

Abstract
Among 4,780 consecutive adult acute lymphoblastic/myeloblastic leukemia patients, we identified 117 (2.4%) patients with mixed-phenotype acute leukemia fulfilling WHO 2008 criteria; these were classified as: Blymphoid+ myeloid (n=64), T-lymphoid+myeloid (n=38), B+T-lymphoid (n=14) and trilineage (n=1). Of 92 patients karyotyped, 59 were abnormal and were classified as: complex (22 of 92), t(9;22)(q34;q11) (14 of 92), monosomy 7 (7 of 92), polysomy 21 (7 of 92), t(v;11q23) (4 of 92), t(10;11)(p15;q21) (3 of 92), while STIL-TAL1 fusion was detected in one (T+My) patient. After investigating common acute leukemia-related mutations in 17 genes, 12 of 31 (39%) patients were found to have at least one mutation, classified with: IKZF1 deletion (4 of 31), and EZH2 (3 of 31), ASXL1 (3 of 31), ETV6 (2 of 31), NOTCH1 (1 of 31), and TET2 (1 of 31) mutations. Array-CGH revealed genomic deletions of CDKN2A (4 of 12), IKZF1 (3 of 12), MEF2C (2 of 12), BTG1 (2 of 12), together with BCOR, EBF1, K-RAS, LEF1, MBNL1, PBX3, and RUNX1 (one of 12 each). Our results indicate that mixed-phenotype acute leukemia is a complex entity with heterogeneous clinical, immunophenotypic, cytogenetic, and molecular genetic features.
Key words: mixed-phenotype acute leukemia, immunophenotype, cytogenetic, mutation, therapy
Introduction
In the majority of patients with acute leukemia, blast cells can be unequivocally assigned to a specific lineage, myeloid, B- or T-lymphoid. However, in approximately 2-5% of patients, lineage origin remains ambiguous even after comprehensive immunophenotyping by flow cytometry (FCM). Historically, a variety of terms have been used to describe these cases, such as mixed lineage leukemia, hybrid acute leukemia, bilineal leukemia, and biphenotypic leukemia.1-5 The 2008 WHO classification of hematopoietic and lymphoid tumors (WHO-2008) modified the diagnostic criteria and introduced a new designation for this entity, this is now termed mixed-phenotype acute leukemia (MPAL).6
The clinical and laboratory features of mixed lineage leukemia, hybrid acute leukemia, bilineal leukemia, or biphenotypic leukemia have been described in a few retrospective studies.1-5,7-13 However, given the dearth of reports so far, the clinical and laboratory features of patients with MPAL remain largely undefined. The only investigation reported to date involved 2 infants, 28 children and 68 adults with MPAL who were reassessed according to WHO-2008.14
Here, we retrospectively analyzed the clinical, immunophenotypic, cytogenetic, and molecular genetic features of patients with MPAL as defined under WHO-2008 criteria.
Design and Methods
Patients
The study was approved by the Ethics Committee of the First Affiliated Hospital of Soochow University (FAHSU), P.R. China, according to the Declaration of Helsinki. It involved 4,780 adult (≥14 years of age) patients presenting with de novo acute leukemia at the FAHSU from January 1998 to August 2011; 117 (2.4%) cases, all ethnic Chinese, fulfilled WHO-2008 criteria for MPAL. Patients with AML-related translocations, chronic myeloid leukemia in blast crisis (CML-BC), myelodysplasia-related changes, or therapy-related AML were excluded from the study.
The combined AML+ALL regimen, MOAP/IOAP/DOAP was used to treat 24 cases as induction therapy: daunorubicin, idarubicin or mitoxantrone and cytarabine plus vincristine or vindesine and prednisone. The CAG regimen was administered to 16 patients, 10 at presentation and 6 refractory to the combined AML+ALL therapy. The CAG regimen consisted of Ara-C 10 mg/m2 injected subcutaneously every 12 h (Days 1-14), aclarubicin 6 mg/m2 infused intravenously (Days 1-8), and G-CSF administered subcutaneously at a dose of 200 μg/m2/day. Eight patients with MPAL underwent allo-HSCT. Donors were double unit cord blood (n=3), matched unrelated (n=2), mismatched related (n=2) and matched sibling (n=1).
