, CA) filled with 25 mL Krebs-Henseleit solution (in mmol/L: NaCl 118.3, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, and glucose 5.6) continuously bubbled with a mixture of 5% CO2 and 95% O2 (pH 7.38 to 7.42) at 37°C, according to the protocol used in our laboratory.23 Vasorelaxation was assessed in vessels preconstricted with phenylephrine (1 μmol/L) in response to isoproterenol, acetylcholine, or nitroprusside, all from 10 nmol/L to 10 μmol/L, freshly prepared on the day of experiment.23 Endothelium-independent vasorelaxation also was tested after incubation (10 μmol/L, 15 minutes) with NG-nitro-l-arginine methyl ester (Sigma-Aldrich, Milan, Italy), a competitive inhibitor of endothelial nitric oxide synthase (eNOS). Concentrations are reported as the final molar value in the organ bath.
Histology
Samples (hearts, kidneys, muscles) were fixed in 10% buffered formalin and processed for paraffin embedding.24 Slides were stained with hematoxylin and eosin for architectural analysis or with Masson's trichrome to assess the presence and extent of interfiber interstitial fibrosis.19,24–25 Percent collagen was calculated from high-resolution, color-calibrated digital images of Masson's trichrome–stained sections with the use of dedicated software (NIH ImageJ64), as described.19 To measure myocyte cross-sectional area (μm2) we used fluorescence-tagged wheat germ agglutinin staining (5.0 μg/mL; with samples incubated in the dark for 10 minutes at 37°C). Images were recorded at 494-nm excitation and 518-nm emission and were evaluated with ImageJ64. Lectin immunohistochemical staining was performed on myocardial and skeletal muscle.21,26
Quantification of Atherosclerotic Lesions
Atherosclerotic lesions in 18-month-old mice were detected by staining with the neutral lipid-targeting lysochrome Oil Red O (Sigma-Aldrich, Milan, Italy). Each aorta was rinsed first in distilled water and then quickly in 60% isopropyl alcohol. Subsequently, vessels were stained for 25 minutes (in a solution of 15 g Oil Red O, 30 mL isopropyl alcohol, and 20 mL distilled water, freshly prepared and filtered) and then were washed. The Oil Red O–stained areas of the inner aortic surfaces were quantified using the free software Fiji. The extent of atherosclerosis was assessed on longitudinally opened aorta and expressed as the percentage of the lipid-accumulating lesion area to the total aortic area analyzed. Acquisition of images and analysis of lesions were performed in a blinded fashion.
In Vitro Assays
Cell Culture
Murine aortic ECs were isolated from 3-month-old CaMK4−/− and CaMK4+/+ animals as previously described.27 Bovine aortic ECs and human embrionic kidney (HEK293) cells were purchased from Lonza (Basel, Switzerland) and American Type Culture Collection (ATCC; Manassas, VA), respectively. Cells were cultured in Dulbecco's modified Eagle medium (Sigma-Aldrich, Milan, Italy) as described.19 All experiments were performed in triplicate to ensure reproducibility. We used ionomycin (1 μmol/L; Sigma-Aldrich) as activator and KN93 (5 μmol/L; Seikagaku Corporation, Tokyo, Japan) as inhibitor of CaMK.5,28
Immunoprecipitation and Immunoblotting
Immunoblot analysis was performed as previously described and validated.21 Blots were probed with mouse monoclonal antibodies against eNOS, phospho-eNOS (peNOS Ser1177), CaMKIV (BD Bioscience, Franklin Lakes, NJ), peNOS Ser114, peNOS Ser615 (Millipore, Billerica, MA), peNOS Thr495, pCaMKIV, CaMKII, pCaMKII, and actin (Santa Cruz Biotechnology, Santa Cruz, CA). Images then were digitalized and densitometry was assessed with dedicated software (Image Quant, GE Healthcare, Piscataway, NJ). Data are presented as arbitrary units after normalization for the total corresponding protein or actin as loading control, as indicated.
eNOS Activity Assay
eNOS activity was detected in CaMK4−/− and CaMK4+/+ murine aortic ECs and in bovine aortic ECs by measuring the conversion of l-[3H]arginine to l-[3H]citrulline at 37°C for 30 minutes with the eNOS assay kit (Calbiochem-Nova Biochem, San Diego, CA), according to the manufacturer's instructions. Unlabeled l-arginine was added to l-[3H]arginine (specific activity, 60 Ci/mmol/L) at a ratio of 3:1. Mouse cerebellum extracts, containing elevated amounts of neuronal NOS, were used as positive controls, whereas samples incubated in the presence of NG-nitro-l-arginine methyl ester (1 mmol/L) were used to determine nonspecific activity.
CaMKIV Activity Assay
The CaMKIV activity assay consisted of 2 reaction steps. Briefly, in the first step, active recombinant full-length CaMKIV (Millipore, Billerica, MA) was incubated at 30°C for 30 minutes with 0.5 mmol/L CaCl2 and 1 μmol/L CaM in 25 μL of a reaction mixture (25 mmol/L 4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid [HEPES], pH 7.5, 0.5 mmol/L MgCl2, 1 mmol/L dithiothreitol, 0.5 mg/L BSA, 1 mmol/L sodium orthovanadate, 0.1 mmol/L cold adenosine triphosphate (ATP), and H2O 0.01% Tween 20). In the second step, a 20-μL aliquot from the first reaction mixture containing active CaMKIV was incubated in 50 μL of a solution containing 0.1 IU of eNOS (Calbiochem-Nova Biochem, San Diego, CA) as substrate and 1 mmol/L EGTA, 0.5 μL [32P]-γATP (3000 Ci/mmol/L) for 30 minutes at 30°C. The reactions were stopped by the addition of sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer, and the whole reaction mixes were separated on 4% to 12% SDS-PAGE (Life Technologies, Grand Island, NY). Then, the gel was dried and peNOS was visualized by phosphorImager (GE Healthcare, Piscataway, NJ).
Alternatively, a partially modified protocol was performed without the use of radioactive ATP. In this case, after PAGE, proteins were blotted on nitrocellulose, and eNOS phosphorylation by CaMKIV was also assessed by Western blot by using the previously mentioned antibodies.
Overlay Blot Assay
Twenty nanograms of CaMKIV and 0.1 IU of eNOS-purified proteins were subjected to SDS-PAGE and transferred on nitrocellulose. The membranes were incubated 2 hours at room temperature in 5% blocking solution. At this time, the filters were incubated with CaMKIV or eNOS-purified protein in phosphorylation solution (final concentrations: 20 μmol/L ATP, 1 mmol/L CaCl2, 20 mmol/L MgCl2, and 4 mmol/L Tris, pH 7.5). After 1 hour of incubation at room temperature, the blots were cooled rapidly on ice; washed twice with NaCl, Tris, and 0.1% Tween 20; and then fixed with 0.5% formaldehyde for 10 minutes. The filters were washed 3 times with 2% glycine and once with NaCl, Tris, and 0.1% Tween 20. CaMKIV or eNOS-bound proteins were detected by chemiluminescence.29
Human Association Study
Study participants were consecutive hypertensive patients referred to the Hypertension Diagnosis and Care Outpatient Clinic of “Federico II” University of Naples. Age-matched unaffected controls were recruited from a database of normotensive blood donors. The matching design was accounted for in the statistical analyses. All subjects were white and born within the Campania region in Southern Italy. We had access to a digital archive for each participant.30 Enrollment criteria for hypertensive status were an age of 18 to 80 years and a confirmed diagnosis of essential hypertension. We considered the average systolic and diastolic BP values, according to European guidelines.16
Analysis of the rs10491334 SNP of CaMK4 was performed on peripheral blood DNA by restriction fragment length polymorphism.30 Genetic analyses were performed by laboratory personnel blinded to sample identity. Patients' lymphocytes were extracted by means of phicoll purification with HISTOPAQUE-1077 (Sigma-Aldrich) from a 20-mL blood sample, which had been anticoagulated with ethylenediaminetetraacetic acid.31
Data Presentation and Statistical Analysis
Data are presented as mean ± standard error (SE) unless otherwise mentioned. To determine the statistical significance of the results, we used 1-way ANOVA or Kruskal-Wallis test, as appropriate (nonparametric analysis was used when a large number of tests increased the risk of a type α error). Survival curves were compared with the log-rank test.
