

Abstract
Background
Barth syndrome is a rare, multisystem disorder caused by mutations in tafazzin that lead to cardiolipin deficiency and mitochondrial abnormalities. Patients most commonly develop an early-onset cardiomyopathy in infancy or fetal life.
Methods and Results
Knockdown of tafazzin (TAZKD) in a mouse model was induced from the start of gestation via a doxycycline-inducible shRNA transgenic approach. All liveborn TAZKD mice died within the neonatal period, and in vivo echocardiography revealed prenatal loss of TAZKD embryos at E12.5-14.5. TAZKD E13.5 embryos and newborn mice demonstrated significant tafazzin knockdown, and mass spectrometry analysis of hearts revealed abnormal cardiolipin profiles typical of Barth syndrome. Electron microscopy of TAZKD hearts demonstrated ultrastructural abnormalities in mitochondria at both E13.5 and newborn stages. Newborn TAZKD mice exhibited a significant reduction in total mitochondrial area, smaller size of individual mitochondria, reduced cristae density, and disruption of the normal parallel orientation between mitochondria and sarcomeres. Echocardiography of E13.5 and newborn TAZKD mice showed good systolic function, but early diastolic dysfunction was evident from an abnormal flow pattern in the dorsal aorta. Strikingly, histology of E13.5 and newborn TAZKD hearts showed myocardial thinning, hypertrabeculation and noncompaction, and defective ventricular septation. Altered cellular proliferation occurring within a narrow developmental window accompanied the myocardial hypertrabeculation-noncompaction.
Conclusions
In this murine model, tafazzin deficiency leads to a unique developmental cardiomyopathy characterized by ventricular myocardial hypertrabeculation-noncompaction and early lethality. A central role of cardiolipin and mitochondrial functioning is strongly implicated in cardiomyocyte differentiation and myocardial patterning required for heart development. (J Am Heart Assoc. 2012;1:jah3-e000455 doi: 10.1161/JAHA.111.000455.)
Keywords: Barth syndrome, cardiolipin, mitochondrial disease, noncompaction cardiomyopathy, tafazzin
Introduction
Barth syndrome (MIM 302060) is a rare X-linked, multisystem disorder characterized by cardiomyopathy, skeletal myopathy, neutropenia, and growth retardation.1–3 The most common presentation is early cardiomyopathy, often occurring in infancy. Indeed, prenatal loss and/or morbidity can occur from fetal cardiomyopathy.4 Patients may exhibit a dilated or hypertrophic cardiomyopathy, endomyocardial fibroelastosis, and/or a developmental cardiomyopathy known as myocardial (left ventricular) noncompaction or hypertrabeculation (“spongy myocardium”). This last cardiomyopathy is considered a hallmark of Barth syndrome, and is also notable in other mitochondrial disorders.5
Barth syndrome is caused by mutations in tafazzin, which lead to severe deficiency and altered molecular species of the phospholipid cardiolipin.6,7 Tafazzin catalyzes cardiolipin remodeling reactions at the final stage of cardiolipin biosynthesis, producing so-called “mature” cardiolipin.8 In tafazzin-deficient cells, cardiolipin levels are lower, while monolysocardiolipin levels are markedly elevated, and the acyl chain composition is shifted toward less unsaturated species.9,10 For reasons not completely understood, normal cardiolipin profiles seem to be critical to the structural integrity and the bioenergetic function of mitochondria.9,10
Recently, a short-hairpin RNA (shRNA) -inducible tafazzin-knockdown (TAZKD) mouse model became available that exhibited an adult-onset cardiomyopathy associated with abnormal cardiolipin profiles and mitochondrial structural abnormalities.11,12 Using a modified induction scheme in the same TAZKD mouse, we found near-universal pre- and perinatal lethality. This early death was caused by a developmental cardiomyopathy, a phenotype that strikingly resembles the noncompaction cardiomyopathy of Barth syndrome. Our results support an important role for cardiolipin in myocardial patterning during heart development.
Methods
Animals and Doxycycline Transgene Induction
All protocols were approved by the Institutional Animal Care and Use Committee of NYU School of Medicine and Langone Medical Center, and conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH). Mice were housed under temperature-controlled conditions under a 12-hour light/dark cycle with free access to drinking water and food. Transgenic mice were generated at TaconicArtemis, GmbH (Köln, Germany) under contract from the Barth Syndrome Foundation, and made freely available to investigators; these mice are now available through The Jackson Laboratory (Bar Harbor, ME). These TAZKD mice have been previously described in detail,11,12 on the basis of a tight inducible shRNA expression system13 (Figure 1). TAZKD transgenic mice used in this study are the heterozygote off-spring of heterozygote (male)-wildtype (female) C57Bl/6 crosses, as per TaconicArtemis’ protocol. The day after overnight mating that a plug was found was designated as E0.5. In brief, fresh solutions containing 2 mg/mL doxycycline (Frontier Scientific D-10056) and 10% sucrose were prepared every other day and kept dark. (The use of 10% sucrose was necessary to improve palatability of the doxycycline solution; so far as we know, such a diet has not been shown to result in abnormal myocardial patterning.) Doxycycline was administered to wildtype female mice on a continuous basis, to make sure doxycycline remained in the maternal system and the embryonic transgene would therefore be induced throughout gestation. However, because the males carry the transgene as heterozygotes, we fed the mating pairs regular water without doxycycline to reduce the possibility of male sterility, which is a known adverse effect in a Drosophila model of tafazzin deficiency.14 Doxycycline was reintroduced into the drinking water of the females on the day following overnight mating (day of plug).
Figure 1.
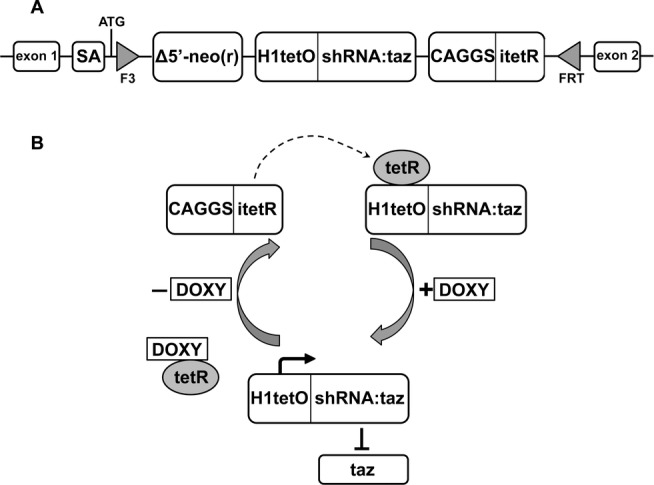
Schematic of taz-specific inducible shRNA-mediated targeting strategy. Doxycycline dose was 2 mg/mL in the drinking water (13). (A) Inserted by recombinase-mediated cassette exchange, the F3-FRT cassette contains the taz-specific shRNA H1-tet-On promoter, a tet repressor (tetR), and a neomycin selection gene; (B) the tetO-shRNA promoter is inactivated by tetR until doxycycline is added to the system, inactivating tetR, and allowing transcription of taz-specific shRNA; the system is reversible. Of several correctly targeted embryonic stem cell clones generated at TaconicArtemis, GmbH, one construct resulted in acceptable taz knockdown, and breeder mice were sent to investigators on request.
We used the same induction protocol as in the original protocol,13 but doxycycline dosing differed from recent investigations (625 mg/kg chow 11; 200 mg/kg chow 12). We estimate our TAZKD mice ingested 3 to 10 times the doxycycline used in studies by Acehan et al11 and Soustek et al,12 because mice ingest on average 15 g/100 g body weight in food and 15 mL/100 g body weight water daily (http://www.lvma.org/mouse.html). To determine the kinetics of tafazzin knockdown and effects on development, doxycycline induction was initiated on different gestational ages as described later.