Immunophenotyping
FCM immunophenotyping was performed on blast populations using FACSCalibur instruments (BD Biosciences, San Jose, CA, USA). All cases were characterized by a panel of antibodies to leukocyte-associated markers, including surface CD2, CD3 (and cytoplasmic cyCD3), CD5, CD7, CD10, CD11b, CD11c, CD13, CD14, CD15, CD19, CD20, CD22, (cyCD22), CD33, CD34, CD45, CD64, CDw65, CD79a, (cyCD79a), CD117, HLA-DR, TdT, and myeloperoxidase (MPO). Surface antigen expression was considered positive when more than 20% of blasts showed a positive signal. For cytoplasmic antigens, the threshold was set at 10%.
Cytogenetic and molecular genetic analysis
R-banding karyotypic analysis was carried out on bone marrow (BM) or peripheral blood (PB) cells at diagnosis. A multiplex reverse transcription-polymerase chain reaction (RT-PCR) strategy was used in 44 patients with MPAL to detect 29 acute leukemia-related fusion genes, as previously described.15
Mutation analysis was perfomed in 31 MPAL patients for whom genomic DNA and RNA were available. A variety of acute leukemia-related mutations were evaluated: ASXL1, CBL, DNMT3A, ETV6, EZH2, FBXW7, FLT3-ITD, FLT3-TKD, IDH1, IDH2, IKZF1, KIT, NOTCH1, NPM1, PHF6, RUNX1, TET2, and WT1.
Array-based comparative genomic hybridization (array-CGH) analysis with Agilent 244k Human CGH Microarrays (Agilent Technologies, Santa Clara, California, USA) was performed in 12 MPAL patients for whom at least 1.5 μg genomic DNA was available. Data were visualized with Agilent Genomic Workbench Lite Edition 6.5 software.
Statistical analysis
Patients' characteristics were analyzed by χ2 or Fisher's exact tests for univariate analysis. P<0.05 was considered significant. All calculations were performed using the SPSS software package (version 13.0).
Results and Discussion
Patients
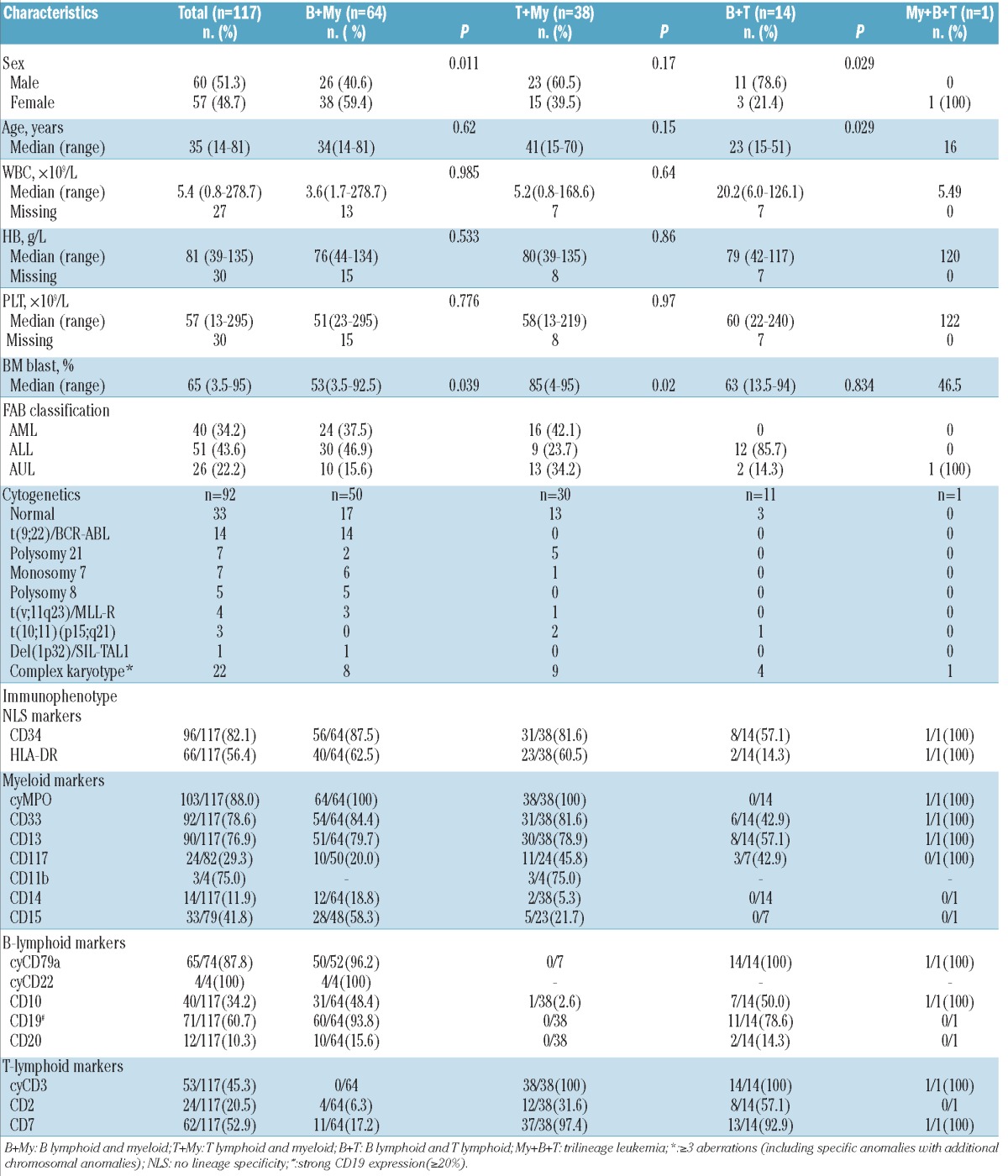