To assess significant differences between genotype classes in the human studies, we used the Student t test for continuous variables and the Fisher exact test for categorical variables. The association between the SNP and hypertension was adjusted for age, sex, heart rate, and body mass index. Statistical significance was set at P<0.05. All the analyses were performed with GraphPad Prism version 5.01 (GraphPad Software, San Diego, CA), Systat 13 (Systat software, Inc, Chicago, IL), and Statistical Package for Social Sciences software version 20.0.0 (IBM SPSS Inc, Armonk, NY).
Results
BP, Organ Damage, and Survival
CaMK4−/− mice developed higher systolic and diastolic BP levels than did CaMK4+/+ littermates, as shown in Table 1. We decided to assess BP-dependent phenotypes, such as target-organ damage, in homozygous mice only. At 6 months, cardiac ultrasound analysis showed that CaMK4−/− mice displayed concentric25,31–32 LV hypertrophy (LVH) due to increased septum and posterior wall thickness, with no significant changes in internal diameter, as depicted in Table 1. At gross analysis, CAMK4−/− hearts were larger both as absolute values (163.1±4.5 versus 136.8±3.1 mg, Figure 1A) and after correction by either body weight (Figure 1B) or tibial length (Figure 1C). Histological analysis showed increased cell size of CAMK4−/− cardiomyocytes (Figure 1D and 1E) and augmented interstitial fibrosis (Figure 1F and 1G), 2 common elements of hypertension-induced LVH.19 In older mice (18 months old), cardiac damage evolved toward dilatation and dysfunction (Table 1) complicated by arrhythmias, as shown by ECG (Figure 2A through 2C), according to the natural history of untreated hypertension.33 Indeed, LVH is associated with alterations in the dispersion of repolarization34 and prolongation of ventricular action potentials.35 These effects result in electrical instability and increase the propensity to develop arrhythmias. When an antihypertensive treatment with furosemide was initiated at 3 months of age, the development of both hypertension and LVH was prevented (Table 2).
Table 1.
Systemic and LV Hemodynamics in CAMK4+/+ and CAMK4−/− Mice
| CaMK4+/+ (n=12) | CaMK4−/− (n=13) | CaMK4+/+ (n=14) | CaMK4−/− (n=16) | CaMK4+/+ (n=12) | CaMK4−/− (n=13) | |
|---|---|---|---|---|---|---|
| 3-Month-Old Mice | 6-Month-Old Mice | 18-Month-Old Mice | ||||
| HR, bpm | 492±28 | 486±36 | 486±18 | 474±16 | 460±32 | 468±28 |
| SBP, mm Hg | 111±1.3 | 113±1.8 | 110±0.6 | 123±0.7* | 111±1.1 | 124±1.3* |
| DBP, mm Hg | 81±0.9 | 82±1.1 | 81±0.3 | 90±0.3* | 86±0.8 | 93±0.9* |
| LVEDD, mm | 3.71±0.42 | 3.73±0.35 | 3.85±0.32 | 3.74±0.29 | 3.89±0.37 | 4.36±0.3* |
| LVESD, mm | 2.08±0.26 | 2.15±0.28 | 2.19±0.27 | 2.37±0.28 | 2.22±0.3 | 3.02±0.46* |
| IVS, mm | 0.72±0.04 | 0.73±0.05 | 0.72±0.03 | 0.88±0.02* | 0.82±0.04 | 0.89±0.03* |
| LVPW, mm | 0.71±0.03 | 0.72±0.04 | 0.69±0.02 | 0.89±0.03* | 0.78±0.03 | 0.92±0.04* |
| LVFS, % | 43.4±4.8 | 42.6±5.5 | 42.4±4.5 | 36.2±6.1* | 37.2±5.8 | 30.2±7.2* |
| LVEF, % | 66.8±8.2 | 66.3±9.1 | 64.8±8.6 | 59.6±7.8 | 56.4±9.4 | 50.1±9.8* |
| +dP/dt, mm Hg/s | 6334±602 | 6211±522 | 6237±531 | 5681±498* | 6004±582 | 5014±578* |
| −dP/dt, mm Hg/s | 6420±542 | 6322±508 | 6302±504 | 5702±486* | 6080±682 | 5112±606* |
Data are mean±SE. HR indicates heart rate; SBP, systolic BP; DBP, diastolic BP; LVEDD, LV end-diastolic diameter; LVESD, LV end-systolic diameter; IVS, interventricular septum; LVPW, LV posterior wall; LVFS, LV fractional shortening; and LVEF, LV ejection fraction.
P<0.05 comparing CaMK4−/− to CaMK4+/+at each time point; otherwise, P not significant.
Figure 1.
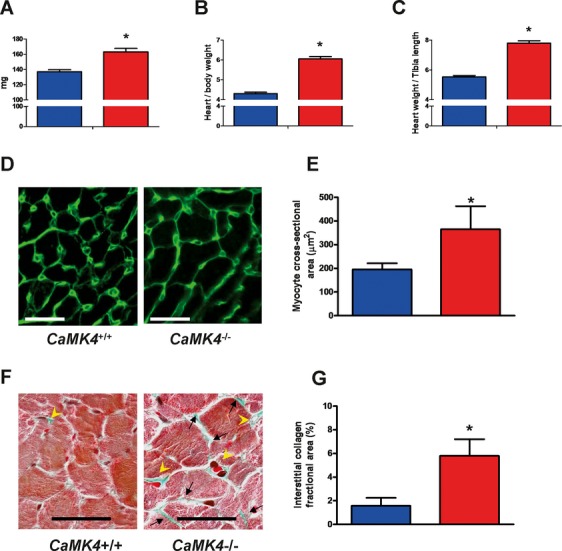
LVH in CaMK4−/− mice. CaMK4−/− 6-month-old mice present cardiac hypertrophy, as determined by an increase in heart weight (A), normalized by body weight (B) or tibial length (C). Fluorescence-tagged wheat germ agglutinin, which binds to saccharides of cellular membranes, showed increased cardiac myocyte cross-sectional area in CaMK4−/− mice (D and E; magnification ×200, scale bar = 15 μm). Masson's trichrome staining (F; magnification ×300, scale bar = 15 μm; interstitial and perivascular collagen deposition indicated by black arrows and yellow arrowheads, respectively) also revealed an increase in fibrosis, quantified as described in Methods (G). Blue bars are CaMK4+/+ mice; red bars, CaMK4−/− mice. *P<0.05 vs CaMK4+/+.
Figure 2.