Genotyping and Quantitative Reverse Transcription Polymerase Chain Reaction
Mice were genotyped by standard polymerase chain reaction using proteinase K digested tail or yolk sacs of mouse embryos. Real-time quantitative reverse transcription polymerase chain reaction was done in standard fashion.15 Briefly, this set of primers identified the TAZ-shRNA (where TAZ is tafazzin) transgene, and amplifies a 381 Bp fragment: 5′-CCATGGAATTCGAACGCTGACGTC-3′/5′-TATGGGCTATGAACTAATGACCC-3′. For quantitative reverse transcription polymerase chain reaction, the sequences of the taz primers used were as follows: (forward) 5′-GCAGACATCTGCTTCACCAA-3′/(reverse) 5′-TGAAGTCCATCCCTTTCTGG-3′. Briefly, total RNA was extracted using the acid-phenol guanidinium method (TriReagent, Sigma). Embryonic and newborn tissues were dissected quickly in iced phosphate-buffered saline and snap-frozen. RNA purification was performed using Ambion's TRIzol Plus RNA Purification Kit according to the manufacturer's instructions (Invitrogen). Total RNA was reverse-transcribed in standard fashion using Roche's Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Science). Polymerase chain reaction was performed in 384-well format, and reactions were performed using an ABI Prism 7900HT Sequence Detection System (Perkin-Elmer Applied Biosystems) available through the NYU Cancer Institute Genomics Facility. To quantitate gene expression, we used the widely used ΔΔCt method, using SDS software (version 2.3, Applied Biosystems); the Ct data of a target transcript were expressed as relative changes between the two experimental conditions, measured within the exponential phase of the polymerase chain reaction. A correction was performed using a passive reference dye (Rox), which is present in the polymerase chain reaction master mix. Reactions with a Ct value >36 or with any evidence of nonspecificity (low melting temperatures or multiple peaks in melting point analysis) were excluded from the analysis. We calculated target gene expression relative to that of a subset of appropriate reference genes (β-actin, GAPDH, histone-3, ribosomal protein S6, and hydroxymethylbilane synthase), using the Bestkeeper algorithm.15
Cardiolipin Analysis
Mouse tissues were homogenized and the homogenates were transferred immediately to glass tubes containing 2 mL methanol and 1 mL chloroform. These tubes were incubated at 37°C for 30 minutes. Lipids were extracted according to Bligh and Dyer,16 extracts were dried under a stream of nitrogen and finally resuspended in 100 μL chloroform/methanol (1:1). For lipid mass spectrometry, we followed the method of Sun et al17 with minor modifications. An aliquot of the extract was diluted 1:10 in 2-propanol/acetonitrile (3:2). A total of 10 μL of the diluted solution were mixed with 10 μL of matrix solution, containing 9-aminoacridine (10 g/L) in 2-propanol/acetonitrile (3:2). One microliter of this mixture was applied to a sample well and was allowed to dry. Mass spectrometry was performed with the MALDI micro-MX instrument from Waters operated in negative ion mode and in reflectron mode. The instrument was calibrated daily with a reference mixture of myristoyl-lyso-phosphatidylglycerol (m/z=455.2415), dioleoyl-phosphatidylglycerol (m/z=773.5338), dilinoleoyl-phosphatidylglycerol (m/z=738.5079), and tetralinoleoyl-cardiolipin (m/z=1447.9650). Voltages were set to the following parameters: pulse voltage 2000 V, detector voltage 2200 V, flight tube voltage 12 kV, reflectron voltage 5.2 kV, and negative anode voltage 3.5 kV. Time-lag focusing delay was set to 700 ns. The nitrogen laser (337 nm) was fired at a rate of 5 Hz and 10 laser shots were acquired per subspectrum. We typically acquired 100 to 200 subspectra per sample in a mass range from 400 to 2000 Da. Data were analyzed with the software MassLynx 4.1.
Prenatal and Postnatal Mouse Echocardiography
For noninvasive imaging, adult mice (including pregnant mice) were anesthetized with isoflurane via nose cone (maintenance: 1% isoflurane mixed with 100% medical oxygen at flow rate 2.5 L/min), with strict thermoregulation (37±1°C) to optimize physiological conditions and reduce hemodynamic variability.18,19 Neonatal and very young juvenile mice (<4 weeks of age), which were too small or fragile for nosecone anesthetic, were held very gently with one hand (permitting self-thermoregulation); the mice remained very calm and still, with no signs to suggest that they experienced any significant discomfort, distress, or pain. In vivo transthoracic imaging for postnatal mice and transabdominal imaging for timed-pregnant mice was used, permitting accurate localization of embryos and as detailed by our laboratory previously.18–20 Two-dimensional, spectral Doppler, and color Doppler flow echocardiography was performed using the Vevo 2100 (Visual Sonics, Toronto, ON, Canada) with a 40 MHz center frequency transducer. To minimize bias, we performed all echocardiographic measurements prior to knowledge of genotype.
Electron Microscopy
Electron micrographic analyses have been previously described,21 and detailed mitochondrial morphometrics were performed. Briefly, heart tissue was preserved by conventional chemical fixation, followed by gradual ethanol dehydration, infiltration, and curing with epoxy resin, and staining with uranylacetate and Sato Lead Stain. The sample blocks were sectioned to 50 to 70 μm thickness. The samples were imaged using a Philips CM12 microscope operated at 120 kV. The following measurements were performed using Image J (NIH): (1) mitochondrial area density (total cross sectional area of mitochondria divided by total cytosol area); (2) mitochondrial size (average cross-sectional area of mitochondria); (3) mitochondrial number (number of mitochondria per cell area, excluding nucleus); (4) cristae density (number of cristae per mitochondrial area); (5) mitochondrial orientation to myofibrillar structures (A, angle between long axes of mitochondria and associated myofibril bundle, measured as [90 minus {the angle between the Z-disk and the long axis of the adjacent associated mitochondrion}]; B, distance between the near edge of the myofibril bundle and the center of the adjacent associated mitochondrion). From multiple animals in each experimental group, multiple measurements from each of multiple random sections from cardiomyocytes were analyzed for each animal.
Histology and Immunohistochemistry
We used standard histological and immunohistochemical methods to determine cellular morphometry, degree of myocardial fibrosis, myocardial cellular proliferation rates, and apoptosis. Standard 6 μm paraffin histological sections were used to determine primarily the presence of myocardial hypertrabeculation. Sections were stained with hematoxylin and eosin for standard morphometric analysis, and Masson's trichrome stain to gauge fibrosis in cardiomyopathic hearts. To determine the degree of myocardial noncompaction/hypertrabeculation, we adapted the protocol of Ishiwata et al22 to determine the degree of hypertrabeculation in the left ventricular lateral wall, left ventricular apex, and right ventricular lateral wall. Random samplings of left ventricular free wall and adjacent intracavitary trabeculations were measured using Image J, and the ratios of trabecular area to left ventricular free wall compact zone area were calculated in the left ventricular lateral wall, left ventricular apex, and right ventricular lateral wall of newborn pups. We quantified total cell proliferation in the heart by Ki-67 immunochemistry. Although many myocardial cells, including cardiomyocytes, are not yet fully differentiated at the stages we are studying,23 cardiomyocytes comprise the great majority of cells in the developing heart.24 Caspase-3 assays were utilized to determine apoptosis in the ventricular myocardium. Positively labeled cells were counted against blue-staining nuclei from several high-power fields, from both the trabecular and the compact myocardium separately.
Statistical Analysis
Data are expressed as mean±SEM. Differences among groups were analyzed using the Mann-Whitney U test for continuous variables and Fisher's exact test for categorical variables. Statistical significance was set at P<0.05.
Results
Tafazzin Knockdown Leads to Prenatal and Perinatal Lethality
We observed a significantly lower number of TAZKD pups born, which had not been reported previously.11,12 Therefore, we focused on in vivo echocardiography at various stages of embryonic development (E12.5, E13.5, E14.5), which showed varying numbers of TAZKD mice dead (Figure 2, Table 1). Furthermore, when we attempted to permit several litters to survive (without tissue harvest) in order to grow several TAZKD mice into adulthood, there were no TAZKD survivors beyond the first week of life. While some TAZKD pups indeed were found dead shortly after birth, others may have been cannibalized by their mothers because of their condition.
Figure 2.
