

Abstract
We report the regioselective and enantioselective allylation of an ester enolate, trimethylsiloxyfuran. This enolate reacts in the 3-position with linear aromatic allylic carbonates or aliphatic allylic benzoates to form the branched substitution products in the presence of a metallacyclic iridium catalyst. This process provides access to synthetically important 3-substituted butenolides in enantioenriched form. Stoichiometric reactions of the allyliridium intermediate imply that the trimethylsiloxyfuran is activated by the carboxylate leaving group.
Asymmetric allylic substitution catalyzed by metallacyclic iridium phosphoramidite complexes 1 and 21 forms enantioenriched materials from readily available allylic esters and a variety of heteroatom2 and carbon3 nucleophiles. However, the carbon nucleophiles that undergo this process are mainly stabilized enolates.3a-h Reactions of unstabilized enolates3i-k have been limited to those derived from methyl ketones. Enolates of esters that undergo iridium catalyzed allylic substitutions possess a second electron-withdrawing group (EWG) to stabilize the enolate.
Trimethylsiloxyfuran 3a is an important ester enolate because it can be used to construct butenolides, a motif found in over 13,000 natural products.4 High diastereo- and enantioselectivity has been achieved from the addition of 3a to carbonyl acceptors with Lewis-acid or organic catalysts.4a, 5 These reactions occurred at C-5 of 3a. Regioselective reactions at C-3 of 3a to form enantioenriched, 3-substituted butenolides are rare.6
One set of palladium-catalyzed kinetic resolutions of methyl substituted allylic acetate with trimethylsiloxyfuran does occur at C-3 of the nucleophile 3a (a of Scheme 1).7 In this case, the regioselectivity at the electrophile was controlled by the properties of the substrate. The attack at the less hindered end of the allyl unit led to the conjugated product.8, 9 This origin of regioselectivity restricts the scope of electrophiles that give one major product. Moreover, the reactions occurred with only unsubstituted trimethylsiloxyfuran. Asymmetric reactions with catalysts based on other metals could provide complementary selectivities to these palladium–catalyzed reactions. However, asymmetric allylic substitutions catalyzed by complexes of other metals with ester enolates lacking a second EWG have not been reported.
SCHEME 1.
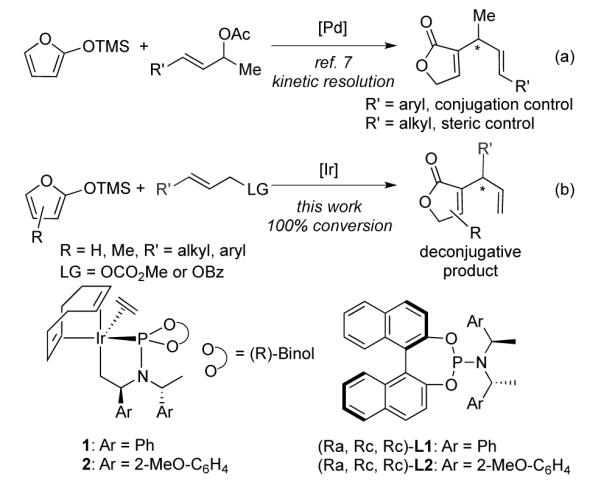
Palladium and iridium catalyzed allylic substitution with trimethylsiloxyfuran
We report an iridium catalyzed allylic substitution between trimethylsiloxyfuran and prochiral electrophiles to form enantioenriched 3-substituted butenolides with high regioselectivity for the more hindered product (b of Scheme 1), including formation of the deconjugated product from reactions of cinnamyl carbonates. The process furnishes 3-substituted butenolides containing an easily functionalized terminal double bond and various aryl and alkyl groups at the stereogenic center. In addition to providing useful butenolides, this process begins to address the challenge of conducting allylic substitution with ester enolates.