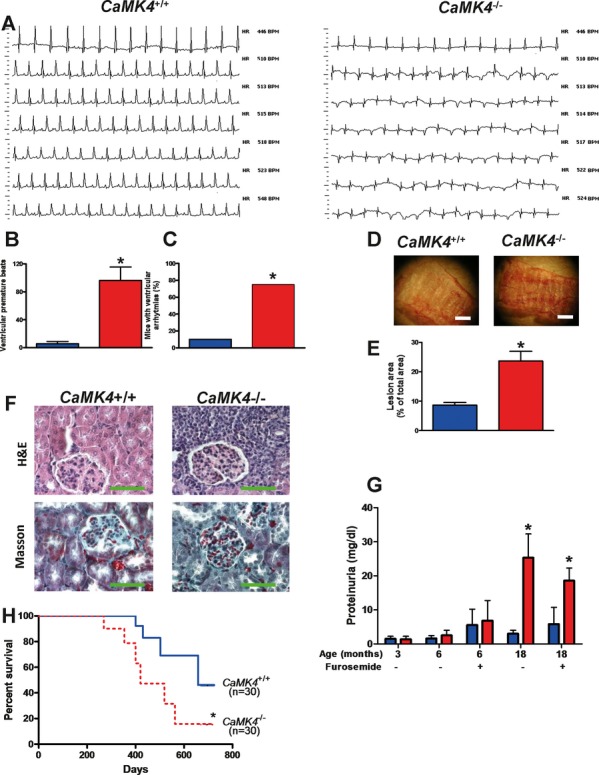
Organ damage in 18-month-old mice. Old CaMK4−/− mice displayed a dilatation of the LV (see also Table 1) and exhibited several spontaneous ventricular arrhythmias (A; representative ECG records at different heart rates). These data are quantified in B and C (n=10 per group; *P<0.05). Of note, heart rate among mutated animals did not significantly differ (see Table 1). D and E, Lipid deposition in vessels of CaMK4+/+ and CaMK4−/− mice (n=12 per group). Representative pictures of oil red O–stained ascending aortas (D; white bar = 600 μm); areas of atherosclerotic lesions were quantified using the free software Fiji and are represented as percentage of lesion area to total aortic area (E; *P<0.05 vs CaMK4+/+). Kidneys (F) from the CaMK4−/− group exhibited increased glomerulosclerosis, inflammatory cell infiltration, and tubulointerstitial fibrosis compared with CaMK4+/+ mice (n=8 per group; representative pictures of hematoxylin and eosin or Masson's trichrome staining; magnification ×60; green bar = 100 μm). Moreover, 18-month-old CaMK4−/− mice presented another typical feature of the hypertensive phenotype (G), showing greater proteinuria than CaMK4+/+ mice (n=12 per group; *P<0.05 vs CaMK4+/+). In all histograms, blue bars are CaMK4+/+ mice; red bars, CaMK4−/− mice. Notably, compared with CaMK4+/+ mice, CaMK4-null mice displayed significantly (*P<0.05) impaired survival, as well (Kaplan-Meier curves; H). Blue line indicates CaMK4+/+ mice; red dotted line, CaMK4−/− mice.
Table 2.
Effect of Long-Term Diuretic Treatment on Systemic and LV Hemodynamics in CAMK4+/+ and CAMK4−/− Mice
| CaMK4+/+ (n=11) | CaMK4−/− (n=16) | CaMK4+/+ (n=10) | CaMK4−/− (n=14) | |
|---|---|---|---|---|
| 6-Month-Old Mice Treated (3 Months) With Furosemide | 18-Month-Old Mice Treated (15 Months) With Furosemide | |||
| HR, bpm | 498±46 | 492±38 | 483±52 | 488±41 |
| SBP, mm Hg | 107±6.8 | 114±1.8 | 103±6.6 | 104±1.3 |
| DBP, mm Hg | 74±8.5 | 83±1.1 | 75±5.9 | 78±1.2 |
| LVEDD, mm | 3.82±0.9 | 3.83±0.4 | 3.9±0.8 | 3.95±0.7 |
| LVESD, mm | 2.21±0.8 | 2.23±0.3 | 2.26±0.9 | 2.28±0.5 |
| IVS, mm | 0.71±0.08 | 0.76±0.04 | 0.78±0.09 | 0.8±0.05 |
| LVPW, mm | 0.69±0.07 | 0.77±0.05 | 0.76±0.08 | 0.79±0.06 |
| LVFS, % | 41.6±8.8 | 39.2±7.3 | 36.9±8.6 | 36.5±7.1 |
| LVEF, % | 62.8±9.7 | 62.5±8.4 | 55.4±8.1 | 55.1±8.9 |
| +dP/dt, mm Hg/s | 6126±684 | 6088±642 | 5522±528 | 5876±627 |
| −dP/dt, mm Hg/s | 6188±747 | 6104±594 | 5596±639 | 5892±582 |
Data are mean±SE. HR indicates heart rate; SBP, systolic BP; DBP, diastolic BP; LVEDD, LV end-diastolic diameter; LVESD, LV end-systolic diameter; IVS, interventricular septum; LVPW, LV posterior wall; LVFS, LV fractional shortening; and LVEF, LV ejection fraction.
Along with increased heart size, CAMK4−/− mice over time developed hypertensive vascular damage, as assessed by Oil Red O staining (Figure 2D), which revealed larger atherosclerotic lesions in aortas from 18-month-old CaMK4−/− mice versus CaMK4+/+ mice (Figure 2E). We also found renal damage, another typical feature of the hypertensive phenotype,16 inasmuch as CaMK4−/− mice displayed increased glomerulosclerosis, inflammatory cell infiltration, and tubulointerstitial fibrosis compared with CaMK4+/+ mice (Figure 2F). The functional correlate to this histological alteration is increased proteinuria in 18-month-old CaMK4−/− mice (Figure 2G). Furosemide-treated CaMK4−/− mice still presented proteinuria (Figure 2G), although they had normal BP levels.
Eventually, CaMK4−/− mice showed significantly reduced lifespan compared with CAMK4+/+ littermates (Figure 2H).
Assessment of Endothelium-Dependent Phenotypes
Endothelium-dependent vasodilation in ex vivo experiments was assessed on isolated aortic rings. After vasoconstriction obtained through 1 μmol/L phenylephrine (Figure 3A), aortic rings from CaMK4−/− mice showed impaired endothelial-dependent vasodilation both to the β-adrenergic agonist isoproterenol and to the muscarinic agonist acetylcholine, which is consistent with our hypothesis of endothelial dysfunction (Figure 3B and 3C), a known hallmark of the hypertensive state.13 Moreover, there was no difference in the vascular smooth muscle cell–mediated response to the nitric oxide donor nitroprusside (Figure 3D), and experiments performed after NG-nitro-l-arginine methyl ester incubation confirmed intact endothelium-independent vasodilation in both CaMK4−/− and CaMK4+/+ vessels (Figure 3E and 3F).
Figure 3.
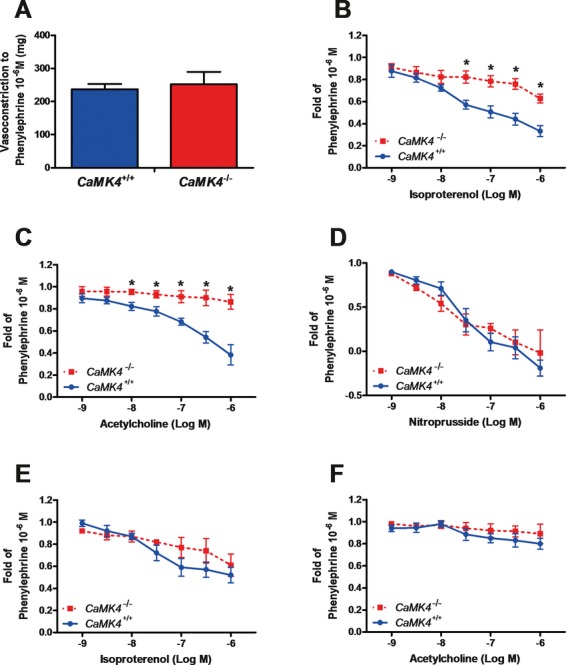
Vascular responses on isolated aortic rings from 6-month-old mice. Vasoconstriction to α1-adrenergic agonist phenylephrine was similar in CaMK4+/+ and CaMK4−/− mice (A). Endothelium-dependent vasorelaxation induced by the β-adrenergic agonist isoproterenol (B) or by the muscarinic agonist acetylcholine (C) was blunted in CaMK4−/− vessels, whereas endothelium-independent vasodilation to nitroprusside was not different between CaMK4+/+ and CaMK4−/− (D). To better explore the role of nitric oxide in endothelial responses, we also evaluated vascular responses in the presence (10 μmol/L) of the specific eNOS inhibitor NG-nitro-l-arginine methyl ester (E and F). *P<0.05 vs CaMK4+/+.