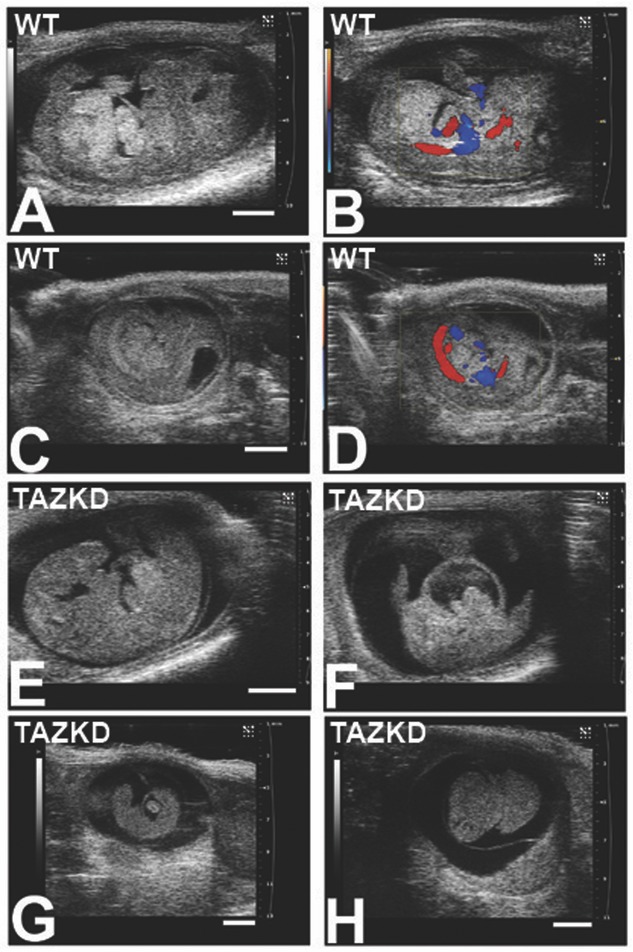
Doxycycline-inducible shRNA-mediated tafazzin knockdown leads to prenatal lethality. In vivo echocardiography shows a spectrum of prenatal loss, and provides rapid morphological phenotyping of live and dead mouse embryos. Sagittal views demonstrate control wildtype (WT) E14.5 mouse embryo, B-mode/2-dimensional imaging (A) and color Doppler blood flow imaging (B); and WT E12.5 mouse embryos, sagittal views (C, D), to show differences in size with E14.5. A dead E14.5 TAZKD mouse embryo (E) demonstrates relatively well-preserved morphology indicating recent death; B-mode and color Doppler imaging showed lack of a heartbeat and blood flow. A transverse view of the chest in the same E14.5 embryo (F) better demonstrates the large pericardial effusion. In early stages of resorption (G), a dead E13.5 TAZKD embryo demonstrates a large pericardial effusion; the small size of the embryo indicates death at least 1 day before imaging. A littermate of (G), a dead E13.5 TAZKD embryo (H) shows further necrosis as indicated by homogeneous gray tissue, closer to complete resorption. Live TAZKD embryos at any stage were indistinguishable from WT live embryos by 2D echo imaging. Scale bars: 2 mm. TAZKD indicates tafazzin knockdown embryos.
Table 1.
Spectrum of Lethality Starting by E12.5
| Stage | Total | WT Alive | WT Dead | TAZKD Alive | TAZKD Dead | P-value (Fisher's) |
|---|---|---|---|---|---|---|
| E12.5 | 14 | 7 | 1 | 3 | 3 | 0.245 |
| E13.5 | 67 | 31 | 2 | 29 | 5 | 0.428 |
| E14.5 | 28 | 18 | 1 | 3 | 6 | 0.001 |
| Newborn | 60 | 35 | 0 | 13 | 12 | <0.0001 |
TAZKD indicates tafazzin knockdown (induced); WT, wildtype.
Mice were harvested at the stages shown in the Table; newborn mice were harvested within hours of birth. Fisher's exact test (two-tailed) was performed on 2×2 contingency tables of WT versus TAZKD mice, alive versus dead, and shows loss of expected Mendelian ratios of living TAZKD mice by E14.5. Additional data from longitudinal echocardiographic imaging of three litters further attested to prenatal and perinatal lethality: a total of 24 embryos (18 alive, six dead [nonresorbed]) were demonstrable at E12.5, but only 10 were born; eight lived (all WT) while two died early on the day of birth (both TAZKD).
Tafazzin Knockdown Leads to Alterations in Cardiolipin and Mitochondrial Ultrastructure
Effective tafazzin knockdown (>70% to 80%) was confirmed in E13.5 embryos and in newborns (Figure 3). Similar to other reports, TAZKD mice exhibited abnormalities in cardiolipin, but ours is the first report of abnormalities during embryonic development. In both TAZKD embryonic and newborn mice, the composition of cardiolipin molecular species was altered with a shift to a higher monolysocardiolipin:cardiolipin ratio (Figure 2); this increased MLCL:CL ratio is diagnostic for Barth syndrome.9
Figure 3.
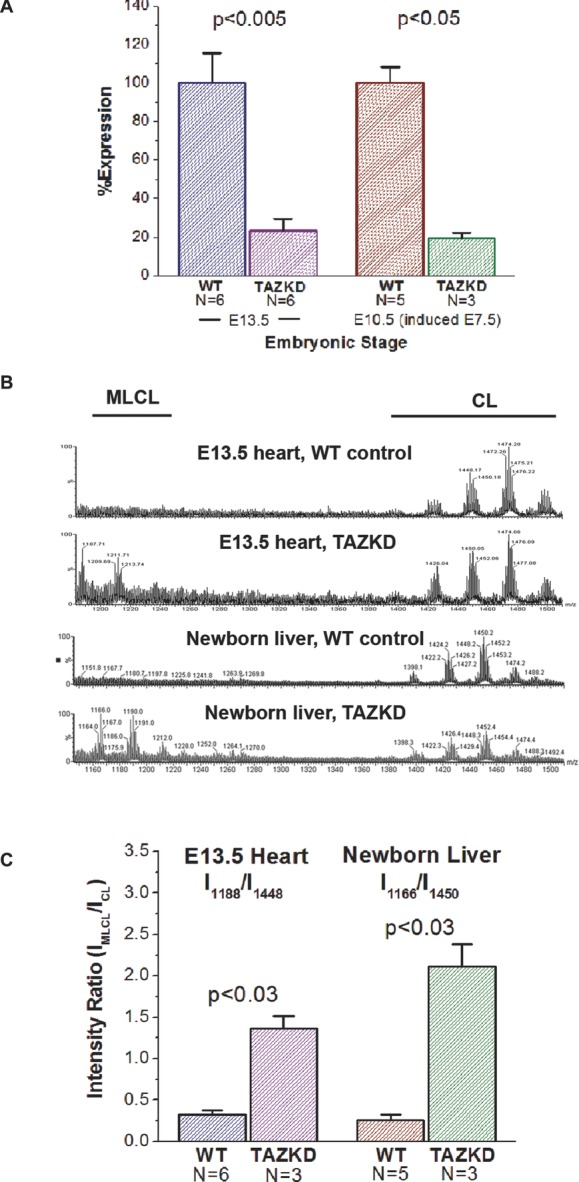
Effective tafazzin knockdown leads to altered cardiolipin profiles during development in TAZKD mice. Quantitative RT-PCR analysis of TAZKD mouse embryos shows tafazzin can be knocked down effectively during development (A). Induction from the start of gestation leads to approximately ∼80% reduction in taz mRNA by E13.5 ([A], left-hand bars). In addition, 3 days' induction (starting at E7.5) with doxycycline leads to the same degree of taz knockdown by E10.5 ([A], right-hand bars]. MALDI mass spectrometry [B] shows altered cardiolipin spectra in TAZKD E13.5 embryonic mouse hearts and newborn mouse livers; TAZKD tissues show a characteristic increase in the ratio of monolysocardiolipin species (MLCL) to cardiolipin (CL), also demonstrated by quantitative analysis of cardiolipin profiles (C). In E13.5 heart, ratios were obtained from molecular species at m/z 1188 (MLCL):1448 (CL), and in newborn liver, at 1166 (MLCL):1450 (CL). TAZKD indicates tafazzin knockdown mice; RT-PCR, reverse transcription polymerase chain reaction.
Quantitative analysis of electron micrographs showed a spectrum of abnormalities in mitochondrial ultrastructure and sarcomeric organization, including vacuolated cristae, a reduction in total mitochondrial area density, a reduction in mitochondrial size, and lower cristae density. Also, newborn TAZKD mice demonstrated a disruption of the normal parallel orientation between mitochondria and sarcomeres (Figures 4 and 5). Overall, these abnormalities indicate abnormal mitochondria and are consistent with delayed cardiomyocyte differentiation.21
Figure 4.