Our initial studies focused on the reaction of trimethylsiloxyfuran 3a with cinnamyl carbonate 4a. Table 1 summarizes the effect of several parameters on this reaction. Reactions catalyzed by complexes 1 and 2 with CsF to activate the silyl enolate formed the 3-allylated product 5a regioselectively.10, 11 The reaction with catalyst 2 occurred in higher yield than the reaction with catalyst 1 (entries 1 and 2). Further assessment of fluoride activators showed that reactions conducted with ZnF2 gave the desired product 5a in a high 85% isolated yield with 99% ee (entry 3). The reaction with soluble fluoride salts, such as TBAF and TBAT, gave only trace amounts of the desired product (entries 4 and 5). To our surprise, this reaction also proceeded to completion without any additives, although higher catalyst loadings were needed in this case (entries 6 and 7). This final observation is consistent with studies on stoichiometric reactions of catalytic intermediates in the absence of additives described later in this paper.
TABLE 1.
Effect of catalyst and fluoride source on the Ircatalyzed allylic substitution of trimethylsiloxyfurana
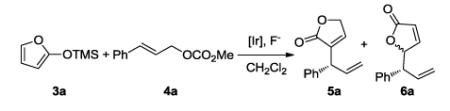
| Entry | Cat.b | Fluoride | Yield(%)c | 5a:6ad | ee(%)e |
|---|---|---|---|---|---|
| 1 | 1 | CsF | 13 | - | - |
| 2 | 2 | CsF | 32 | 20:1 | 99 |
| 3 | 2 | ZnF2 | 87(85) | 20:1 | 99 |
| 4f | 2 | TBAF | trace | - | - |
| 5 | 2 | TBAT | trace | - | - |
| 6 | 2 | None | 40 | 20:1 | 99 |
| 7g | 2 | None | 89 | 20:1 | 99 |
See supporting information for experimental details.
1 mol % Ir catalyst was used unless otherwise noted.
The yield was determined by 1H NMR analysis with mesitylene as the internal standard. The value in parentheses corresponds to the isolated yield.
The ratio was determined by 1H NMR analysis of crude reaction mixtures.
The ee was determined by chiral HPLC.
1-phenylallylalcohol was formed in 69% yield.
2 mol % Ir catalyst was used.
The scope of the Ir-catalyzed asymmetric allylation of trimethylsiloxyfuran in the presence of ZnF2 is summarized in Table 2. The reaction of the electron-rich, 4-methoxy cinnamyl carbonate 4b afforded the desired substitution product in good yield with exceptional enantioselectivity and high regioselectivity. The reactions of electron-poor substrates 4c-e furnished the corresponding products in high yield with excellent regio- and enantioselectivities, although 3-substituted cinnamyl carbonate 4e required 2 mol % catalyst loading for full conversion.
TABLE 2.
Iridium-catalyzed allylic substitution of trimethylsiloxyfuran 3aa
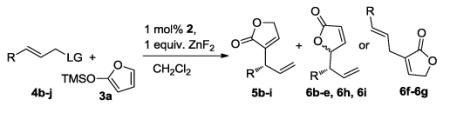
| Entry | R (4) | LG | Yield(%)b | 5/6 c | eed (%) |
|---|---|---|---|---|---|
| 1 | 4-MeO-C6H4 (4b) | OCO2Me | 5b, 70 | 20:1 | 98 |
| 2 | 4-F- C6H4 (4c) | OCO2Me | 5c, 83 | 20:1 | 95 |
| 3 | 4-Cl- C6H4 (4d) | OCO2Me | 5d, 78 | 20:1 | 97 |
| 4e | 3-F- C6H4 (4e) | OCO2Me | 5e, 91 | 20:1 | 97 |
| 5f | n-Propyl (4f) | OCO2Me | 5f, 18 | 10:1 | 99 |
| 6f,g | n-Propyl (4g) | OBz | 5f, 80 | 10:1 | 96 |
| 7 f,g | Methyl (4h) | OBz | 5g, 71 | 10:1 | 97 |
| 8 g | Cyclohexyl (4i) | OBz | 5h, 60 | 8:1 | 94 |
| 9 | 1-propenyl (4j) | OCO2Me | 5i, 90 | 1:1 | - |
| 10h | 1-propenyl (4j) | OCO2Me | 5i, 83 | 3:1 | 99 |
See supporting information for experimental details.
Isolated yield
Ratio was determined by 1H NMR analysis of the crude reaction mixtures.
ee was determined by chiral HPLC analysis.
2 mol % Ir catalyst 2 was used.