Additionally, we assessed in vivo the angiogenic response to ischemia after femoral artery removal, a phenotype that is largely under the control of the endothelium.21,23,36 CaMK4−/− mice displayed impaired angiogenesis after 21 days of peripheral ischemia, as assessed by laser Doppler (Figure 4A and 4B), Doppler ultrasound (Figure 4C), dyed microbeads (Figure 4D), and capillary density (Figure 4E and 4F).
Figure 4.
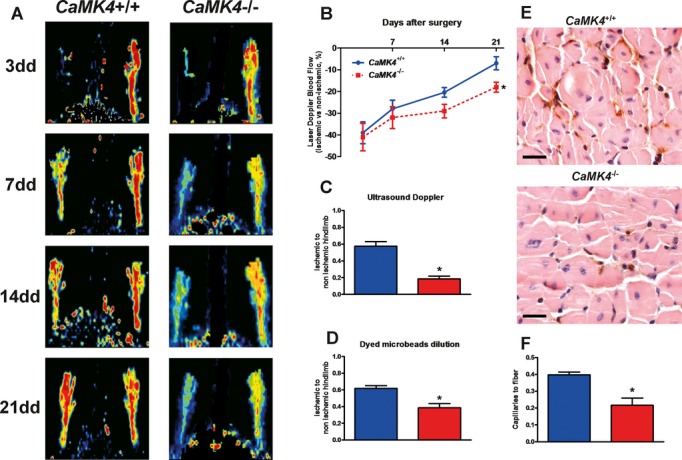
Decreased neoangiogenic responses in CaMK4−/− mice during chronic ischemia in vivo (n=6 per group). Determination of laser Doppler blood flow (A and B) on postoperative days 3, 7, 14, and 21 showed a deficit in ischemic hindlimb perfusion, compared with the contralateral hindlimb, that was significantly increased in CaMK4−/− vs CaMK4+/+ mice (*P<0.05, repeated measurements, ANOVA; laser Doppler blood flow data are expressed as percent of ischemic to nonischemic limb). Ultrasound Doppler flowmetry of posterior tibial artery (C), performed 3 weeks after femoral artery removal, confirmed the attenuated blood flow restoration in CaMK4−/− mice (*P<0.05 vs CaMK4+/+). This result was mirrored by the dyed-bead perfusion analysis (D; *P<0.05 vs CaMK4+/+). Lectin staining of capillaries in the ischemic hindlimb (E; magnification ×20, black bar = 100 μm) showed that chronic ischemia produced a greater rarefaction of the capillary density, evaluated as number of capillaries corrected for the number of muscle fibers (F), in CaMK4−/− compared with CaMK4+/+ mice (*P<0.05). In all histograms, blue bars are CaMK4+/+ mice; red bars, CaMK4−/− mice.
Development of Heart Failure After Myocardial Infarction
Increased BP and reduced angiogenesis are expected to precipitate the evolution of the heart failure phenotype after myocardial damage.17,37 Indeed, 8 weeks after myocardial cryoinfarction, CaMK4−/− mice presented larger LV dilatation and a greater decrease in cardiac function than did CAMK4+/+ mice, as shown in both Table 3 and Figure 5A. This feature also was accompanied by reduced capillary density in the peri-infarct area (Figure 5B and 5C).
Table 3.
Systemic and LV Hemodynamics in CAMK4+/+ and CAMK4−/− Mice 8 Weeks After Myocardial Infarction
| CaMK4+/+ (n=12) | CaMK4−/− (n=14) | |
|---|---|---|
| HR, bpm | 498±20 | 482±18 |
| SBP, mm Hg | 100±0.5 | 108±0.6* |
| DBP, mm Hg | 74±0.3 | 82±0.4* |
| LVEDD, mm | 4.16±0.12 | 4.6±0.11* |
| LVESD, mm | 3.15±0.08 | 3.71±0.13* |
| IVS, mm | 0.66±0.04 | 0.85±0.05* |
| LVPW, mm | 0.68±0.03 | 0.86±0.03* |
| LVFS, % | 24.74±2.9 | 17.8±2.3* |
| LVEF, % | 49.02±4.8 | 35.8±3.4* |
| +dP/dt, mm Hg/s | 3784±402 | 3004±382* |
| −dP/dt, mm Hg/s | 3918±386 | 3128±394* |
Data are mean±SE. Mice were 8 months old (myocardial infarction was induced in 6-month-old animals). HR indicates heart rate; SBP, systolic BP; DBP, diastolic BP; LVEDD, LV end-diastolic diameter; LVESD, LV end-systolic diameter; IVS, interventricular septum; LVPW, LV posterior wall; LVFS, LV fractional shortening; and LVEF, LV ejection fraction.
P<0.05; otherwise, P not significant.
Figure 5.
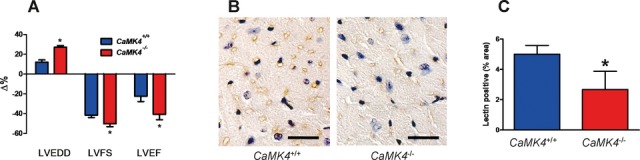
Cardiac evaluation after cryoinjury. Changes in echocardiographic parameters (A) 8 weeks after myocardial infarction. (LVEDD indicates LV end-diastolic diameter; LVFS, LV fractional shortening; and LVEF, LV ejection fraction. n=12 per group. *P<0.05 vs CaMK4+/+). Immunohistochemical analysis (B; lectin staining, magnification ×60, black bar = 30 μm) of myocardium in mice 8 weeks after infarction. The peri-infarct area demonstrated lower capillary density, as confirmed by quantitative analysis (C); blue bars are CaMK4+/+ mice; red bars, CaMK4−/− mice. *P<0.05 vs CaMK4+/+.
Interaction Between CaMKIV and eNOS
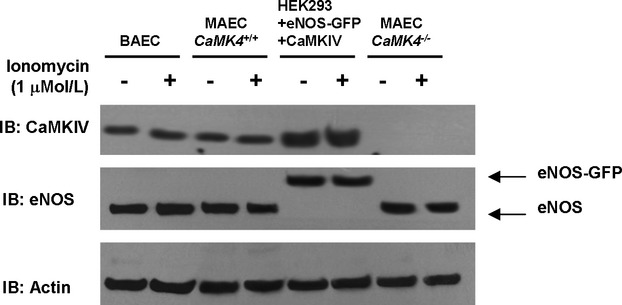
Because endothelium dysfunction can be mimicked by altered production or removal of nitric oxide, we assessed the effect of CaMK4 knockout on eNOS activation. Indeed, calcium-induced eNOS phosphorylation on Ser1177 is impaired in CaMK4−/− ECs. (Figures 6A and 7A through 7C). This impairment associates with a reduction in CaMKIV expression and activity in CaMK4−/− EC, whereas activation of CaMKII remains unaffected (Figure 6A). This finding also is endorsed by a reduction in eNOS activity (Figure 8). Interestingly, transgenic restoration of CaMKIV expression in CaMK4−/− ECs also corrects calcium-induced eNOS activation (Figures 6B, 7D, and 7E). CaMKIV and eNOS can coimmunoprecipitate in either naïve or overexpressing cells (Figures 6C and 9). The physical interaction between the 2 proteins can be replicated in an overlay assay with the use of purified CaMKIV and eNOS, which indicates that the interaction is not mediated by a third component (Figure 6D). The result of this physical interaction is the incorporation in eNOS of 32P (Figure 6E). Also, in vitro, purified CaMKIV can phosphorylate eNOS directly on Ser1177 and Ser615, 2 phosphorylation sites that are regulatory for enzyme activity, but not on other phosphorylation sites of eNOS, namely Ser114 and Thr495 (Figures 6F and 7F through 7I).
Figure 6.