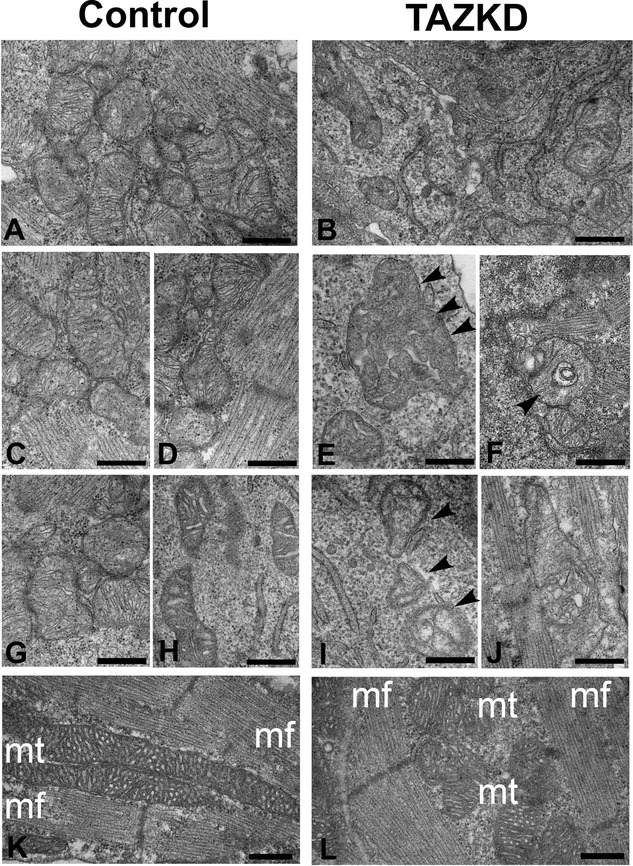
Electron micrographic analysis shows a spectrum of mitochondrial abnormalities similar to human Barth syndrome. We compared E13.5 embryonic wildtype (A, C, D, G, H) and TAZKD (B, E, F, I, J) cardiomyocytes, focusing on the mitochondria and regions around the mitochondria. Examples of abnormal mitochondrial ultrastructure include reduced mitochondrial density (A, B); abnormal cristae morphology (B, E, F, I, J), including disruption of cristae (B, E) and decreased cristae matrix density (I); and abnormally large mitochondria (E, J) (see arrowheads for selected abnormal mitochondria). Disruption of the normal alignment of mitochondria (mt) with myofibrils (mf) is shown in representative newborn control (wildtype) versus TAZKD cardiomyocytes (K, L). Scale bars: 500 nm. TAZKD indicates tafazzin knockdown.
Figure 5.
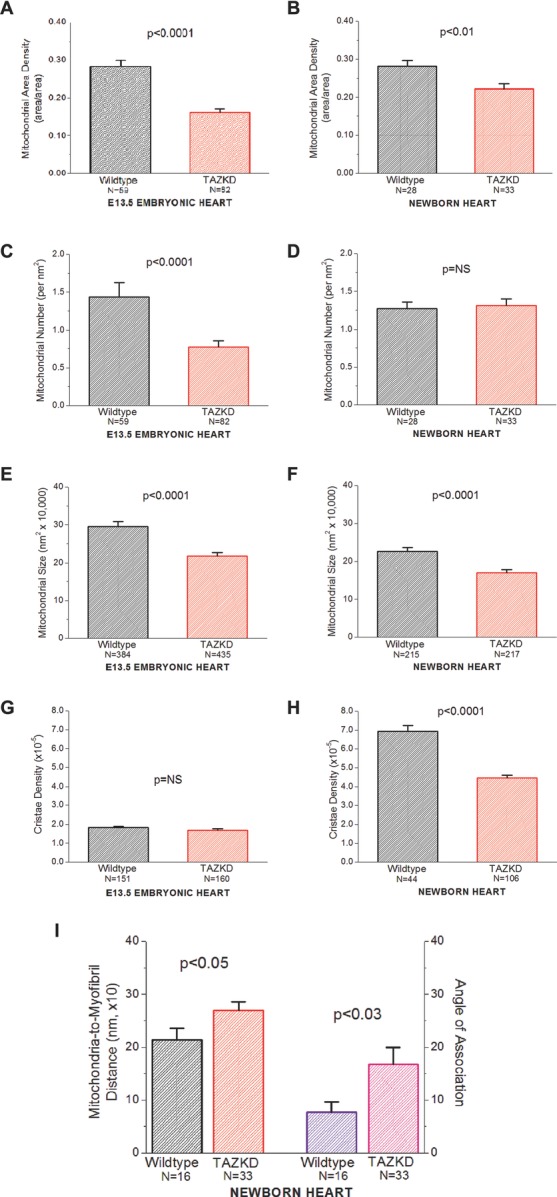
Morphometric analysis of mitochondria reveals distinct abnormalities in TAZKD mice. Mitochondrial structures were analyzed from electron micrographs taken of E13.5 embryonic hearts (A, C, E, G) and newborn hearts (B, D, F, H, I). TAZKD cardiomyocytes demonstrate statistically significant decreases in mitochondrial area density (embryonic, newborn), mitochondrial number per unit area (embryos), mitochondrial size (embryonic, newborn), and cristae density (newborns). Newborn myocardium exhibits disruptions in the spatial relations between mitochondria and myofibrils, as indicated by increased distances and a less-parallel geometry between mitochondria and myofibrils (I) (NB: Embryonic myocardium does not exhibit myofibrillar units well-developed enough for analysis). In A, B, C, F, and I, the N shown are the number of fields used for data analysis; in E, F, G, and H, N is the number of mitochondria analyzed. TAZKD indicates tafazzin knockdown.
TAZKD Mouse Hearts Exhibit Distinct Functional Abnormalities at the Embryonic Stage
We studied whether cardiomyopathy could explain the prenatal and perinatal deaths. As reflected by normal cardiac dimensions and fractional area change, systolic function was preserved in both TAZKD mouse embryos and newborn pups (Tables 2 and 3). However, peak dorsal aortic velocity was significantly lower in the TAZKD than control wildtype embryos, which might suggest subtle systolic dysfunction, and this was even lower in more severely affected TAZKD embryos with an “a-kick” (see later). Heart rates, atrioventricular conduction (PR interval), and umbilical arterial blood flows (a surrogate of placental health) were not significantly different between TAZKD and controls, although heart rates in TAZKD embryos trended lower in more severely affected embryos and as gestation progressed (Table 2).
Table 2.
In Vivo UBM-Derived Embryonic Hemodynamic Measurements
| Embryonic Stage | ||||
|---|---|---|---|---|
| Functional Parameter | E12.5 | E13.5 | E13.5 (TAZKD+a-kick) | E14.5 |
| N (Total Numbers With Usable Data; Not All Measurements Could Be Obtained in All Embryos) | ||||
| Wildtype | 8 | 16 | 16 | 6 |
| TAZKD | 5 | 16 | 3 | 3 |
| Heart Rate, beats/min | ||||
| Wildtype | 132±13 | 136±7 | 136±7 | 126±6 |
| TAZKD | 140±24 | 143±13 | 101±14 | 109±8 |
| End-Diastolic Area, mm2 (From 4-Chamber View) | ||||
| Wildtype | 1.765±0.101 | 1.969±0.057 | 1.969±0.057 | 2.503±0.107 |
| TAZKD | 1.695±0.054 | 1.832±0.072 | 1.720±0.315 | 2.501±0.114 |
| Fractional Area Change, % | ||||
| Wildtype | 36.2±1.8 | 42.1±1.7 | 42.1±1.7 | 44.9±2.9 |
| TAZKD | 45.2±1.3* | 45.5±1.3 | 46.0±3.3 | 52.5±1.2 |
| PR Interval, ms | ||||
| Wildtype | 98±12 | 119±6 | 119±6 | 120±6 |
| TAZKD | 97±24 | 121±7 | 151±12 | 96 |
| Isovolumic Contraction Time, ms | ||||
| Wildtype | 27±8 | 40±4 | 40±4 | 33±4 |
| TAZKD | 43±6 | 51±5 | 59±11 | 21 |
| Isovolumic Relaxation Time, ms | ||||
| Wildtype | 61±18 | 53±8 | 53±8 | 44±1 |
| TAZKD | 45±5 | 42±9 | 23±19 | 20 |
| DA Peak Velocity, mm/s | ||||
| Wildtype | 100±13 | 103±8 | 103±8 | 126±11 |
| TAZKD | 85±22 | 78±8* | 59±12* | 115±29 |
| Atrial “kick” Wave, N (%) | ||||
| Wildtype | 0 | 0 | 0 | 0 |
| TAZKD | 1 (20%) | 3 (19%) | (3) | 3 (100%) |
| DA ET, ms | ||||
| Wildtype | 174±12 | 172±6 | 172±6 | 186±6 |
| TAZKD | 156±14 | 158±9 | 187±6 | 196±9 |
| UA Peak Velocity, mm/s | ||||
| Wildtype | 75±9 | 75±5 | 75±5 | 103±9 |
| TAZKD | 67±12 | 67±6 | 60±12* | 91±5 |
| UV Peak Velocity, mm/s | ||||
| Wildtype | 45±7 | 48±5 | 48±5 | 68±7 |
| TAZKD | 39±4 | 37±4 | 27±5* | 43±2* |
| UV Trough Velocity, mm/s | ||||
| Wildtype | 15±3 | 17±2 | 17±2 | 21±4 |
| TAZKD | 6±3 | 11±2* | 4±2† | 5±3* |
| UV Peak/Trough Ratio | ||||
| Wildtype | 0.34±0.03 | 0.34±0.02 | 0.34±0.02 | 0.31±0.03 |
| TAZKD | 0.16±0.07* | 0.29±0.04 | 0.20±0.12 | 0.13±0.08 |
Data are expressed as mean±SEM.