The products 6 are 3-substituted linear products.
The reaction was conducted with 3 mol % Ir catalyst 2 at 50 °C.
2 mol % [(dbcot)IrCl]2 was used.
Reactions of aliphatic allylic electrophiles occurred with some changes to the reaction conditions. Propyl-substituted allylic carbonate 4f gave only 18% yield of branched product 5f with a 10:1 ratio of 5f to linear product 6f (entry 5). However, the reaction of the corresponding aliphatic allylic benzoate 4g gave the branched allylation product 5f in good yield and regioselectivity (10:1) with high enantioselectivity (entry 6) in the presence of 3 mol % catalyst 2 at 50 °C.
These conditions were suitable for a range of aliphatic benzoates. For example, crotyl substrate 4h reacted to give the substitution product in 71% yield with excellent enantioselectivity (entry 7). Furthermore, the branched product formed selectively, even when the aliphatic substituent was branched at the carbon adjacent to the allyl unit (entry 8). The allylic substitution of dienyl benzoate 4j with catalyst 2 yielded the product as a 1:1 mixture of constitutional isomers (entry 9), but occurred with moderate regioselectivity and high enantioselec-tivity when dibenzo[a,e]cyclooctatetraene (DBCOT) was used as a supporting ligand instead of cyclooctadiene (COD).2
The reactions of 3, 4, and 5-methyl trimethylsiloxyfurans (3b-d) revealed the effect of furanyl substituents on the catalytic process. The reaction of 3-methyl trimethylsiloxyfuran 3b gave two products (Scheme 2). The 5-substituted linear product 6j was isolated in 28% yield and 94% ee. The 3-substituted product was obtained as inseparable diastereomers 5j and 5j’. Heating the mixture of diastereomers in dichloromethane at 40 °C for 1 h led to a single diastereomer 5j containing adjacent quaternary and tertiary stereogenic centers and linear product 6j. Product 6j formed by a Cope rearrangement of one of the diastereomers.12 Thus, the 3-substituted product 5j was ultimately isolated as a single diastereomer in 97% ee by a one-pot process involving asymmetric allylation and Cope rearrangement (Scheme 2, bottom).13
SCHEME 2.

Iridium-catalyzed allylic substitution with 3-methyl substituted trimethylsiloxyfuransa
a (a) 2 mol % (S, S, S)-2, ZnF2, CH2Cl2, 12 h; (b) 40 oC, CH2Cl2, 1 h
The reaction of 4-methyl trimethylsiloxyfuran 3c with cinnamyl carbonate 4a in the presence of the catalyst (S, S, S)-2 also gave products from allylic substitution (Scheme 3). The reaction of 3c produced the 3-substituted product in 47% yield, which consisted of double bond isomers 5k and 5k’, and the undesired 5-substituted product in 38% yield. We were able to convert isomer 5k’ to isomer 5k with a catalytic amount of O-desmethyl quinine under mild conditions.14 Thus, the overall process furnished the 3-substituted product 5k in 45% yield with 93% ee. Likewise, the reaction of 5-methyl trimethylsiloxyfuran 3d gave disubstituted butenolides 5l in an overall 88% yield with 6:1 diastereoselectivity in 99% ee after isomerization of the diastereomeric mixture of 5l’ to isomer 5l in the presence of O-desmethyl quinine.14
SCHEME 3.