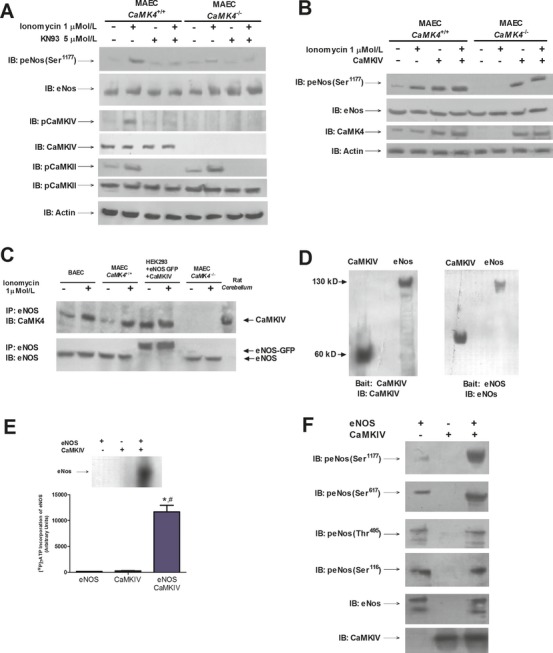
Interaction between CaMKIV and eNOS. eNOS phosphorylation (Ser1177) is enhanced by ionomycin, echoing the phosphorylation of CaMKIV, and is prevented by the CaMK inhibitor KN93 (A). Notably, eNOS activation was less evident in CaMK4−/− MAEC, where CaMK4 was not expressed (A). Transgenic restoration of CaMKIV expression in CaMK4−/− ECs corrected calcium-induced eNOS activation (B). The interaction between CaMKIV and eNOS was demonstrated by performing immunoprecipitation (IP) experiments in different cellular settings, both in basal conditions and after stimulation with ionomycin (C). Such interaction is shown in BAEC and CaMK4+/+ MAEC but not in CaMK4−/− MAEC. In a nonendothelial cell type, HEK293, we confirmed the interaction after reconstituting the system by using a plasmid encoding CaMKIV and a plasmid encoding eNOS linked to GFP (C; rat cerebellum was used as CaMKIV-positive control). The input protein levels are shown in Figure 8. Overlay assay with purified CaMKIV (left blot) or eNOS (right blot) as bait (D). CaMKIV induced eNOS [32P]-γATP incorporation (E). Purified CaMKIV induced eNOS phosphorylation on Ser1177 and Ser615 but not on Ser114 and Thr495 (F). *P<0.05 vs eNOS, *P<0.05 vs CaMKIV; representative images from triplicate experiments are shown. Densitometric analyses are reported in Figure 7. MAEC indicates murine aortic ECs; BAEC, bovine aortic ECs.
Figure 7.
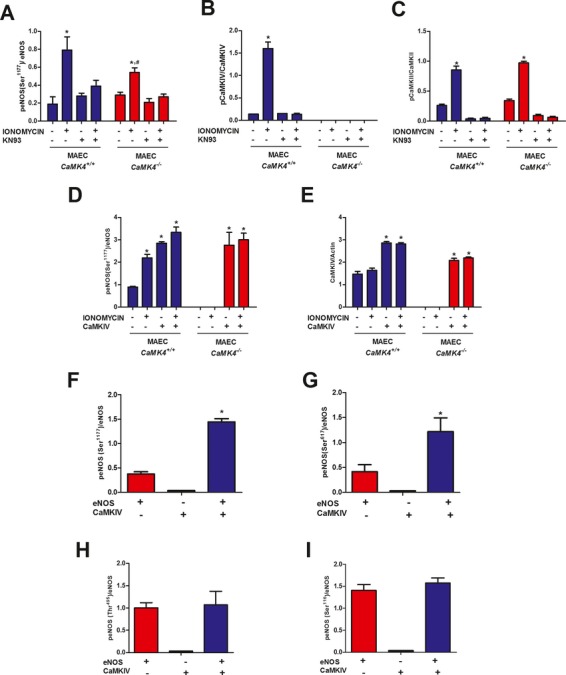
Quantification of blot results presented in Figure 6. Relative protein quantification levels for Figure 6A (A–C) and Figure 6B (D–E). Blue bars are CaMK4+/+; red bars, CaMK4−/−. *P<0.05 vs untreated cells; #P<0.05 vs MAEC CaMK4+/+. MAEC indicates murine aortic ECs. Densitometric analyses for Figure 6F (F–H) showing CaMKIV-mediated eNOS phosphorylation on Ser1177 and Ser615 but not on Ser114 and Thr495. Red bars indicate eNOS; black bars, CaMKIV; and blue bars, eNOS and CaMKIV. *P<0.05 vs eNOS.
Figure 8.
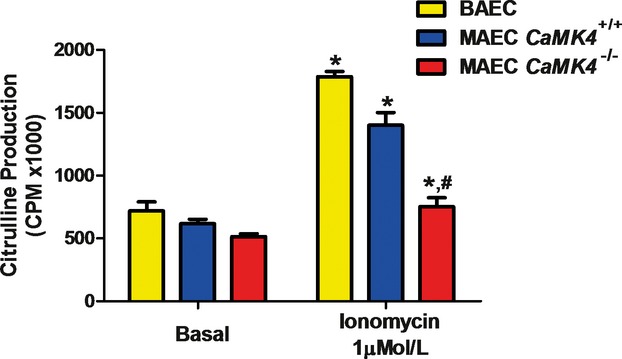
eNOS activity in ECs. eNOS activity, assessed by arginine–citrulline conversion, after stimulation by ionomycin (1 μmol/L) was blunted in CaMK4−/− MAEC. *P<0.05 vs basal; #P<0.05 vs CaMK4+/+. MAEC indicates murine aortic ECs; CPM, counts per minute.
Figure 9.
Input Western blots of immunoprecipitation assay represented in Figure 6C. To confirm that equal amounts of proteins were present in the cell lysates used for immunoprecipitation as depicted in Figure 6C, we performed Western blotting on 30 μg of proteins of corresponding cell lysates with the same antibodies used in the experiment represented in Figure 6C, raised respectively against CaMKIV and eNOS. Furthermore, actin was detected to confirm equal amount of proteins.
Role of rs10491334 T/C Polymorphism of Human CaMK4 Gene in Hypertensive Patients
The results gained in the CaMK4−/− mouse show a role for this kinase in the setup of hypertension. To confirm the possible relevance of this finding in humans, we studied the frequencies of the rs10491334 T/C SNP.6 This polymorphism associates with a reduction in the cellular expression of the kinase.38 We studied 2 populations of normotensive (n=457) and hypertensive subjects (n=730). Clinical characteristics of these individuals are reported in Table 4. We found a higher occurrence of the polymorphism among the hypertensive patients that fell short of statistical significance (normotensive patients: 31.32%; hypertensive patients: 41.64%; P=0.0594, Pearson χ2 analysis). Thus, we dichotomized our hypertensive population according to European Guidelines into categories of severe (Grade 2/3) and not-severe (Grade 1) diastolic hypertension (cutoff: diastolic BP=100 mm Hg)16 and found a significantly larger frequency of the T variant of the rs10491334 SNP in patients with severe hypertension than in patients with diastolic BP<100 mm Hg (54.42% versus 38.41%; P<0.05, Pearson χ2 analysis), as shown in Table 5. Intriguingly, hypertensive patients homozygous for the polymorphic T allele showed reduced expression levels of CaMKIV in circulating peripheral blood lymphocytes (Figure 10).
Table 4.