Among 4,780 patients admitted to our institute since 1998, we identified 117 adult patients fulfilling current WHO-2008 criteria for MPAL (60 males and 57 females). The proportion of MPAL patients in this cohort (2.4%) was consistent with that of the 0.9-2.6% of acute leukemia cases according to the meta-analysis performed by Weinberg et al.5 Median age was 35 years (range 14-81 years). Based on FAB criteria, 40 (34%) patients showed AML morphologies, mainly M1 and M5. A total of 51 patients (44%) were classified ALL-L1, the dominant subtype. The remaining 26 (22%) cases resisted classification by morphology and were categorized as acute undifferentiated leukemia. Main patients' characteristics are summarized in Table 1.
Table 1.
Clinical, morphological, immunological and cytogenetic characteristics of 117 cases with MPAL.
Immunophenotype
Immunophenotyping data (Table 1) showed that 64 of 117 cases (55%) had combined B+My, 38 (33%) combined T+My, 14 (12%) combined B+T, and one (0.9%) a trilineage (My+B+T), immunophenotype. MPAL cases with B+T immunophenotypes were more commonly seen in males; 11 of 14 (79%), when compared to 23 of 38 (61%) with T+My (P=0.376) and 26 of 64 (41%) with B+My immunophenotypes (P=0.01). Immunophenotyping data are summarized in Table 1.
Our immunophenotyping data revealed significant gender differences in MPAL subtypes, with more male in B+T and T+My groups but female predominance among B+My patients. This contrasts with data reported by Matutes et al.14 Further studies are needed to clarify this and, in particular, whether ethnic or genetic factors might contribute to these discrepancies in gender distribution of MPAL cases.
It is presumed that MPAL arises from multipotent stem cells, capable of differentiating into myeloid and lymphoid progenitors during the development of acute leukemia. In this study, a marker of early hematopoietic cells, CD34, was strongly positive in 82% cases, reinforcing the view that MPAL cells originate at early stages of hematopoietic differentiation.