DA indicates dorsal aorta; ET, ejection time; TAZKD, tafazzin knockdown; UA, umbilical artery; UV, umbilical vein.
“E13.5 (TAZKD + a-kick)” represents the statistical comparisons between E13.5 wildtype and only those TAZKD embryos exhibiting an abnormal dorsal aortic “a-kick” (N=3). Notably, of the E14.5 litters imaged and assayed, all TAZKD embryos either were dead or exhibited an abnormal “atrial kick’ in the dorsal aorta. Values without SEM represent available data from one embryo only. TAZKD compared with WT.
P<0.05;
P<0.01.
Table 3.
In Vivo UBM-Derived Newborn Hemodynamic Measurements
| Newborn | ||
|---|---|---|
| Functional Parameter | Wildtype | TAZKD |
| N | 16 | 9 |
| Weight, g | 1.15±0.05 | 1.01±0.04* |
| Heart rate, beats/min | 260±13 | 244±3 |
| PR interval, ms | 76±3 | 82±3 |
| Isovolumic contraction time, ms | 24±5 | 31±11 |
| Isovolumic relaxation time, ms | 47±1 | 50±6 |
| Diastolic septal thickness, mm | 0.27±0.01 | 0.27±0.01 |
| Diastolic LV dimension, mm | 1.34±0.03 | 1.31±0.04 |
| Diastolic LV posterior wall, mm | 0.26±0.01 | 0.26±0.01 |
| Systolic septal thickness, mm | 0.39±0.01 | 0.39±0.01 |
| Systolic LV dimension, mm | 0.92±0.03 | 0.93±0.03 |
| Systolic LV posterior wall, mm | 0.40±0.03 | 0.35±0.01* |
| LV shortening fraction,% | 31±2 | 29±1 |
| LV end-diastolic area, mm2 | 1.413±0.070 | 1.375±0.058 |
| LV fractional area change,% | 50.8±1.4 | 49.4±1.1 |
Data are expressed as means±SEM.
LV indicated left ventricular; TAZKD, tafazzin knockdown.
NB: Because of technical challenges, only a limited subset of data could be obtained on newborn pups. It should be noted that newborn pups are not anesthetized during imaging. There were no significant differences between wildtype and TAZKD newborn echo functional parameters; although systolic LV posterior wall achieved statistical significance (*P<0.05), the large number of comparisons in this table and the absence of other differences in cardiac morphometry and function cast doubt as to the relevance of this finding.
Weight was significantly lower in the TAZKD newborn pups (P<0.05).
Notably, flow in the dorsal aorta of several TAZKD embryos showed a distinct peak immediately preceding the normal antegrade flow envelope; this was shown to originate from atrial contraction by Doppler interrogation of the outflow tract (Figure 6). The presence of an atrial systolic kick in the aorta indirectly suggests ventricular diastolic dysfunction, with a left ventricle so stiff that the atrial contraction, exceeding aortic pressure, is transmitted into the aorta. Although we did not validate the Doppler findings by other means, this contention is supported by the umbilical venous flow patterns, which showed lower peak and trough velocities, suggesting increased “downstream” impedance and therefore diastolic dysfunction in the heart. Importantly, diastolic dysfunction preceded overt systolic dysfunction, but there was no evidence of progressive ventricular dilatation. An A-wave in a downstream artery is a well-known echocardiographic indicator of abnormally elevated ventricular stiffness,25 and patients with myocardial noncompaction almost always exhibit diastolic dysfunction.26 That atrial pressures can exceed aortic pressures is plausible in the mouse embryo, because intracardiac and intraarterial pressures are very low.22 Diastolic dysfunction with preserved systolic function has been demonstrated in the friend of GATA-2 (FOG-2) null mouse that also develops noncompaction.22 Although another measure of diastolic function, isovolumic relaxation time, appeared normal in TAZKD mice, it should be noted that the cardiac valves are not yet completely mature in mouse embryos and are unlikely to behave like the mature, thin, and delicate valves of an adult heart that lead to “normal” isovolumic time intervals.27
Figure 6.
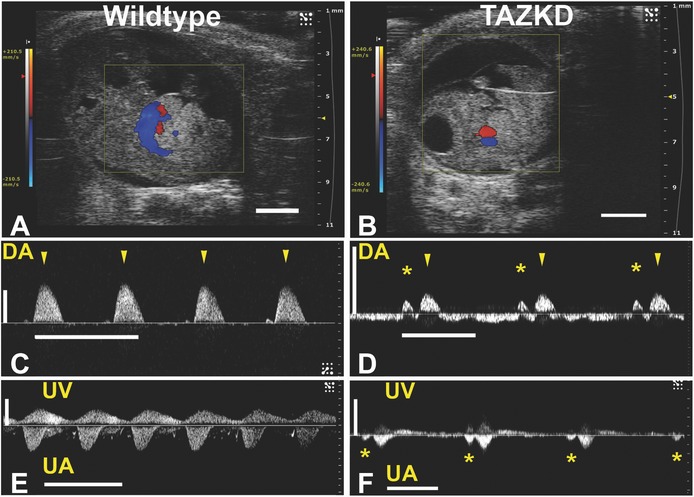
Cardiac dysfunction is evident in some TAZKD embryonic mice. Some TAZKD embryos exhibit severe diastolic dysfunction with a unique “atrial kick” in the dorsal aorta, at E12.5, 13.5, and 14.5, as revealed by spectral Doppler analysis of dorsal aortic blood flow; images are of an E13.5 wildtype embryo (A, C, E) and TAZKD embryo (B, D, F). Sagittal views of control wildtype (A) and TAZKD (B) embryos with superimposed color Doppler map show blood flow, but no obvious abnormalities such as hydrops (scale bars = 2 mm). Normal all-antegrade dorsal aortic (DA) blood flow is seen in the wildtype embryo (C), with peak aortic pulsations indicated by the arrowheads. Blood flow in the TAZKD dorsal aorta (D) shows an “atrial kick” (asterisks) occurring just before the peak aortic pulsations (arrowheads). In umbilical artery (UA) and umbilical vein (UV), the TAZKD embryo (F) shows propagation of the atrial kick even into the umbilical artery (asterisks), not evident in the wildtype embryo (E). Also note peak aortic velocities in TAZKD embryos (D, F) are lower than in WT littermates (C, E). Time (horizontal) scale bars for panels C to F = 0.5 s; velocity (vertical) scale bar = 100 mm/s. TAZKD indicates tafazzin knockdown.
While diastolic dysfunction may lead to pre-/perinatal lethality in this model, we cannot exclude a malignant arrhythmia leading to sudden cardiac death, a well-established feature of Barth syndrome.3 Indeed, we speculate the normal newborn TAZKD echo data (Table 3) may represent a survival bias, and therefore they died not from cardiac dysfunction, but even more likely from malignant arrhythmia.
TAZKD Mouse Hearts Develop Myocardial Hypertrabeculation-Noncompaction Cardiomyopathy Because of Aberrations in Early (<E13.5) Embryogenesis
To further understand the cause of the diastolic dysfunction, we examined histological sections of embryonic and newborn mouse hearts. TAZKD mice showed myocardial hypertrabeculation with noncompaction throughout development. Hypertrabeculation-noncompaction was also associated with defects in ventricular septation (Figure 7). There was no evidence of myocardial fibrosis (data not shown). Uninduced control mice (wildtype and TAZ transgenic) showed normal survival and heart structure (data not shown); therefore, our findings could not be attributed to the presence of the transgene itself.
Figure 7.