Characteristics of the Normotensive and Hypertensive Populations
| Normotensive Subjects (n=457) | Hypertensive Subjects (n=730) | |
|---|---|---|
| Age, y | 52.91±3.4 | 55.84±2.9 |
| Sex (male/female), n | 288/169 | 452/278 |
| HR, bpm | 76.8±9.3 | 72.8±8.2 |
| SBP, mm Hg | 131.67±0.84 | 147.7±0.72* |
| DBP, mm Hg | 77.06±0.51 | 96.69±0.48* |
| Body mass index, kg/m2 | 26.03±0.27 | 27.19±0.21 |
| Smoking (current or former), % | 53.6 | 54.1 |
| Glycemia, mmol/L | 4.8±0.72 | 5.2±0.88 |
| Diabetes, % | 4.7 | 5.9 |
| Total cholesterol, mmol/L | 4.35±0.12 | 4.98±0.18 |
| High-density lipoprotein cholesterol, mmol/L | 1.24±0.11 | 1.21±0.14 |
| Low-density lipoprotein cholesterol, mmol/L | 2.96±0.13 | 3.25±0.19 |
| Triglycerides, mmol/L | 1.46±0.09 | 1.53±0.11 |
| Dyslipidemia, % | 37.8 | 41.2 |
| LV mass index, g/m2 | 92.6±4.5 | 115.34±4.8* |
| CaMK4 rs10491334 polymorphism (T-allele frequency), % | 31.32 | 41.64 |
Data are mean±SE; n, or %, as indicated. HR indicates heart rate; SBP, systolic BP; and DBP, diastolic BP.
P<0.05; otherwise, P not significant.
Table 5.
Characteristics of the Hypertensive Patients, Subdivided Into 2 Populations According to a DBP Cutoff of 100 mm Hg
| Patients With DBP <100 mm Hg (n=583) | Patients With DBP ≥100 mm Hg (n=147) | |
|---|---|---|
| Age, y | 54.9±3.6 | 56.2±3.8 |
| Sex, male/female, n | 371/212 | 81/66 |
| HR, bpm | 71.6±9.6 | 72.2±11.3 |
| SBP, mm Hg | 142.86±1.58 | 154.46±1.42* |
| DBP, mm Hg | 91.89±0.74 | 108.69±0.86* |
| Body mass index, kg/m2 | 26.85±0.63 | 27.71±1.74 |
| Smoking (current or former), % | 55.4 | 53.3 |
| Glycemia, mmol/L | 5.4±0.77 | 5.1±0.98 |
| Diabetes (%) | 5.7 | 6.1 |
| Total cholesterol, mmol/L | 4.77±0.22 | 5.01±0.28 |
| High-density lipoprotein cholesterol, mmol/L | 1.22±0.15 | 1.17±0.19 |
| Low-density lipoprotein cholesterol, mmol/L | 3.32±0.17 | 3.05±0.23 |
| Triglycerides, mmol/L | 1.52±0.13 | 1.55±0.16 |
| Dyslipidemia, % | 43.1 | 39.8 |
| LV mass index, g/m2 | 112.6±4.5 | 116.86±7.3 |
| CaMK4 rs10491334 polymorphism (T-allele frequency), % | 38.41 | 54.42* |
Data are mean±SE; n, or %, as indicated. HR indicates heart rate; SBP, systolic BP; and DBP, diastolic BP.
P<0.05; otherwise, P not significant.
Figure 10.
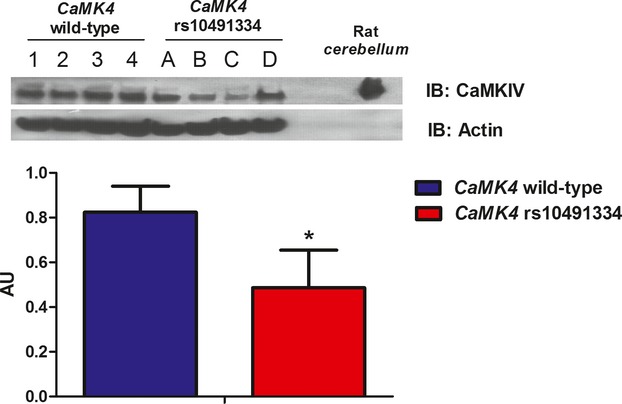
Expression levels of CaMKIV in circulating lymphocytes of hypertensive patients. Western blot analysis of CaMKIV on peripheral blood lymphocytes showed that CaMKIV levels were higher in subjects with the CaMK4 wild-type genotype (1, 2, 3, and 4 represent samples from 4 different individuals) than in subjects homozygous for the polymorphic CaMK4 rs10491334 variant (A, B, C, and D represent samples from 4 different individuals). Rat cerebellum was used as CaMKIV-positive control. Data from immunoblots (IB; representative images from 5 experiments are shown) were quantified by densitometric analysis. CaMKIV levels were normalized to actin densitometry. *P<0.05 vs CaMK4+/+. AU indicates arbitrary units.
Discussion
In the present study, we provide compelling evidence for a fundamental and previously unrecognized role of CaMKIV in the regulation of vascular function. Indeed, the phenotype of the CaMK4−/− mouse indicates that this kinase is extremely important for endothelial function. A paramount finding of this study is that the loss of CaMK4 results in the development of hypertension, accompanied by its typical hallmarks: endothelial dysfunction, target-organ damage, and reduced survival rate.16,32 Interestingly, furosemide-treated mice did not display LVH, which suggests that LVH is indeed the result of increased BP rather than being genetically determined by CaMK4 gene removal.13 We used a loop diuretic to obtain an effective BP decrease with minimum effect on vascular function.16 Moreover, other diuretics, such as thiazides, have metabolic implications not present in furosemide treatment16 that could confuse the cardiovascular phenotype of our CaMK4−/− mice further. In our model, endothelial dysfunction could be either primitive to hypertension or, alternatively, secondary to the hypertensive state of CaMK4−/− mice. We rule out this second hypothesis on the basis of 2 pieces of evidence: First, diuretic treatment resulted in normalization of hemodynamic-dependent LVH but did not correct proteinuria, which is a characteristic of endothelial dysfunction.39 Second, in a hemodynamic-independent setup36 (ie, in isolated ECs), CaMK4 removal causes endothelial dysfunction as assessed by reduction of eNOS activity, which is corrected only after gene replacement. Endothelial dysfunction is known to induce hypertension, as demonstrated in eNOS−/− mice, which show absent endothelial-dependent vasorelaxation and increased BP.13
Our study is the first to demonstrate the interaction of CaMKIV and eNOS. Previous reports had suggested the importance of CaMKs in endothelium-dependent relaxation.40–42 In addition, 2 independent groups had provided evidence that a nonselective CaMK inhibitor could significantly decrease bradykinin-induced eNOS activity12 and prevent eNOS phosphorylation28 in rat and porcine aortic ECs, respectively. Our study characterizes the close relationship between CaMKIV and eNOS in the endothelium. Indeed, CaMKIV can phosphorylate eNOS directly in Ser1177 and Ser615, 2 sites that are known to induce eNOS activation. Although we have not investigated all phosphorylation sites of eNOS, these 2 appear to be relevant for the described mechanism.
It is remarkable that CaMKII does not seem to supersede CaMKIV loss. Although CaMKIV and CaMKII often are considered mutually exchangeable, these 2 kinases present differences in tissue distribution and regulation43–44 and cannot be considered isoforms.5 For the present study, we used mice generated in the Means' laboratory in the 1990s.9 The cardiovascular phenotype of this mouse was never before investigated as extensively as we have in the present report. A hint of the higher BP of CaMK4−/− mice can be found in the article of Colomer and colleagues,4 showing that after constriction of the thoracic aorta, BP gradients were higher in CaMK4-null than in wild-type mice. Other reports have failed to describe cardiovascular parameters of these mice. Interestingly, though, CaMK4−/− mice present cognitive disorders that are typical of patients in the advanced phases of untreated hypertension,45 such as erasure of long-term memory.10 This phenotype has been ascribed to the loss of CaMK4 signaling in neurons of CaMK4−/− mice but also can be worsened by chronic exposure to increased BP levels.
To find a correlation between our observations in this genetically modified mouse and the human condition, we took advantage of the DNA Bank associated with the Campania Salute database of >5000 hypertensive patients.30 Previously, the Framingham study had identified an association marker for high diastolic BP in the rs10491334 SNP of the human CaMK4 gene.6 Our study confirms this finding: We performed an association analysis with a candidate gene approach and found a significant correlation between the rs10491334 SNP and diastolic BP levels among hypertensive patients. Furthermore, in the present work, we show that this polymorphism associates with a reduction in the cellular expression levels of CaMKIV, similar to that observed in other populations.38 These data are highly suggestive of the intrinsic regulatory nature of CaMKIV in hypertension.