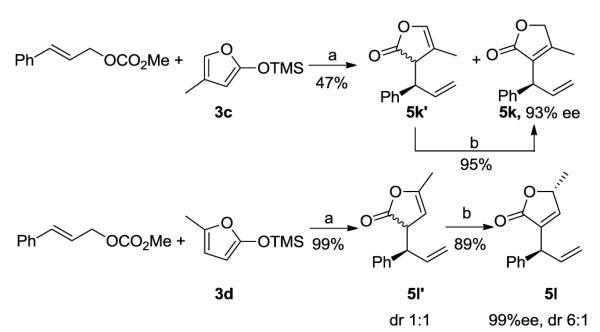
Iridium-catalyzed allylic substitution with 4- and 5-methyl substituted trimethylsiloxyfuransa
a (a) 2 mol % (S, S, S)-2, ZnF2, CH2Cl2, 12 h; (b) 20 mol % O-desmethyl quinine, CH2Cl2, 12 h
The products of these substitutions contained two double bonds that underwent further functionalizations (Scheme 4). For example, the terminal double bond of 5a underwent cross metathesis with vinyl boronate15 in the presence of the Grubbs-Hoveyda 2nd generation catalyst.16 The resulting vinyl boro-nate 7a underwent Suzuki-Miyaura coupling with bromoben-zene or 5-bromo-2-methylpyridine to yield 7b or 7c in the presence of Pd(Qphos)(crotyl)Cl. Compound 5g was converted to 7d by a cross metathesis with styrene catalyzed by Schrock’s catalyst.17 In addition, the electron deficient double bond in 5l served as a site for conjugate additions.18 Me2CuLi reacted with 5l to furnish product 7e containing four contiguous stereogenic centers.
SCHEME 4.
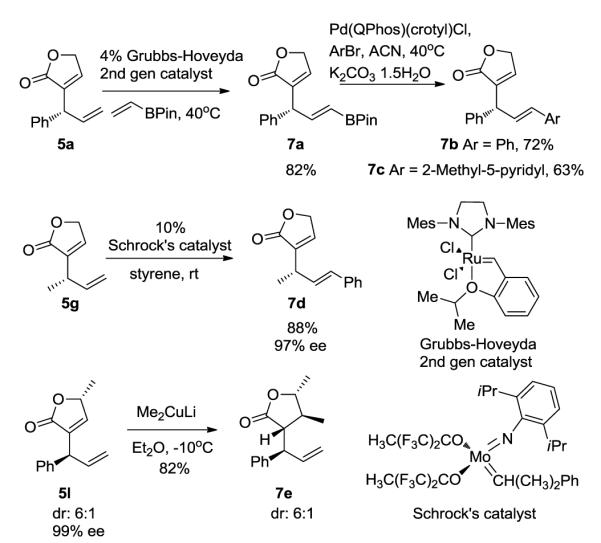
Derivatization of the butenolide producta
a See supporting information for experimental details. All yields reported here are isolated yields. Diastereomeric ratio was determined from 1H NMR spectra of the crude reaction mixtures. ee was determined by chiral HPLC analysis.
Stoichiometric reactions were conducted with the Irallylcomplex19 to gain insight into the mode of addition and effect of additives on this addition step. The isolated Ir-allyl complex did not react with 1.5 equiv of trimethylsiloxyfuran in the presence of ZnF2 alone (a of Scheme 5).20 However, the iridium complex reacted immediately in the presence of 1.0 equiv of NBu4OAc with or without ZnF2 to form the product 5a in 92% yield and 99% ee with the same absolute configuration as the product of the catalytic reaction (b of Scheme 5). This observation implies that the carbonate or benzoate generated in situ in the catalytic reaction activates the siloxyfuran. The R absolute configuration of the product of this stoichiometric reaction suggests that nucleophilic attack occurs at the face anti to the iridium fragment.
SCHEME 5.
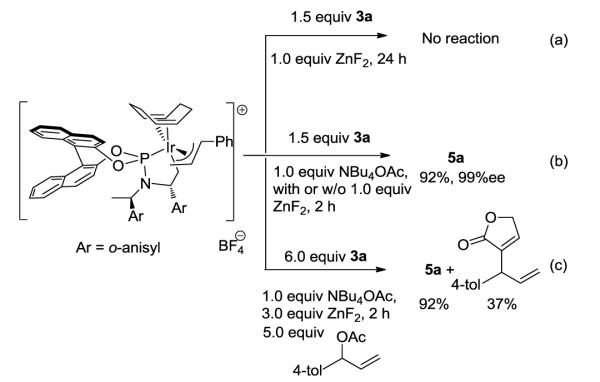
Stoichiometric reactions of the cinnamyl iridium intermediate with trimethylsiloxyfuran
To determine if the acetate anion activated the trimethylsiloxyfuran directly or if it reacted with Ir-allyl complex to release an allylic acetate that reacted in a subsequent catalytic process, the reaction of the allyliridium complex with trimethylsiloxyfuran 3a was conducted in the presence of 5.0 equiv of p-tolyl allylic acetate (c of Scheme 5). If the acetate anion led to the release of the free allylic acetate, little product from the allyl group on iridium would be observed in the presence of excess of the tolyl-substituted allylic acetate. In the event, the reaction of the Ir-allyl complex in the presence of excess of tolyl-substituted acetate formed phenyl-substituted product 5a in 92% yield. (The p-tolyl product formed in only 37% yield based on p-tolyl allylic acetate.) This yield of phenyl-substituted product 5a was the same as that formed in the absence of p-tolyl allylic acetate, and the yield of 5a was much higher than that of p-tolyl-substituted product. These observations imply that the trimethylsiloxyfuran reacts directly with the Ir-allyl complex when activated by a carboxylate, not by initial release of an allylic ester.
