

SUMMARY
Intravenous (i.v.) administration of encephalitogenic peptide can effectively prevent experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis; however, the underlying cellular and molecular mechanisms are not fully understood. In this study, we induced i.v. tolerance to EAE by administration of MOG35-55 peptide and determined the effect of this approach on intracellular signaling pathways of the IL-23/IL-17 system, which is essential for the pathogenesis of MS/EAE. In tolerized mice, phosphorylation of Janus kinase (Jak)/signal transducer and activator of transcription (STAT)-1, -4, ERK1/2 and NF-κBp65 were significantly reduced in splenocytes and the central nervous system. MOG i.v. treatment led to significantly lower production of IL-17, and administration of exogenous IL-17 slightly broke immune tolerance, which was associated with reduced activation of STAT4 and NF-κB. Suppressed phosphorylation of these pathway molecules was primarily evident in CD11b+ and small numbers of CD4+, CD8+ and CD11c+ cells. More importantly, adoptive transfer of CD11b+ splenocytes of tolerized mice effectively delayed onset and reduced clinical severity of actively induced EAE. This study correlates MOG i.v. tolerance with modulation of Jak/STATs signaling pathways and investigates novel therapeutic avenues for the treatment of EAE/MS.
Keywords: MOG i.v., EAE/MS, cytokine, intracellular signaling
INTRODUCTION
Multiple sclerosis (MS) is a chronic autoimmune disease of the central nervous system (CNS) that mainly affects young adults, and is generally diagnosed between the ages of 20 and 40 years [1]. Experimental autoimmune encephalomyelitis (EAE), induced by immunization of susceptible mouse strains with myelin oligodendrocyte glycoprotein peptides (MOG35-55) or other myelin components, provides a useful animal model for human multiple sclerosis. Self-reactive encephalitogenic CD4+ T cells are critically involved in the initiation and development of EAE. In addition to the role of self-reactive effector CD4+ T cells in the progression of EAE, other cell types such as macrophages, resident microglia and astrocytes also contribute to immunopathogenesis in MS/EAE. When activated, these cells can act as antigen-presenting cells (APCs), producing pro-inflammatory cytokines and chemokines in the local environment [2-3].
Thus far, there are no effective therapeutics that can stop the progression of MS or significantly restore neurological function. Soluble peptides representing encephalitogenic epitopes administered orally and via other routes have proved effective in suppressing EAE in mouse models during the induction phase of EAE [4-6]; however, the effectiveness of this strategy was elusive in clinical trials involving MS patients. Administration of a mixture of multiple encephalitogenic peptides has proved more effective than administration of a single peptide [7]. Several mechanisms have been proposed to explain the phenomenon of such immune tolerance. Evidence from animal models suggests that immune tolerance is associated with anergy and deletion of self-reactive T cells and generation of regulatory T cells, which can be induced by partial or complete blockade of costimulatory signaling [8-10]. In addition, immunomodulatory cytokines TGF-β and IL-10 are strongly associated with regulatory T cells and immune tolerance [9]. The transcription factor Foxp3 is critical to CD4+CD25+ regulatory T cell differentiation, probably through inhibition of NF-κB activation and nuclear translocation [11]. Thus, T cell anergy, deletion, generation of regulatory T cells and production of anti-inflammatory cytokines mediate immune tolerance in physiological and pathological conditions.
IL-17 is produced by a number of cell types including a subset of CD4+ T cells, Th17 cells, which have been linked with autoimmune and chronic inflammatory conditions [12-13]. Recently it has been found that IL-23-driven Th17 cells are encephalitogenic while TGF-β + IL-6-driven Th17 cells do not support EAE development. This is partially explained by IL-10 expression of the latter population [14]. IL-17 knockout mice show a significant, but not complete, reduction in severity of EAE, indicating an important, although not absolute, role of this cytokine in the pathogenesis of EAE [13]. Indeed, recent studies showed that both Th1 and Th17 cell responses are required for EAE development [15-16]. Suppression of IL-17 by immunoregulatory cytokines, such as newly defined IL-27, effectively suppresses EAE at least partially via upregulation of IL-10 [17-18]. However, whether inhibition of IL-17 is also involved in tolerance induction against EAE is not known.