Our study follows the groove of the identification of the physiological implications of CaMKs in the cardiovascular system.1–3 Some authors have investigated the effects of CaMKIV in the heart by overexpressing it in cardiomyocytes, leading to cardiac hypertrophy.46 This notion was challenged by a more recent study showing that mice null for CaMK4 still developed LVH.4 Our data reconcile these opposing views by suggesting that dysfunctional CaMKIV, albeit not expressed in the heart, might partake in cardiac organ damage in the context of the hypertensive state.
Conclusion
Our findings establish that CaMKIV plays a relevant role in the regulation of the vascular tone by a mechanism that involves eNOS activation through phosphorylative events. Impairment of CaMK-mediated activation of eNOS, as in CaMK4 gene deletion, induces hypertension, as demonstrated by the fact that CAMK4−/− mice display a hypertensive phenotype that leads to typical organ damage. Extending our observations to the clinical scenario, we show that in hypertensive patients a CaMK4 polymorphism that causes reduced expression of the protein identifies a subset of patients with higher BP levels. Altogether, our results point to CaMKIV as a novel potential biological target for therapeutic interventions in hypertension.
Acknowledgments
We thank Prof Jean-Luc Balligand, Catholic University of Louvain, Belgium, for providing the plasmid encoding for eNOS-GFP. Author contributions: Dr Santulli conceived the project, performed experiments, analyzed data, and wrote the paper; Drs Cipolletta and Monaco and A.S. Maione performed in vitro experiments; Dr Sorriento, C. Del Giudice, and A. Anastasio performed experiments; Drs Condorelli, Puca, Trimarco, and Illario analyzed data and contributed to discussion; and Dr Iaccarino supervised the project, analyzed data, and wrote the paper.
Sources of Funding
This work was supported by grants from Italian Society of Arterial Hypertension and Italy Health Ministry (PRIN2009 to Dr Iaccarino, PRIN2008 to Dr Trimarco, and PRIN2009 to Dr Illario).
Disclosures
None.
References
- 1.Kushnir A, Shan J, Betzenhauser MJ, Reiken S, Marks AR. Role of CaMKIIdelta phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proc Natl Acad Sci USA. 2010;107:10274-10279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L, Maier LS. Reactive oxygen species–activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555-565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang T, Brown JH. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc Res. 2004;63:476-486 [DOI] [PubMed] [Google Scholar]
- 4.Colomer JM, Mao L, Rockman HA, Means AR. Pressure overload selectively up-regulates Ca2+/calmodulin-dependent protein kinase II in vivo. Mol Endocrinol. 2003;17:183-192 [DOI] [PubMed] [Google Scholar]
- 5.Wayman GA, Tokumitsu H, Davare MA, Soderling TR. Analysis of CaM-kinase signaling in cells. Cell Calcium. 2011;50:1-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levy D, Larson MG, Benjamin EJ, Newton-Cheh C, Wang TJ, Hwang SJ, Vasan RS, Mitchell GF. Framingham Heart Study 100K Project: genome-wide associations for blood pressure and arterial stiffness. BMC Med Genet. 2007;8suppl 1S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, Wozniak DF, Nardi A, Arvin KL, Holtzman DM, Linden DJ, Zhuo M, Muglia LJ, Chatila TA. Impaired synaptic plasticity and cAMP response element–binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci. 2000;20:6459-6472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyano O, Kameshita I, Fujisawa H. Purification and characterization of a brain-specific multifunctional calmodulin-dependent protein kinase from rat cerebellum. J Biol Chem. 1992;267:1198-1203 [PubMed] [Google Scholar]
- 9.Ribar TJ, Rodriguiz RM, Khiroug L, Wetsel WC, Augustine GJ, Means AR. Cerebellar defects in Ca2+/calmodulin kinase IV–deficient mice. J Neurosci. 2000;20:RC107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei F, Qiu CS, Liauw J, Robinson DA, Ho N, Chatila T, Zhuo M. Calcium calmodulin–dependent protein kinase IV is required for fear memory. Nat Neurosci. 2002;5:573-579 [DOI] [PubMed] [Google Scholar]
- 11.Iaccarino G, Ciccarelli M, Sorriento D, Cipolletta E, Cerullo V, Iovino GL, Paudice A, Elia A, Santulli G, Campanile A, Arcucci O, Pastore L, Salvatore F, Condorelli G, Trimarco B. AKT participates in endothelial dysfunction in hypertension. Circulation. 2004;109:2587-2593 [DOI] [PubMed] [Google Scholar]
- 12.Schneider JC, El Kebir D, Chereau C, Lanone S, Huang XL, De Buys Roessingh AS, Mercier JC, Dall'Ava-Santucci J, Dinh-Xuan AT. Involvement of Ca2+/calmodulin–dependent protein kinase II in endothelial NO production and endothelium-dependent relaxation. Am J Physiol Heart Circ Physiol. 2003;284:H2311-H2319 [DOI] [PubMed] [Google Scholar]
- 13.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239-242 [DOI] [PubMed] [Google Scholar]
- 14.Santulli G. Thrombolysis outcomes in acute ischemic stroke patients with prior stroke and diabetes mellitus. Neurology. 2012;78:840. [DOI] [PubMed] [Google Scholar]
- 15.Position of the American Heart Association on research animal use. Circulation. 1985;71:849A-850A [PubMed] [Google Scholar]
- 16.Mancia G, De Backer G, Dominiczak A, Cifkova R, Fagard R, Germano G, Grassi G, Heagerty AM, Kjeldsen SE, Laurent S, Narkiewicz K, Ruilope L, Rynkiewicz A, Schmieder RE, Struijker Boudier HA, Zanchetti A, Vahanian A, Camm J, De Caterina R, Dean V, Dickstein K, Filippatos G, Funck-Brentano C, Hellemans I, Kristensen SD, McGregor K, Sechtem U, Silber S, Tendera M, Widimsky P, Zamorano JL, Kjeldsen SE, Erdine S, Narkiewicz K, Kiowski W, Agabiti-Rosei E, Ambrosioni E, Cifkova R, Dominiczak A, Fagard R, Heagerty AM, Laurent S, Lindholm LH, Mancia G, Manolis A, Nilsson PM, Redon J, Schmieder RE, Struijker-Boudier HA, Viigimaa M, Filippatos G, Adamopoulos S, Agabiti-Rosei E, Ambrosioni E, Bertomeu V, Clement D, Erdine S, Farsang C, Gaita D, Kiowski W, Lip G, Mallion JM, Manolis AJ, Nilsson PM, O'Brien E, Ponikowski P, Redon J, Ruschitzka F, Tamargo J, van Zwieten P, Viigimaa M, Waeber B, Williams B, Zamorano JLThe Task Force for the Management of Arterial Hypertension of the European Society of Hypertension The Task Force for the Management of Arterial Hypertension of the European Society of Cardiology 2007 Guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J. 2007;28:1462-1536 [DOI] [PubMed] [Google Scholar]
- 17.Santulli G. Coronary heart disease risk factors and mortality. JAMA. 2012;307:1137. [DOI] [PubMed] [Google Scholar]
- 18.Donaldson C, Eder S, Baker C, Aronovitz MJ, Weiss AD, Hall-Porter M, Wang F, Ackerman A, Karas RH, Molkentin JD, Patten RD. Estrogen attenuates left ventricular and cardiomyocyte hypertrophy by an estrogen receptor–dependent pathway that increases calcineurin degradation. Circ Res. 2009;104:265-275211p following 275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sorriento D, Santulli G, Fusco A, Anastasio A, Trimarco B, Iaccarino G. Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-kappaB–dependent hypertrophic gene expression. Hypertension. 2010;56:696-704 [DOI] [PubMed] [Google Scholar]