In summary, we report an iridium-catalyzed asymmetric allylic substitution reaction with a silyl ketene acetal. The reaction between a variety of aromatic and aliphatic allylic carbonates or benzoates and trimethylsiloxyfuran proceeded smoothly to furnish 3-substituted butenolides in excellent regio- and enantioselectivity. Moreover, methyl substituted trimethylsiloxyfurans react regioselectively to form enantioenriched products. These allylation products can be converted to an array of organic building blocks by reactions at one or the other of the alkene units of the product. Stoichiometric reactions of the allyliridium intermediate imply that the reaction proceeds by anti attack on the coordinated allyl ligand, as reported previously for iridium catalyzed allylic substitution with carbon and heteroatom nucleophiles,19a but that the siloxyfuran is activated by coordination of the carboxylate leaving group. Further studies to expand the scope of the reaction to encompass additional silyl ketene acetals are underway in this laboratory.
Supplementary Material
ACKNOWLEDGMENT
We thank the NIH (GM-58108) for support of this work, Johnson-Matthey for gifts of [Ir(COD)Cl]2, and Dr. Klaus Ditrich and BASF for gifts of chiral amines. We thank Dr. Phong V. Pham and Dr. Tyler W. Wilson for helpful discussions.
Footnotes
ASSOCIATED CONTENT Experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1) (a).Tosatti P, Nelson A, Marsden SP. Org. Biomol. Chem. 2012;10:3147. doi: 10.1039/c2ob07086c. [DOI] [PubMed] [Google Scholar]; (b) Liu W-B, Xia J-B, You S-L. Top. Organomet. Chem. 2012;38:155. [Google Scholar]; (c) Hartwig JF, Pouy MJ. Top. Organomet. Chem. 2011;34:169. [Google Scholar]; (d) Hartwig JF, Stanley LM. Acc. Chem. Res. 2010;43:1461. doi: 10.1021/ar100047x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Helmchen G. Iridium-catalyzed asymmetric allylic substitutions. Wiley-VCH Verlag GmbH & Co.; KGaA: 2009. p. 211. [Google Scholar]
- (2) (a).Stanley LM, Hartwig JF. J. Am. Chem. Soc. 2009;131:8971. doi: 10.1021/ja902243s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shu C, Leitner A, Hartwig JF. Angew. Chem. Int. Ed. 2004;43:4797. doi: 10.1002/anie.200460276. [DOI] [PubMed] [Google Scholar]; (c) Ohmura T, Hartwig JF. J. Am. Chem. Soc. 2002;124:15164. doi: 10.1021/ja028614m. [DOI] [PubMed] [Google Scholar]; (d Seehafer K, Helmchen G. J. Am. Chem. Soc. 2011;133:2072. doi: 10.1021/ja109953v. [DOI] [PubMed] [Google Scholar]; (e) Lyothier I, Defieber C, Carreira EM. Angew. Chem. Int. Ed. 2006;45:6204. doi: 10.1002/anie.200602408. [DOI] [PubMed] [Google Scholar]; (f) Shu C, Hartwig JF. Angew. Chem. Int. Ed. 2004;43:4794. doi: 10.1002/anie.200460214. [DOI] [PubMed] [Google Scholar]; (g) Ueda M, Hartwig JF. Org. Lett. 2010;12:92. doi: 10.1021/ol9023248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3) (a).Dübon P, Schelwies M, Helmchen G. Chem.