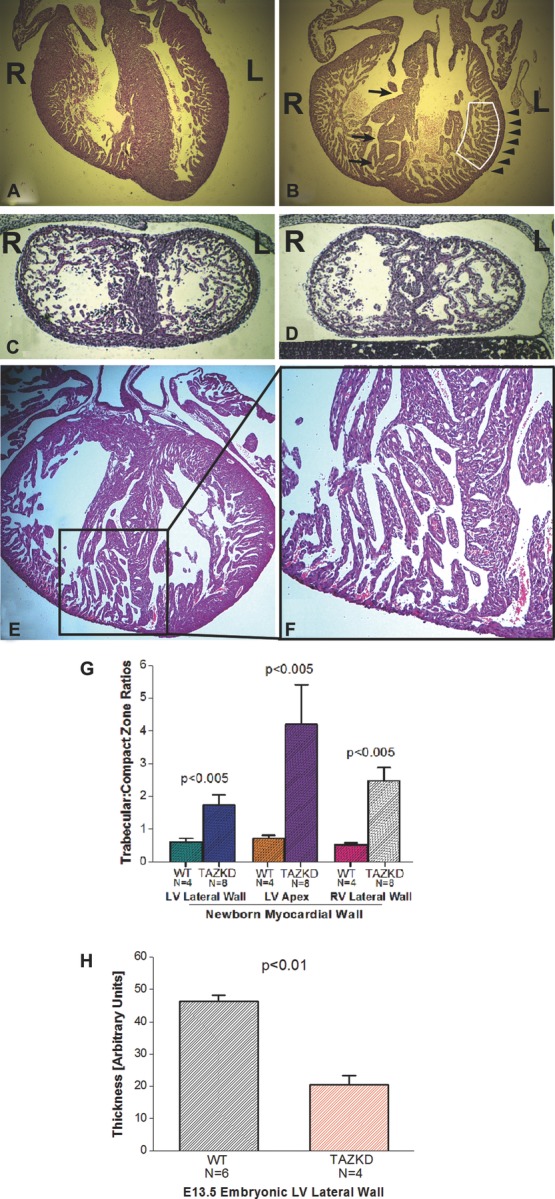
TAZKD mice exhibit thinner myocardium with prominent trabeculations suggestive of myocardial noncompaction. “4-chamber” views of newborn WT control (A) and TAZKD (B) hearts show deeper trabecular recesses and more abundant trabeculae (boxed area), thinner myocardium (arrowheads), and a less well-developed and more perforate interventricular septum in TAZKD hearts (arrows) (R = right, L = left). Coronal sections of E13.5 embryonic WT control heart (C) and TAZKD (D) hearts also demonstrate a thinner myocardium with more prominent trabeculae that occupy more of the left ventricular lumen in TAZKD hearts; again, the septum appears more porous. Tafazzin knockdown in these mice (A to D) was induced from start of gestation. Induction of TAZKD starting at E10.5 (E) recapitulates the cardiomyopathic phenotype, as represented by this newborn TAZKD heart (enlargement of apex in [F]). TAZKD newborn pups show a significantly higher degree of hypertrabeculation-noncompaction and myocardial thinning in all areas, but most notably the LV apex (G). In E13.5 TAZKD littermate embryos (H), significant myocardial thinning is also evident. Because this embryonic stage still exhibits a normally prominent trabecular myocardium, quantitation of trabecular-compact zone myocardium thicknesses was not performed. TAZKD indicates tafazzin knockdown; WT, wildtype.
Development of trabeculations followed by myocardial compaction are steps in myocardial patterning critical to embryonic heart functioning and development of the cardiac conduction system. The sequence in the mouse occurs specifically from E10.5 to E14.5.28,29 To determine the approximate developmental window of the origins of cardiomyopathy in our model, we varied the timing of doxycycline induction. First, we determined that taz mRNA knockdown reaches steady state (>70%) within 72 hours of doxycycline induction. Then, we demonstrated that induction with doxycycline at the start of gestation, at E7.5, and at E10.5 recapitulated the hypertrabeculation-noncompaction cardiomyopathy (Figure 8), whereas induction at a later stage (E13.5 and E14.5) failed to result in either early lethality or a structural cardiomyopathy. Therefore, the developmental cardiomyopathy hinges specifically on tafazzin dysfunction in the embryonic period following E10.5 but not extending past E13.5, suggesting tafazzin plays a critical role in heart development during this window.
Figure 8.
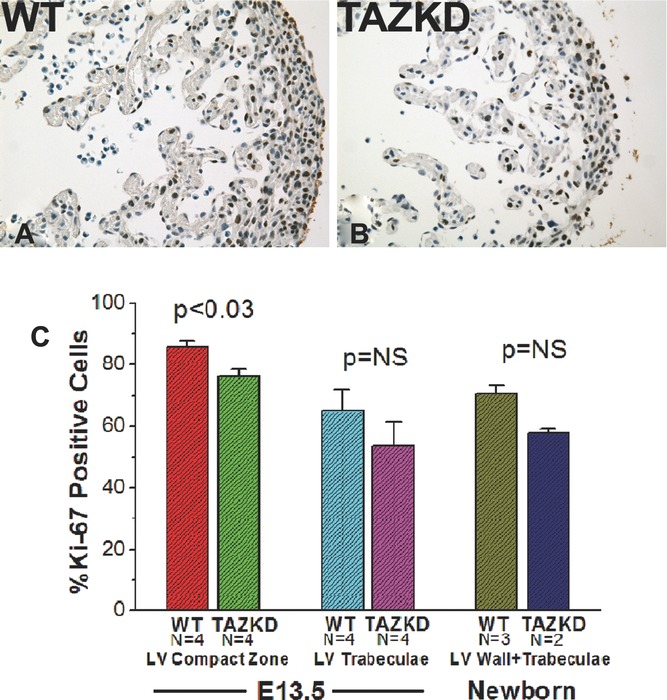
Myocardial noncompaction cardiomyopathy is accompanied by abnormal cellular proliferation. Cellular proliferation immunostaining shows Ki-67-positive nuclei (proliferating cells) in brown color (A, E13.5 wildtype [WT] embryo; B, TAZKD embryo). TAZKD mice exhibit significantly lower cellular proliferation rates (C). Because in normal embryonic development, trabecular and compact zone myocardium exhibit differential proliferation rates, we measured cellular proliferation separately in E13.5 trabecular and compact zones (C, left-hand and middle columns); TAZKD embryos exhibit significantly lower proliferation rates in the compact zone, but the differences were not significant in the trabecular zone. TAZKD indicates tafazzin knockdown.
Molecular mechanisms underlying myocardial hypertrabeculation-noncompaction are unknown, but several mouse models point to cell cycling alterations.30–34 We therefore determined differences in cellular proliferation and apoptosis in E13.5 and newborn hearts. There were no differences in apoptosis at any stage (data not shown), consistent with other models of hypertrabeculation-noncompaction.34 Cellular proliferation was decreased in TAZKD mice; furthermore, trabecular myocardium showed lower proliferation rates than compact myocardium (Figure 8). Taken together, our data indicate myocardial hypertrabeculation-noncompaction is associated with abnormal cell cycling starting in the broad developmental window when the myocardium should be compacting.
Discussion
These data are the first to demonstrate a developmental cardiomyopathy in this murine model of tafazzin knockdown. Notably, myocardial hypertrabeculation with noncompaction is consistently seen in this model. The constellation of abnormalities highlights this TAZKD mouse as a promising model to study the human Barth syndrome. Importantly, the data point to important aspects of mitochondrial functioning in the development of myocardial patterning and tissue architecture.
Prior reports of this model have demonstrated similar biochemical and mitochondrial abnormalities, but only in adult mice; cardiomyopathy did not develop until well into adulthood.11,12 We believe our model was induced by a higher-than-previously reported dosing protocol of doxycycline induction. Seibler et al13 showed the dose of doxycycline influenced especially the speed with which the knockdown achieved steady-state levels. Prior studies11,12 did not demonstrate tafazzin knockdown during embryogenesis, so their data cannot be compared directly with ours. When we carried our doxycycline induction protocol in young postnatal mice, tafazzin was knocked down to 3.8±0.68% of control levels in heart, lower than Soustek et al12 but similar to Acehan et al11; we speculate the kinetics of induction were also faster because of our higher dosage, generating the embryonic phenotype.