- 20.Perino A, Ghigo A, Ferrero E, Morello F, Santulli G, Baillie GS, Damilano F, Dunlop AJ, Pawson C, Walser R, Levi R, Altruda F, Silengo L, Langeberg LK, Neubauer G, Heymans S, Lembo G, Wymann MP, Wetzker R, Houslay MD, Iaccarino G, Scott JD, Hirsch E. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma. Mol Cell. 2011;42:84-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iaccarino G, Ciccarelli M, Sorriento D, Galasso G, Campanile A, Santulli G, Cipolletta E, Cerullo V, Cimini V, Altobelli GG, Piscione F, Priante O, Pastore L, Chiariello M, Salvatore F, Koch WJ, Trimarco B. Ischemic neoangiogenesis enhanced by beta2-adrenergic receptor overexpression: a novel role for the endothelial adrenergic system. Circ Res. 2005;97:1182-1189 [DOI] [PubMed] [Google Scholar]
- 22.Ciccarelli M, Santulli G, Campanile A, Galasso G, Cervero P, Altobelli GG, Cimini V, Pastore L, Piscione F, Trimarco B, Iaccarino G. Endothelial alpha1-adrenoceptors regulate neo-angiogenesis. Br J Pharmacol. 2008;153:936-946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santulli G, Ciccarelli M, Palumbo G, Campanile A, Galasso G, Ziaco B, Altobelli GG, Cimini V, Piscione F, D'Andrea LD, Pedone C, Trimarco B, Iaccarino G. In vivo properties of the proangiogenic peptide QK. J Transl Med. 2009;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santulli G, Lombardi A, Sorriento D, Anastasio A, Del Giudice C, Formisano P, Beguinot F, Trimarco B, Miele C, Iaccarino G. Age-related impairment in insulin release: the essential role of beta2-adrenergic receptor. Diabetes. 2012;61:692-701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knoll R, Iaccarino G, Tarone G, Hilfiker-Kleiner D, Bauersachs J, Leite-Moreira AF, Sugden PH, Balligand JL. Towards a re-definition of ‘cardiac hypertrophy’ through a rational characterization of left ventricular phenotypes: a position paper of the Working Group ‘Myocardial Function’ of the ESC. Eur J Heart Fail. 2011;13:811-819 [DOI] [PubMed] [Google Scholar]
- 26.Santulli G, Basilicata MF, De Simone M, Del Giudice C, Anastasio A, Sorriento D, Saviano M, Del Gatto A, Trimarco B, Pedone C, Zaccaro L, Iaccarino G. Evaluation of the anti-angiogenic properties of the new selective alphaVbeta3 integrin antagonist RGDechiHCit. J Transl Med. 2011;9:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lembo G, Iaccarino G, Vecchione C, Barbato E, Morisco C, Monti F, Parrella L, Trimarco B. Insulin enhances endothelial alpha2-adrenergic vasorelaxation by a pertussis toxin mechanism. Hypertension. 1997;30:1128-1134 [DOI] [PubMed] [Google Scholar]
- 28.Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:e68-e75 [DOI] [PubMed] [Google Scholar]
- 29.Fusco A, Santulli G, Sorriento D, Cipolletta E, Garbi C, Dorn GW, II, Trimarco B, Feliciello A, Iaccarino G. Mitochondrial localization unveils a novel role for GRK2 in organelle biogenesis. Cell Signal. 2011;24:468-475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanni F, Santulli G, Izzo R, Rubattu S, Zanda B, Volpe M, Iaccarino G, Trimarco B. The Pl(A1/A2) polymorphism of glycoprotein IIIa and cerebrovascular events in hypertension: increased risk of ischemic stroke in high-risk patients. J Hypertens. 2007;25:551-556 [DOI] [PubMed] [Google Scholar]
- 31.Santulli G, Campanile A, Spinelli L, Assante di Panzillo E, Ciccarelli M, Trimarco B, Iaccarino G. G protein–coupled receptor kinase 2 in patients with acute myocardial infarction. Am J Cardiol. 2011;107:1125-1130 [DOI] [PubMed] [Google Scholar]
- 32.Iaccarino G, Keys JR, Rapacciuolo A, Shotwell KF, Lefkowitz RJ, Rockman HA, Koch WJ. Regulation of myocardial betaARK1 expression in catecholamine-induced cardiac hypertrophy in transgenic mice overexpressing alpha1B-adrenergic receptors. J Am Coll Cardiol. 2001;38:534-540 [DOI] [PubMed] [Google Scholar]
- 33.Nattel S. Effects of heart disease on cardiac ion current density versus current amplitude: important conceptual subtleties in the language of arrhythmogenic ion channel remodeling. Circ Res. 2008;102:1298-1300 [DOI] [PubMed] [Google Scholar]
- 34.Jin H, Chemaly ER, Lee A, Kho C, Hadri L, Hajjar RJ, Akar FG. Mechanoelectrical remodeling and arrhythmias during progression of hypertrophy. FASEB J. 2010;24:451-463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volk T, Nguyen TH, Schultz JH, Faulhaber J, Ehmke H. Regional alterations of repolarizing K+ currents among the left ventricular free wall of rats with ascending aortic stenosis. J Physiol. 2001;530:443-455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sorriento D, Santulli G, Del Giudice C, Anastasio A, Trimarco B, Iaccarino G. Endothelial cells are able to synthesize and release catecholamines both in vitro and in vivo. Hypertension. 2012;60:129-136 [DOI] [PubMed] [Google Scholar]
- 37.Iaccarino G, Lefkowitz RJ, Koch WJ. Myocardial G protein–coupled receptor kinases: implications for heart failure therapy. Proc Assoc Am Physicians. 1999;111:399-405 [DOI] [PubMed] [Google Scholar]
- 38.Malovini A, Illario M, Iaccarino G, Villa F, Ferrario A, Roncarati R, Anselmi CV, Novelli V, Cipolletta E, Leggiero E, Orro A, Rusciano MR, Milanesi L, Maione AS, Condorelli G, Bellazzi R, Puca AA. Association study on long-living individuals from southern Italy identifies rs10491334 in the CAMKIV gene that regulates survival proteins. Rejuvenation Res. 2011;14:283-291 [DOI] [PubMed] [Google Scholar]
- 39.Ott C, Schneider MP, Delles C, Schlaich MP, Schmieder RE. Reduction in basal nitric oxide activity causes albuminuria. Diabetes. 2011;60:572-576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gadano AC, Sogni P, Yang S, Cailmail S, Moreau R, Nepveux P, Couturier D, Lebrec D. Endothelial calcium-calmodulin dependent nitric oxide synthase in the in vitro vascular hyporeactivity of portal hypertensive rats. J Hepatol. 1997;26:678-686 [DOI] [PubMed] [Google Scholar]
- 41.Liu G, Han J, Profirovic J, Strekalova E, Voyno-Yasenetskaya TA. Galpha13 regulates MEF2-dependent gene transcription in endothelial cells: role in angiogenesis. Angiogenesis. 2009;12:1-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.You J, Peng W, Lin X, Huang QL, Lin JY. PLC/CAMK IV-NF-kappaB involved in the receptor for advanced glycation end products mediated signaling pathway in human endothelial cells. Mol Cell Endocrinol. 2010;320:111-117 [DOI] [PubMed] [Google Scholar]
- 43.Matthews RP, Guthrie CR, Wailes LM, Zhao X, Means AR, McKnight GS. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol. 1994;14:6107-6116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bok J, Wang Q, Huang J, Green SH. CaMKII and CaMKIV mediate distinct prosurvival signaling pathways in response to depolarization in neurons. Mol Cell Neurosci. 2007;36:13-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kilander L, Nyman H, Boberg M, Hansson L, Lithell H. Hypertension is related to cognitive impairment: a 20-year follow-up of 999 men. Hypertension. 1998;31:780-786 [DOI] [PubMed] [Google Scholar]
- 46.Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, Overbeek P, Richardson JA, Grant SR, Olson EN. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest. 2000;105:1395-1406 [DOI] [PMC free article] [PubMed] [Google Scholar]