Cytogenetics
Of the 92 patients with available karyotypic data, 33 (36%) had no detectable chromosomal abnormality, while the remaining 59 (64%) showed abnormal karyotypes. Complex karyotypic abnormalities (≥3 aberrations) described the most prolific subclass, found in 22 cases (24%). The t(9;22)(q34;q11)/BCR-ABL1 fusion was present in 14 patients (15%), all with B+My phenotypes. Monosomy 7 was detected in 7 of 92 cases (7.6%). Polysomy 21 was unexpectedly found in 7 of 92 (7.6%) cases. Trisomy 8 occurred in 5 (5.4%) cases, all with B+My phenotypes. t(v;11q23)/MLL rearrangements were seen in 4 (4.3%) patients, of whom 2 had t(11;19)(q23;p13), while one patient each carried t(4;11)(q21;q23) and t(9;11)(p22;q23), respectively. t(10;11)(p15;q21) was present in 3 (3.3%) cases, including 2 with T+My and one with B+T phenotypes. In 3 cases, translocations not previously reported in leukemia were found: t(7;9)(q32;p24) with B+My phenotype, t(2;9)(q13;q34) with B+T phenotype, and der(9)t(9;11)(p21;q12) with T+My phenotype. A novel fusion between NUP98 and IQCG gene was identified in a MPAL patient with T+My phenotype harboring t(3;11)(q29q13;p15)del(3)(q29), as previously described.16 STIL-TAL1 fusion via 1p32 microdeletion was identified by multiplex RT-PCR analysis in one case with B+My immunophenotype. Cytogenetic data are summarized in Table 1.
Of the patients for whom cytogenetic data were available, 64% showed abnormal karyotypes. Similar to previous reports, the cytogenetic groups most commonly described in MPAL or biphenotypic leukemia, namely those with complex karyotypes, t(9;22)(q34;q11) and t(v;11q23), were also detected in our study in a substantial proportion of MPAL cases: 24%, 15%, and 4.3% of patients, respectively.
Importantly, we identified four recurrent cytogenetic changes for the first time in a significant number of MPAL patients: monosomy 7 in 7.6%, polysomy 21 in 7.6%, t(10;11)(p15;q21) in 3.3%, and 1p32 deletion effecting STILTAL fusion in 1.1%. Monosomy 7 is one of the most frequent chromosome abnormalities observed in patients with myelodysplastic syndromes and acute myeloid leukemia (AML), in which it is associated with a relatively poor prognosis. 17 It may also be found in patients with chronic myeloid leukemia in blast crisis (CML-BC) or Philadelphia positive acute lymphoblastic leukemia (ALL). Acquired isolated trisomy 21 is a frequent cytogenetic abnormality in myeloid malignancies, and has been reported as an anomaly accompanying t(12;21)(p13;q22) in childhood B-cell ALL. Several studies in leukemia patients with trisomy 21 showed frequent mutations of RUNX1, often duplicated as a consequence of the trisomy.18 However, mutations of RUNX1 were absent in all 4 MPAL patients screened for RUNX1 mutation in this study. t(10;11)(p15;q21), resulting in PICALM-MLLT10 fusion, is a recurrent chromosomal translocation seen in AML or ALL patients showing T+My or B+T phenotype. STIL-TAL1 fusion results from a 1p32 microdeletion and is detected in 10-25% of T-cell ALL. This is the first report indicting these four cytogenetic alterations in the pathogenesis of MPAL.
Frequencies and distribution of gene mutations
In the past decade, with the advent of the novel genomic microarray technologies and next-generation sequencing, numerous genetic mutations which escape conventional cytogenetic detection have increasingly been reported in patients with AML, ALL, and other hematologic malignancies. However, as yet no studies have tried to delineate the distribution and clinical significance of these genetic mutations in patients with MPAL.
Frequencies and distribution of gene mutations
In the past decade, with the advent of the novel genomic microarray technologies and next-generation sequencing, numerous genetic mutations which escape conventional cytogenetic detection have increasingly been reported in patients with AML, ALL, and other hematologic malignancies. However, as yet no studies have tried to delineate the distribution and clinical significance of these genetic mutations in patients with MPAL.
In the present study, we evaluated a variety of acute leukemia-related mutations. After excluding known polymorphisms and silent mutations, a total of 12 mutations (39%) were documented (Table 2) including: IKZF1 deletions in 4 of 31 (13%) patients, and mutations affecting EZH2 in 3 of 31 (9.7%), ASXL1 in 2 of 31 (6.5%), and ETV6, NOTCH1, and TET2 mutations in one of 31 (3.2%) patients each. Although mutations of CBL, DNMT3A, FBXW7, FLT3, IDH1, IDH2, KIT, NPM1, PHF6, RUNX1, and WT1 have been frequently detected in patients with AML or ALL, none were found in this study. Collectively, our findings suggest that MPAL may carry a spectrum of molecular aberrations that differ from other acute leukemias.