In our model, myocardial hypertrabeculation-noncompaction developed during a critical developmental window, consistent with what we know about the normal compaction process.28,29 Therefore, the cardiomyopathy observed by others11,12 appears to represent a different process than the developmental cardiomyopathy reported here. We acknowledge that because none of our mice survived beyond the early postnatal period, the model does not perfectly mirror human Barth syndrome. Nevertheless, the hallmark phenotype of hypertrabeculation-noncompaction is more similar to human Barth syndrome than reported previously.11,12
We cannot entirely exclude off-target effects of this transgenic knockdown strategy, similar to previous reports,11,12 because only one shRNA knockdown mouse line was available. Nevertheless, recapitulating the developmental cardiomyopathy seen in Barth syndrome, and the combination of findings—molecular, biochemical, ultrastructural, and functional—make off-target effects of our knockdown strategy unlikely. Nevertheless, additional problems, such as abnormal ventricular septation and myocardial proliferation, may be due to off-target effects. Furthermore, this model relies on induction with doxycycline. Doxycycline may be associated with placental anomalies and fetal loss, especially when introduced into food but seemingly not water.35 However, we detected no gross losses in our wildtype mice. Doxycycline may attenuate the pathophysiology of cardiac failure through matrix metalloproteinase inhibitor mechanisms,36 although the very presence of hypertrabeculation-noncompaction with early lethality in our model argues against a significant protective effect of doxycycline.
Myocardial Hypertrabeculation-Noncompaction
Myocardial noncompaction is a poorly understood developmental cardiomyopathy,5,37 now recognized as a distinct cardiomyopathy.38 Patients with Barth syndrome present as early as fetal life with cardiomyopathy,4 and conversely, noncompacted ventricular myocardium also presents with a clinical spectrum starting in fetal and neonatal life.39 Ventricular septal defects are often associated with noncompaction, both in human beings and in animal models.5,40 Mitochondrial disorders are strongly associated with myocardial noncompaction.5,38,41,42 Nevertheless, not all mitochondrial disorders lead to hypertrabeculation-noncompaction, and therefore, it seems likely only certain aspects of mitochondrial functioning contribute to this condition.
The early diastolic dysfunction is notable. Because there was no fibrosis or abnormal myocardial hypertrophy, we speculate alterations in cardiolipin involve abnormal myocardial physiology, such as altered calcium homeostasis (see later), that precede the overt hypertrabeculation-noncompaction. Interestingly, no severe systolic dysfunction was observed; but then, the model reported by Acehan et al11 and Soustek et al12 exhibited only a mild cardiomyopathy in adulthood without overt heart failure. We speculate we did not observe the “natural history” of this mouse, perhaps because of a lethal arrhythmia or cannibalization of unhealthy-looking pups by the mother; also, while human infants with severe cardiomyopathy are treated, mice are not.
Hypertrabeculation-noncompaction may be the result of dysregulated cellular proliferation in the myocardium,5,30,37,43 although the precise molecular mechanisms remain elusive. Preceding compaction, formation of normal trabecular myocardium plays a critical role in normal myocardial development, and depends on Notch signaling, while exhibiting differential cardiomyocyte proliferation in different layers of the developing heart.44 Our data suggest a dysregulation of this differential cardiomyocyte proliferation: specifically, a thin compact zone exiting earlier from the cell cycle while the trabecular zone continues to proliferate at a more normal level may explain our hypertrabeculation phenotype. We did not distinguish different cell types for this experiment, and therefore cannot exclude nonmyocytes as the main contributor to the decreases in cell proliferation; nonmyocytes may exert as-yet unknown influences on myocardial patterning. These questions will be investigated in ongoing studies.
The Role of Mitochondria in Heart Development
The role of mitochondria in the development of a structural cardiomyopathy is intriguing. Emerging evidence suggests mitochondrial disorders account for a substantial proportion of not simply cardiomyopathies, but specifically myocardial hypertrabeculation-noncompaction and other abnormalities in the development of the heart and other organs.45,46
Although investigators have focused logically on bioenergetics, more recent evidence also indicates a strong interrelationship between mitochondria, calcium homeostasis via the sarcoplasmic reticulum and sarcomeric organization.47 We now provide additional evidence that altered cardiolipin biology leads to a disruption of the normal spatial relation between mitochondria and sarcomeres in the newborn heart. We do not know at present how this disruption will mechanistically contribute to the developmental cardiomyopathy, but speculate excitation-contraction coupling may well be damaged. We acknowledge other mechanisms may also be intertwined, including the possible role of reactive oxygen species and bioenergetics of the developing heart.48
Conclusions and Future Directions
This is the first report of a murine model of tafazzin knockdown exhibiting a developmental cardiomyopathy that closely mimics the human Barth syndrome. Taken together, our data indicate certain aspects of mitochondrial functioning are necessary for proper myocardial patterning, and provide additional evidence of a causative role of mitochondrial dysfunction in the pathogenesis of myocardial hypertrabeculation-noncompaction. Although our data here offer no specific mechanistic insights, aspects of mitochondrial function that are candidates for study include the bioenergetic pathways and calcium homeostasis pathways, as well as the roles of reactive oxygen species.44,45,47
Acknowledgments
None.
Sources of Funding
This work was supported by NIH grants HL091154 (M.R.), HL078788 (M.S.), and 1U54GM094598 (D.L.S.), and grants from the Barth Syndrome Foundation (Y.X., C.K.L.P.). The Vevo 2100 was supported by a NIH Shared Instrumentation Grant (1S10RR026881-01; PI Dr. Glenn Fishman, NYU). We also gratefully acknowledge the support of the Histopathology and Immunohistochemistry Cores, supported partially by the NYU Cancer Institute Center Support Grant NIH/NCI P30CA16087-31.
Disclosures
None.
References
- 1.Barth PG, Wanders RJA, Vreken P, Janssen EAM, Lam J, Baas F. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome) (MIM 302060). J Inherit Metab Dis. 1999;22:555-567 [DOI] [PubMed] [Google Scholar]
- 2.Kelley RI, Cheatham JP, Clark BJ, Nigro MA, Powell BR, Sherwood GW, Sladky JT, Swisher WP. X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methyglutaconic aciduria. J Pediatr. 1991;119:738-747 [DOI] [PubMed] [Google Scholar]
- 3.Spencer CT, Bryant RM, Day J, Gonzalez I, Colan SD, Thompson WR, Berthy J, Redfearn SP, Byrne BJ. Cardiac and clinical phenotype in Barth syndrome. Pediatrics. 2006;118:e337-e346 [DOI] [PubMed] [Google Scholar]
- 4.Steward CG, Newbury-Ecob RA, Hastings R, Smithson SF, Tsai-Goodman B, Quarrell OW, Kulik W, Wanders R, Pennock M, Williams M, Cresswell JL, Gonzalez LL, Brennan P. Barth syndrome: an X-linked cause of fetal cardiomyopathy and stillbirth. Prenat Diagn. 2010;30:970-976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Towbin JA. Left ventricular noncompaction: a new form of heart failure. Heart Fail Clin. 2010;6:453-469 [DOI] [PubMed] [Google Scholar]
- 6.Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJA, Barth PG. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Comm. 2000;279:378-382 [DOI] [PubMed] [Google Scholar]
- 7.Schlame M, Towbin JA, Heerdt PM, Jehle R, DiMauro S, Blanck TJJ. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann Neurol. 2002;51:634-637 [DOI] [PubMed] [Google Scholar]
- 8.Xu Y, Malhotra A, Ren M, Schlame M. The enzymatic function of tafazzin. J Biol Chem. 2006;281:39217-39224 [DOI] [PubMed] [Google Scholar]
- 9.Houtkooper RH, Turkenburg M, Poll-The BT, Karall D, Pérez-Cerdá C, Morrone A, Malvagia S, Wanders RJ, Kulik W, Vaz FM. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim Biophys Acta. 2009;1788:2003-2014 [DOI] [PubMed] [Google Scholar]