-Eur. J. 2008;14:6722. doi: 10.1002/chem.200800495. [DOI] [PubMed] [Google Scholar]; (b) Schelwies M, Dübon P, Helmchen G. Angew. Chem. Int. Ed. 2006;45:2466. doi: 10.1002/anie.200503945. [DOI] [PubMed] [Google Scholar]; (c) Förster S, Tverskoy O, Helmchen G. Synlett. 2008:2803. [Google Scholar]; (d) Gnamm C, Förster S, Miller N, Brödner K, Helmchen G. Synlett. 2007:790. [Google Scholar]; (e) Dahnz A, Helmchen G. Synlett. 2006:697. [Google Scholar]; (f) Kanayama T, Yoshida K, Miyabe H, Takemoto Y. Angew. Chem. Int. Ed. 2003;42:2054. doi: 10.1002/anie.200250654. [DOI] [PubMed] [Google Scholar]; (g) Bartels B, Garcia-Yebra C, Helmchen G. Eur. J. Org. Chem. 2003:1097. [Google Scholar]; (h) Janssen JP, Helmchen G. Tetrahedron Lett. 1997;38:8025. [Google Scholar]; (i) Weix DJ, Hartwig JF. J. Am. Chem. Soc. 2007;129:7720. doi: 10.1021/ja071455s. [DOI] [PubMed] [Google Scholar]; (j) He H, Zheng X-J, Li Y, Dai L-X, You S-L. Org. Lett. 2007;9:4339. doi: 10.1021/ol7019394. [DOI] [PubMed] [Google Scholar]; (k) Graening T, Hartwig JF. J. Am. Chem. Soc. 2005;127:17192. doi: 10.1021/ja0566275. [DOI] [PubMed] [Google Scholar]; (l) Polet D, Rathgeb X, Falciola CA, Langlois J-B, El HS, Alexakis A. Chem.--Eur. J. 2009;15:1205. doi: 10.1002/chem.200801879. [DOI] [PubMed] [Google Scholar]; (m) Liu W-B, Zheng C, Zhuo C-X, Dai L-X, You S-L. J. Am. Chem. Soc. 2012;134:4812. doi: 10.1021/ja210923k. [DOI] [PubMed] [Google Scholar]; (n) Zhuo C-X, Liu W-B, Wu Q-F, You S-L. Chem. Sci. 2012;3:205. [Google Scholar]; (o) Wu Q-F, He H, Liu W-B, You S-L. J. Am. Chem. Soc. 2010;132:11418. doi: 10.1021/ja105111n. [DOI] [PubMed] [Google Scholar]
- (4) (a).Brown SP, Goodwin NC, MacMillan DWC. J. Am. Chem. Soc. 2003;125:1192. doi: 10.1021/ja029095q. [DOI] [PubMed] [Google Scholar]; (b) Carmen Zafra-Polo M, Figadère B, Gallardo T, Tormo J, Cortes D. Phytochemistry. 1998;48:1087. [Google Scholar]; (c) Kitson RRA, Millemaggi A, Taylor RJK. Angew. Chem. Int. Ed. 2009;48:9426. doi: 10.1002/anie.200903108. [DOI] [PubMed] [Google Scholar]
- (5) (a).Casiraghi G, Zanardi F, Appendino G, Rassu G. Chem. Rev. 2000;100:1929. doi: 10.1021/cr990247i. [DOI] [PubMed] [Google Scholar]; (b) Singh RP, Foxman BM, Deng L. J. Am. Chem. Soc. 2010;132:9558. doi: 10.1021/ja103331t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jiang Y-Q, Shi Y-L, Shi M. J. Am. Chem. Soc. 2008;130:7202. doi: 10.1021/ja802422d. [DOI] [PubMed] [Google Scholar]; (d) Szlosek M, Figadère B. Angew. Chem. Int. Ed. 2000;39:1799. doi: 10.1002/(sici)1521-3773(20000515)39:10<1799::aid-anie1799>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- (6).For a substrate controlled example, see: Fujioka H, Matsunaga N, Kitagawa H, Nagatomi Y, Kondo M, Kita Y. Tetrahedron: Asymmetry. 1995;6:2117.; for a racemic example, see: Boukouvalas J, Loach RP. J. Org. Chem. 2008;73:8109. doi: 10.1021/jo8015924.