Table 2.
Characteristics of 12 MPAL patients with gene mutation(s).
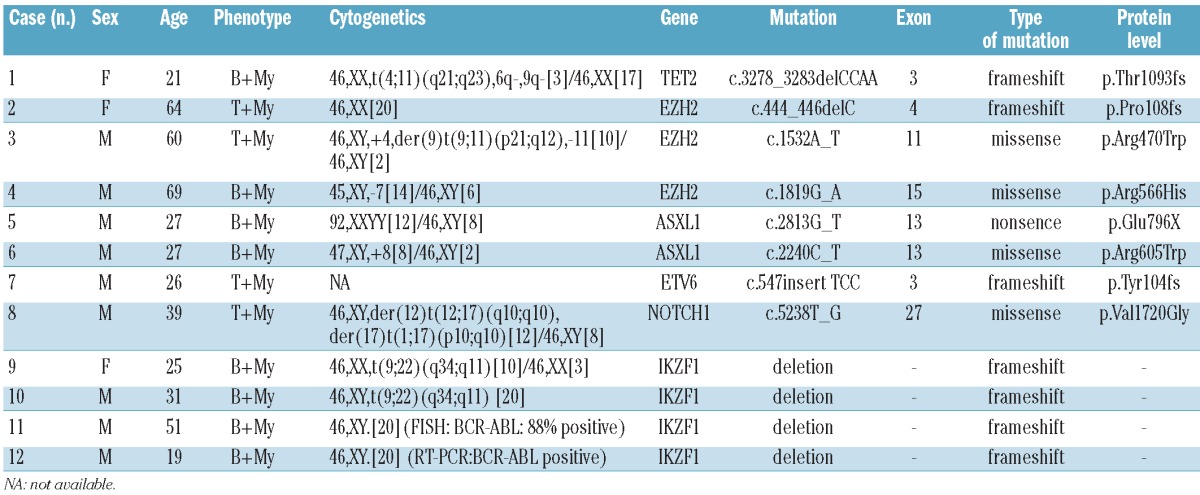
With respect to karyotype, cytogenetic abnormalities were detected in 10 of 11 patients with available data. Among these, IKZF1 deletions were found exclusively in 4 of 8 (50%) patients with t(9;22)(q34;q11)/BCR-ABL1 fusion and B+My phenotype. To evaluate the clinical impact of gene mutations on patients with MPAL, we compared the prognosis of 24 MPAL patients undergoing mutation screening. However, no significant difference on overall survival (P=0.158) between patients with and without gene mutations was found.
Array-based comparative genomic hybridization analysis of 12 MPAL samples
Genomic microarrays provide powerful tools enabling global high-resolution analysis of submicroscopic chromosome deletions, amplifications, and unbalanced chromosome rearrangements. They are widely used in the molecular genetic studies of hematologic malignancies and have helped identify many important cryptic leukemia-related genetic alterations, such as IKZF1 deletion, MYB duplication, LEF1 deletion, SET-NUP214 fusion, PAX5 deletion, and TET2 mutation. This is the first molecular genomic study of MPAL or biphenotypic leukemia. We performed array-CGH analysis on 12 MPAL samples and found that all had one or more genomic abnormalities (Online Supplementary Table S1). A total of 68 genomic alterations were detected, with a mean of 5.7 genomic alterations per sample. We identified new cryptic copy number changes in MPAL containing interesting potential candidate genes such as deletions of the CDKN2A (4 of 12), IKZF1 (3 of 12), MEF2C (2 of 12), together with one each (8.3%) of BCOR, EBF1, KRAS, LEF1, MBNL1, PBX3 and RUNX1.
Among these, deletion of CDKN2A and mutations of KRAS are found in a wide variety of solid tumors and hematologic malignancies. The BTG1, EBF1 and IKZF1 genes are associated with B-cell development and are frequently deleted in patients with B-cell precursor ALL.19,20 Mutations of BCOR and RUNX1 genes have been repeatedly found in patients with AML.21,22 Deletion of LEF1 and transcriptional deregulation of MEF2C have already been identified in T-ALL.23,24 All of the cryptic gene lesions listed above are described here for the first time in MPAL patients.