Signal transducers and activators of transcription (STATs), NF-κB and ERK1/2 are known to regulate cytokine-dependent signal transduction pathways in autoimmune inflammation. Activation of STATs 1-6 is significantly enhanced in brain and spinal cord of EAE mice compared to healthy control mice [19-20]. Cytokine networks exert their pro- and anti-inflammatory effects through multiple downstream signaling pathways. JAK/Stat4 and NF-κB are critical members of the pathways involved in pro-inflammatory processes [21-22]. STAT4-/- mice or mice receiving STAT4-/- donor T cells failed to develop EAE [23-24]. Though the cytokine profiles and downstream molecules of the cytokine signal transduction pathway have been investigated in EAE, the involvement of these molecules in i.v. autoantigen-induced immune tolerance has not been fully defined. In this study, we analyzed the cytokine profiles and JAK/STAT signaling pathways after MOG i.v. treatment in the induction phase of EAE. We observed decreased IL-17 and IFN-γ, but increased IL-27 and IL-10 in the MOG i.v. induced tolerant state. In addition, we found that JAK/STAT4 and NF-κB signaling pathways were significantly up-regulated in EAE, while their activation was significantly suppressed in MOG i.v.-induced immune tolerance. The regulatory roles and contributions of these pathways to immune tolerance were further investigated.
RESULTS
MOG i.v. inhibits IL-17 and suppresses EAE
To induce an immune tolerance mouse model of EAE, mice were s.c. immunized with MOG35-55 peptide in CFA and injected i.v. with MOG35-55 (150 μg/injection) at days 0, 3 and 6 post immunization (p.i.) (EAE + MOG i.v.) or PBS as control (EAE + PBS i.v.). The initial onset of EAE in non-tolerized mice was observed at day 12-14 p.i., reaching the maximum clinical score after 5-6 days with no recovery. However, only very mild clinical signs of EAE were observed in MOG i.v. treated mice (Fig. 1A). Consistent with clinical signs, demyelination and extensive inflammatory infiltrates were mostly observed in white matter of spinal cords of PBS-treated EAE mice, while normal morphology was preserved in MOG i.v. treated mice (data not shown). To quantitatively analyze cytokine profiles in immune tolerance, we harvested supernatants of splenocytes from control EAE or tolerized mice after 3 days of MOG35-55 peptide stimulation and measured the expression level of a range of cytokines by ELISA. Our results showed that IL-17 and IFN-γ were elevated in spleen cultures of EAE + PBS i.v. mice (2102.8 ± 306.2 pg/ml and 1503.5 ± 123.5 pg/ml, respectively), compared to naïve controls; however IL-17 and IFN-γ production was significantly inhibited in the EAE + MOG i.v. group (649.5 ± 69.3 pg/ml and 621.1 ± 141.3 pg/ml, respectively) compared to EAE + PBS i.v. (Fig. 1B). IL-4 and IL-5 production was undetectable in both groups (data not shown). In addition, we observed elevated IL-27p28 (1152.5 ± 208.3 vs. 425.5 ± 80.6 pg/ml) and IL-10 (129.4 ± 16.8 pg/ml vs. 8.4 ± 12.0 pg/ml) in tolerized mice compared to EAE controls (Fig. 1B). Taken together, these results suggest an association of decreased IL-17 and IFN-γ but enhanced IL-10 and IL-27 with immune tolerance.
Fig. 1. MOG35-55 i.v. inhibited IL-17 and the development of EAE.