- (7).Mao B, Ji Y, Fañanás-Mastral M, Caroli G, Meetsma A, Feringa BL. Angew. Chem. Int. Ed. 2012;51:3168. doi: 10.1002/anie.201109075. [DOI] [PubMed] [Google Scholar]
- (8) (a).Hayashi T, Yamamoto A, Hagihara T. J. Org. Chem. 1986;51:723. [Google Scholar]; (b) Branchadell V, Moreno-Mañas M, Pajuelo F, Pleixats R. Organometallics. 1999;18:4934. [Google Scholar]
- (9).Consistent with this origin of regioselectivity, we found that the palladium catalyzed reaction of cinnamyl acetate with 3a failed to deliver the desired product 5a. Instead, a mixture of 3-cinnamyl-2-furanone and 6a formed in a combined 30% yield. See supporting information for details.
- (10).The regioselectivity for reaction at the butenolide is distinct from several processes forming products from attack at C-5. Selectivities from reactions at C-5 have been rationalized by initial [4+2] cycloadditions with an alkene or carbonyl group. In our case, the siloxyfuran activated by an anionic group is more likely reacting as an enolate, which tends to react with electrophiles at the C-3.
- (11) (a).For proposed [4+2] mechanism to rationalize C-5 selectivity, see: Cho C-W, Krische MJ. Angew. Chem. Int. Ed. 2004;43:6689. doi: 10.1002/anie.200461381.; Brown DW, Campbell MM, Taylor AP, Zhang X.-a. Tetrahedron Lett. 1987;28:985.; for a computational study, see: López CS, Ålvarez R, Vaz B, Faza ON, de Lera ÅR. J. Org. Chem. 2005;70:3654. doi: 10.1021/jo0501339. For a C-3 Aldol reaction of lithium enolate, see 11b.
- (12).For a similar aza-Cope rearrangement, see: Kawatsura M, Tsuji H, Uchida K, Itoh T. Tetrahedron. 2011;67:7686.
- (13).The stereochemistry of 5j and 6j was tentatively assigned by analogy to the aza-Cope analogue in ref 12.
- (14).Wu Y, Singh RP, Deng L. J. Am. Chem. Soc. 2011;133:12458. doi: 10.1021/ja205674x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15) (a).Nicolaou KC, Li A, Edmonds DJ, Tria GS, Ellery SP. J. Am. Chem. Soc. 2009;131:16905. doi: 10.1021/ja9068003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Njardarson JT, Biswas K, Danishefsky SJ. Chem. Commun. 2002:2759. doi: 10.1039/b209941a. [DOI] [PubMed] [Google Scholar]
- (16).Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J. Am. Chem. Soc. 2000;122:8168. [Google Scholar]
- (17).Compound 7d was determined to have an S configuration by comparing the optical rotation of this material to a literature value in reference 7.
- (18) (a).Perlmutter P. Conjugate Addition Reactions in Organic Synthesis. Pergamon; Oxford, UK: 1992. [Google Scholar]; (b) Rosso GB, Pilli RA. Tetrahedron Lett. 2006;47:185. [Google Scholar]
- (19) (a).Madrahimov ST, Markovic D, Hartwig JF. J. Am. Chem. Soc. 2009;131:7228. doi: 10.1021/ja902609g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Raskatov JA, Spiess S, Gnamm C, Broedner K, Rominger F, Helmchen G. Chem.--Eur. J. 2010;16:6601. doi: 10.1002/chem.200903465. [DOI] [PubMed] [Google Scholar]
- (20).To understand the effect of ZnF2, 19F NMR experiments were conducted. The 19F NMR spectrum of the catalytic reaction (Table 1, entry 3) contained a peak at −156.9 ppm, which matches the 19F NMR resonance of TMSF. The same species was found in the mixture of ZnF2 and TMSOAc, but not in the mixture of ZnF2 and trimethylsiloxyfuran. These observations suggest that ZnF2 promotes the allylic substitution reaction by reacting with the trimethylsilylcarbonate formed in the reaction to release the carbonate, which then activates the trimethylsiloxyfuran.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.