Treatment and clinical response of MPAL patients
For the moment, the optimal therapy for patients with MPAL has still not been defined. There is little consensus as to whether induction therapy should follow AML and/or ALL chemotherapy, and as to whether HSCT might be effective. We compared the relative efficacies of the combination of AML+ALL therapy and of the CAG regimen in induction therapy on 34 MPAL patients at presentation. MOAP/IOAP/DOAP was administered to 24 MPAL patients, of whom 14 (58%) achieved complete remission (CR), while 2 died from severe infection. The remaining 10 patients received induction therapy according to the CAG regimen of whom 7 (70%) achieved CR. There was no significant difference in CR rates between the two groups (P=0.802). However, interestingly, 5 of the 6 patients (83%) who failed to respond to MOAP/IOAP/DOAP therapy achieved CR after receiving the CAG regimen. Furthermore, the overall survival rate in 8 patients with MPAL receiving allo-HSCT was longer than that in the 13 patients receiving consolidation chemotherapy (22.0 vs. 9.0 months; P=0.004). Therefore, it seems patients receiving allo-HSCT had the better outcomes. However, owing to the limited numbers of patients who received allo-HSCT in this study, further investigation involving more cases is needed to determine its role in MPAL therapy.
In summary, this study demonstrates that adult MPAL is a complex entity with heterogeneous clinical, immunophenotypic and genetic characteristics. We observed widespread differences in the cytogenetic and molecular features of adult MPAL versus AML or ALL. We also identified gene mutations or cryptic copy number alterations housing potential oncogene targets. Further genetic studies with the novel genomic technologies, such as next-generation sequencing, may help define the leukemogenic mechanisms of patients with MPAL. In addition, it will be necessary to conduct multi-center co-operative studies to determine optimal regimens for induction and consolidation therapy and to define the role of HSCT in MPAL.
Acknowledgments
Funding: this work was supported by grants from National Key Scientific Projects of China (2011CB933501), the Priority Academic Program Development of Jiangsu Higher Education Institutions, Jiangsu Province's Key Provincial Talents Program, the National Natural Science Foundation of China (81070416), and the Foundation of Jiangsu Province Health Department (H200915).
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures: The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Mirro J, Zipf TF, Pui CH, Kitchingman G, Williams D, Melvin S, et al. Acute mixed lineage leukemia: clinicopathologic correlations and prognostic significance. Blood. 1985;66(5):1115-23 [PubMed] [Google Scholar]
- 2.Ruiz-Argüelles GJ, Lobato-Mendizábal E, Marín-López A. The incidence of hybrid acute leukaemias. Leuk Res. 1988;12(9):707-9 [DOI] [PubMed] [Google Scholar]
- 3.Weir EG, Ali Ansari-Lari M, Batista DA, Griffin CA, Fuller S, Smith BD, et al. Acute bilineal leukemia: a rare disease with poor outcome. Leukemia. 2007;21(11):2264-70 [DOI] [PubMed] [Google Scholar]
- 4.Matutes E, Morilla R, Farahat N, Carbonell F, Swansbury J, Dyer M, et al. Definition of acute biphenotypic leukemia. Haematologica. 1997;82(1):64-6 [PubMed] [Google Scholar]
- 5.Weinberg OK, Arber DA. Mixed-phenotype acute leukemia: historical overview and a new definition. Leukemia. 2010;24(11):1844-51 [DOI] [PubMed] [Google Scholar]
- 6.Borowitz MJ, Bene MC, Harris NL, Porwit A, Matutes E. Acute leukemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon, 2008, pp 150-5 [Google Scholar]
- 7.Zhang Y, Wu D, Sun A, Qiu H, He G, Jin Z, et al. Clinical characteristics, biological profile, and outcome of biphenotypic acute leukemia: a case series. Acta Haematol. 2011;125(4):210-8 [DOI] [PubMed] [Google Scholar]