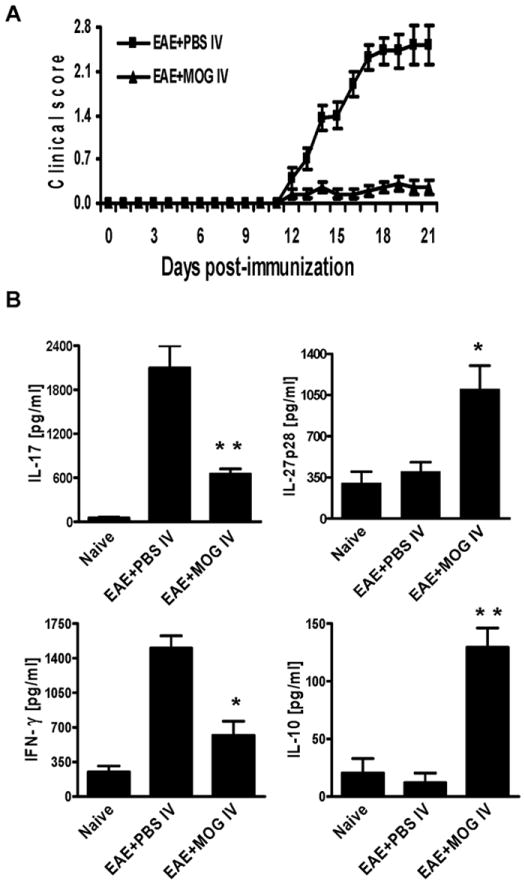
EAE was induced in B6 mice by immunization with MOG35-55/CFA and injection of pertussis toxin on days 0 and 2. At days 0, 3, and 6 p.i., mice were i.v. injected with 150 μg MOG35-55 peptide or the same volume of PBS to serve as EAE control mice. (A). Daily clinical scores of each mouse group (n = 10 each group; pooled from two independent experiments). (B) Cytokine expression in culture supernatants. 5 ×106 splenocytes in duplicate from mice in (A) were stimulated with 10 μg/ml MOG35-55 for 72 hrs. Cell-free supernatants were collected and analyzed for the expression of IL-17, IFN-γ, IL-27p28 and IL-10 by ELISA. Data were pooled from two independent experiments and presented as mean value/group ± s.e.m. (n =10 each group). * p<0.05; ** p<0.01 as comparison between EAE + PBS i.v. and EAE + MOG i.v. mice.
Exogenous IL-17 partially blocks MOG-i.v. induced tolerance in EAE
As Th17 cells play an important role in EAE pathogenesis we profiled these cells in each group by intracellular flow cytometric analysis. We found that following reactivation with MOG35-55 in vitro, propotions of Th17 cells (CD4+IL-17+) were significantly decreased in spleen cells from i.v. tolerized mice (Fig. 2A), which was in agreement with our findings by ELISA. Thus, MOG i.v-induced immune tolerance and EAE suppression are associated with inhibition of self-reactive Th17 cells. To determine whether immune tolerance and subsequent EAE suppression by MOG i.v. treatment is associated with inhibition of self-reactive T cell proliferation we analyzed autoantigen–induced T cell proliferation and the effect of exogenous IL-17 on this process. We found that MOG-specific T cell proliferation was substantially inhibited in mice treated with MOG i.v. compared to EAE controls. Addition of exogenous IL-17 partially, but significantly, reversed this inhibition, suggesting that suppression of IL-17 may play a role in inhibition of proliferation (Fig. 2B). To further define the immunological role of IL-17 suppression in immune tolerance, we administered exogenous IL-17 i.p. in parallel with i.v. injection of MOG35-55. We found that exogenous IL-17 exacerbated EAE and significantly, albeit partially, abrogated the therapeutic effects of MOG i.v. on EAE. (Fig. 2C, Table 1).
Fig. 2. Exogenous IL-17 partially negates the therapeutic effect of MOG i.v.