- 10.Schlame M, Ren M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim Biophys Acta. 2009;1788:2080-2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Acehan D, Vaz F, Houtkooper RH, James J, Moore V, Tokunaga C, Kulik W, Wansapura J, Toth MJ, Strauss A, Khuchua Z. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J Biol Chem. 2011;286:899-908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soustek MS, Falk D, Mah C, Toth M, Schlame M, Lewin A, Byrne B. Characterization of a transgenic short hairpin RNA-induced murine model of tafazzin deficiency. Hum Gene Ther. 2011;22:865-871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seibler J, Kleinridders A, Küter-Luks B, Niehaves S, Brüning JC, Schwenk F. Reversible gene knockdown in mice using a tight, inducible shRNA expression system. Nucleic Acids Res. 2007;35:e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malhotra A, Edelman-Novemsky I, Xu Y, Plesken H, Ma J, Schlame M, Ren M. Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proc Natl Acad Sci USA. 2009;106:2337-2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrell MD, Harbi S, Hoffman JF, Zavadil J, Coetzee WA. Large-scale analysis of ion channel gene expression in the mouse heart during perinatal development. Physiol Genomics. 2007;28:273-283 [DOI] [PubMed] [Google Scholar]
- 16.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911-917 [DOI] [PubMed] [Google Scholar]
- 17.Sun G, Yang K, Zhao Z, Guan S, Han X, Gross RW. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometric analysis of cellular glycerophospholipids enabled by multiplexed solvent dependent analyte-matrix interactions. Anal Chem. 2008;80:7576-7585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collis LP, Meyers MB, Zhang J, Phoon CKL, Sobie EA, Coetzee WA, Fishman GI. Expression of a sorcin missense mutation in the heart modulates excitation-contraction coupling. FASEB J. 2007;21:475-487 [DOI] [PubMed] [Google Scholar]
- 19.Nomura-Kitabayashi A, Phoon CKL, Kishigami S, Rosenthal J, Yamauchi Y, Abe K, Yamamura K, Samtani R, Lo CW, Mishina Y. Outflow tract cushions perform a critical valve-like function in the early embryonic heart requiring BMPRIA-mediated signaling in cardiac neural crest. Am J Physiol Heart Circ Physiol. 2009;297:H1617-H1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji RP, Phoon CKL. Non-invasive localization of NFATc1-/- mouse embryos by ultrasound biomicroscopy-Doppler allows genotype-phenotype correlation. J Am Soc Echocardiogr. 2005;18:1415-1421 [DOI] [PubMed] [Google Scholar]
- 21.Acehan D, Khuchua Z, Houtkooper RH, Malhotra A, Kaufman J, Vaz FM, Ren M, Rockman HA, Stokes DL, Schlame M. Distinct effects of tafazzin deletion in differentiated and undifferentiated mitochondria. Mitochondrion. 2009;9:86-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishiwata T, Nakazawa M, Pu WT, Tevosian SG, Izumo S. Developmental changes in ventricular diastolic function correlate with changes in ventricular myoarchitecture in normal mouse embryos. Circ Res. 2003;93:857-865 [DOI] [PubMed] [Google Scholar]
- 23.Zhang F, Pasumarthi KBS. Ultrastructural and immunocharacterization of undifferentiated myocardial cells in the developing mouse heart. J Cell Mol Med. 2007;11:552-560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Phyiol Heart Circ Physiol. 2007;293:H1883-H1891 [DOI] [PubMed] [Google Scholar]
- 25.Gatzoulis MA, Clark AL, Cullen S, Newman CG, Redington AN. Right ventricular diastolic function 15 to 35 years after repair of tetralogy of Fallot. Restrictive physiology predicts superior exercise performance. Circulation. 1995;91:1775-1781 [DOI] [PubMed] [Google Scholar]
- 26.Ichida F, Hamamichi Y, Miyawaki T, Ono Y, Kamiya T, Akagi T, Hamada H, Hirose O, Isobe T, Yamada K, Kurotobi S, Mito H, Miyake T, Murakami Y, Nishi T, Shinohara M, Seguchi M, Tashiro S, Tomimatsu H. Clinical features of isolated noncompaction of the ventricular myocardium: long-term clinical course, hemodynamic properties, and genetic background. J Am Coll Cardiol. 1999;34:233-240 [DOI] [PubMed] [Google Scholar]
- 27.Phoon CKL, Turnbull DH. Ultrasound biomicroscopy-Doppler in mouse cardiovascular development. Physiol Genomics. 2003;14:3-15 [DOI] [PubMed] [Google Scholar]
- 28.Wessels A, Sedmera D. Developmental anatomy of the heart: a tale of mice and man. Physiol Genomics. 2003;15:165-176 [DOI] [PubMed] [Google Scholar]
- 29.Savolainen SM, Foley JF, Elmore SA. Histology atlas of the developing mouse heart with emphasis on E11.5 to E18.5. Toxicol Pathol. 2009;37:395-414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen H, Zhang W, Li D, Cordes TM, Payne MC, Shou W. Analysis of ventricular hypertrabeculation and noncompaction using genetically engineered mouse models. Pediatr Cardiol. 2009;30:626-634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pashmforoush M, Lu JT, Chen H, St. Amand T, Kondo R, Pradervand S, Evans SM, Clark B, Feramisco JR, Giles W, Ho SY, Benson DW, Sliberbach M, Shou W, Chien KR. Nkx2-5 pathways and congenital heart disease: loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell. 2004;117:373-386 [DOI] [PubMed] [Google Scholar]
- 32.Mysliwiec MR, Bresnick EH, Lee Y. Endothelial Jarid2/Jumonji is required for normal cardiac development and proper Notch1 expression. J Biol Chem. 2011;286:17193-17204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen H, Shi S, Acosta L, Li W, Lu J, Bao S, Chen Z, Yang Z, Schneider MD, Chein KR, Conway SJ, Yoder MC, Haneline LS, Franco D, Shou W. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. 2004;131:2219-2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li D, Hallett MA, Zhu W, Rubart M, Liu Y, Yang Z, Chen H, Haneline LS, Chan RJ, Schwartz RJ, Field LJ, Atkinson SJ, Shou W. Dishevelled-associated activator of morphogenesis 1 (Daam1) is required for heart morphogenesis. Development. 2011;138:303-315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moutier R, Tchang F, Caucheteux SM, Kanellopoulos-Langevin C. Placental anomalies and fetal loss in mice, after administration of doxycycline in food for Tet-system activation. Transgenic Res. 2003;12:369-373 [DOI] [PubMed] [Google Scholar]
- 36.Castro MM, Kandasamy AD, Youssef N, Schulz R. Matrix metalloproteinase inhibitor properties of tetracyclines: Therapeutic potential in cardiovascular diseases. Pharmacol Res. 2011;64:551-560 [DOI] [PubMed] [Google Scholar]
- 37.Zaragoza MV, Arbustini E, Narula J. Noncompaction of the left ventricle: primary cardiomyopathy with an elusive genetic etiology. Curr Opin Pediatr. 2007;19:619-627 [DOI] [PubMed] [Google Scholar]
- 38.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807-1816 [DOI] [PubMed] [Google Scholar]
- 39.Menon SC, O'Leary PW, Wright GB, Rios R, MacLellan-Tobert SG, Cabalka AK. Fetal and neonatal presentation of noncompacted ventricular myocardium: expanding the clinical spectrum. J Am Soc Echocardiogr. 2007;20:1344-1350 [DOI] [PubMed] [Google Scholar]
- 40.Shou W, Aghdasi B, Armstrong DL, Guo Q, Bao S, Charng M-J, Mathews LM, Schneider MD, Hamilton SL, Matzuk MM. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489-492 [DOI] [PubMed] [Google Scholar]
- 41.Scaglia F, Towbin JA, Craigen WJ, Belmont JW, O'Brian Smith E, Neish SR, Ware SM, Hunter JV, Fernbach SD, Vladutiu GD, Wong L-JC, Vogel H. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114:925-931 [DOI] [PubMed] [Google Scholar]
- 42.Tang S, Batra A, Zhang Y, Ebenroth ES, Huang T. Left ventricular noncompaction is associated with mutations in the mitochondrial genome. Mitochondrion. 2010;10:350-357 [DOI] [PubMed] [Google Scholar]
- 43.Chen Q, Chen H, Zheng D, Kuang C, Fang H, Zhou B, Zhu W, Bu G, Jin T, Wang Z, Zhang X, Chen J, Field LJ, Rubart M, Shou W, Chen Y. Smad7 is required for the development and function of the heart. J Biol Chem. 2009;284:292-300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grego-Bessa J, Luna-Zurita L, del Monte G, Bolós V, Melgar P, Arandilla A, Garratt AN, Zang H, Mukouyama Y-s, Chen H, Shou W, Ballestar E, Esteller M, Rojas A, Pérez-Pomares JM, de la Pompa JL. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12:415-429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao X, Chen Y. Mitochondria and calcium signaling in embryonic development. Semin Cell Dev Biol. 2009;20:337-345 [DOI] [PubMed] [Google Scholar]
- 46.Porter GA, Jr, Hom JR, Hoffman DL, Quintanilla RA, de Masy Bentley KL, Sheu S-S. Bioenergetics, mitochnodria, and cardiac myocyte differentiation. Prog Pediatr Cardiol. 2011;31:75-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luykanenko V, Chikando A, Lederer WJ. Mitochondria in cardiomyocyte Ca2+ signaling. Int J Biochem Cell Biol. 2009;41:1957-1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hom J, Quintanilla R, Hoffman D, Bentley K, Molkentin J, Sheu S-S, Porter G., Jr The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Dev Cell. 2011;21:469-478 [DOI] [PMC free article] [PubMed] [Google Scholar]