- 8.Al-Seraihy AS, Owaidah TM, Ayas M, El-Solh H, Al-Mahr M, Al-Ahmari A, et al. Clinical characteristics and outcome of children with biphenotypic acute leukemia. Haematologica. 2009;94(12):1682-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu XQ, Wang JM, Lü SQ, Chen L, Yang JM, Zhang WP, et al. Clinical and biological characteristics of adult biphenotypic acute leukemia in comparison with that of acute myeloid leukemia and acute lymphoblastic leukemia: a case series of a Chinese population. Haematologica. 2009;94(7):919-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubnitz JE, Onciu M, Pounds S, Shurtleff S, Cao X, Raimondi SC, et al. Acute mixed lineage leukemia in children: the experience of St Jude Children's Research Hospital. Blood 2009;113(21):5083-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Killick S, Matutes E, Powles RL, Hamblin M, Swansbury J, Treleaven JG, et al. Outcome of biphenotypic acute leukemia. Haematologica. 1999;84(8):699-706 [PubMed] [Google Scholar]
- 12.Legrand O, Perrot JY, Simonin G, Baudard M, Cadiou M, Blanc C, et al. Adult biphenotypic acute leukaemia: an entity with poor prognosis which is related to unfavourable cytogenetics and P-glycoprotein overexpression. Br J Haematol. 1998;100(1):147-55 [DOI] [PubMed] [Google Scholar]
- 13.Owaidah TM, Beihany AA, Lqbal MA, Elkum N, Roberts GT. Cytogenetics, molecular and ultrastructural characteristics of biphenotypic acute leukemia identified by the EGIL scoring system. Leukemia. 2006;20(4):620-6 [DOI] [PubMed] [Google Scholar]
- 14.Matutes E, Pickl WF, Van't Veer M, Morilla R, Swansbury J, Strobl H, et al. Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood. 2011;117(11):3163-71 [DOI] [PubMed] [Google Scholar]
- 15.Strehl S, König M, Mann G, Haas OA. Multiplex reverse transcriptase-polymerase chain reaction screening in childhood acute myeloblastic leukemia. Blood. 2001;97(3):805-8 [DOI] [PubMed] [Google Scholar]
- 16.Pan Q, Zhu YJ, Gu BW, Cai X, Bai XT, Yun HY, et al. A new fusion gene NUP98-IQCG identified in an acute T-lymphoid/myeloid leukemia with a t(3;11)(q29q13;p15)del (3)(q29) translocation. Oncogene. 2008;27(24):3414-23 [DOI] [PubMed] [Google Scholar]
- 17.Hasle H, Alonzo TA, Auvrignon A, Behar C, Chang M, Creutzig U, et al. Monosomy 7 and deletion 7q in children and adolescents with acute myeloid leukemia: an international retrospective study. Blood. 2007;109(11):4641-7 [DOI] [PubMed] [Google Scholar]
- 18.Larsson N, Lilljebjörn H, Lassen C, Johansson B, Fioretos T. Myeloid malignancies with acquired trisomy 21 as the sole cytogenetic change are clinically highly variable and display a heterogeneous pattern of copy number alterations and mutations. Eur J Haematol. 2012;88(2):136-43 [DOI] [PubMed] [Google Scholar]
- 19.Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116(23):4874-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lundin C, Hjorth L, Behrendtz M, Nordgren A, Palmqvist L, Andersen MK, et al. High frequency of BTG1 deletions in acute lymphoblastic leukemia in children with down syndrome. Genes Chromosomes Cancer. 2012;51(2):196-206 [DOI] [PubMed] [Google Scholar]
- 21.Grossmann V, Tiacci E, Holmes AB, Kohlmann A, Martelli MP, Kern W, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118(23):6153-63 [DOI] [PubMed] [Google Scholar]
- 22.Tang JL, Hou HA, Chen CY, Liu CY, Chou WC, Tseng MH, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009;114(26):5352-61 [DOI] [PubMed] [Google Scholar]
- 23.Gutierrez A, Sanda T, Ma W, Zhang J, Grebliunaite R, Dahlberg S, et al. Inactivation of LEF1 in T-cell acute lymphoblastic leukemia. Blood. 2010;115(14):2845-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagel S, Meyer C, Quentmeier H, Kaufmann M, Drexler HG, MacLeod RA, et al. MEF2C is activated by multiple mechanisms in a subset of T-acute lymphoblastic leukemia cell lines. Leukemia. 2008;22(3):600-7 [DOI] [PubMed] [Google Scholar]