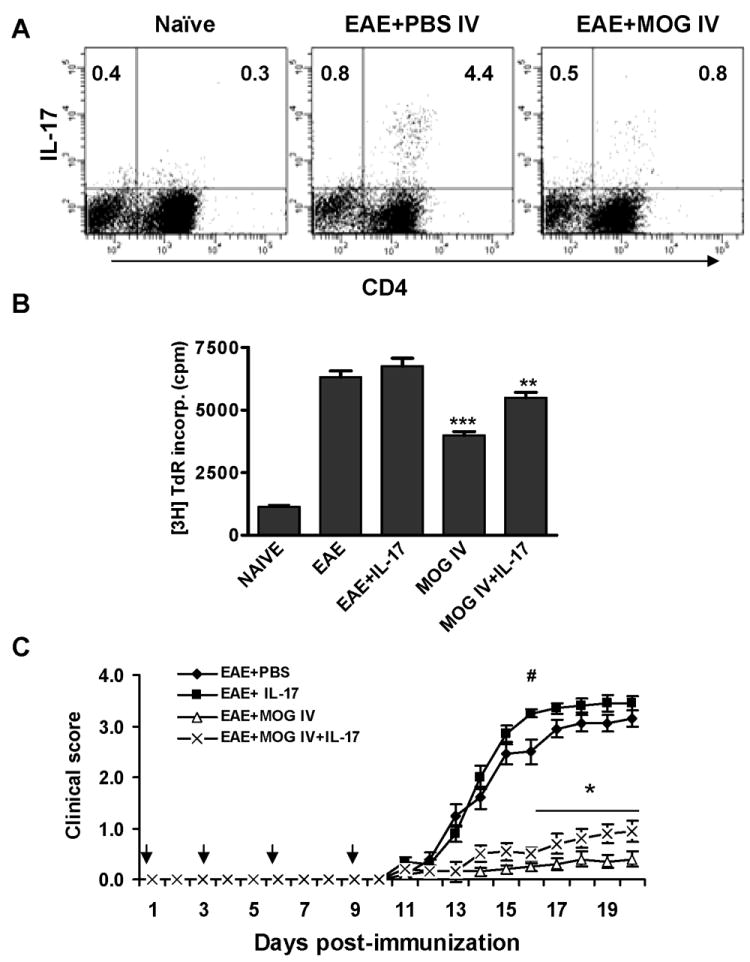
EAE was induced in C57BL/6 mice by immunization with MOG35-55/CFA and administration of pertussis toxin on days 0 and 2. At days 0, 3, and 6 p.i., mice were i.v. injected with 150 μg MOG35-55 peptide (EAE + MOG i.v.) to induce i.v. tolerance. (A) Th17 cell proportions in total splenocytes from control EAE or tolerized mice that were restimulated with MOG35-55 for three days before staining. (B) Proliferation assay of auto-reactive T cells. 5 ×105 splenocytes from mice at 21 days p.i. were stimulated with 10 μg/ml MOG35-55 with or without recombinant IL-17 (1 ng/ml) for 60 hrs, pulsed with 1 μCi 3H thymidine/well for 18 hrs, and thymidine incorporation was measured. Data were pooled from two independent experiments and presented as mean counts per minute (cpm) in each group ± s.e.m. (n =10 each group). ** p<0.01; *** p<0.001 as compared to EAE +PBS i.v. mice. (C) Simultaneously with MOG i.v. injection, exogenous IL-17 (200 ng/mouse; EAE+MOG i.v. + IL-17) or PBS (control; EAE + MOG i.v.) was injected i.p. Immunized mice were treated with the same doses of IL-17 (EAE + IL-17), or PBS as control (EAE + PBS). Data were pooled from two independent experiments and presented as mean clinical score/group ± s.e.m. (n =10 each group). * p<0.05 for comparison between EAE + MOG-i.v. and EAE + MOG-i.v. + IL-17 mice; # p<0.05 for comparison between EAE + PBS and EAE + IL-17 mice.
Table 1.
Clinical parameters of EAE after i.v. tolerance and administration of exogenous IL-17.
| Mice | Incidence (%)a | Mean day of disease onset (± SEM) b | Mean maximum score (± SEM) | Mean cumulative score (± SEM) c | Area under curve (± SEM) |
|---|---|---|---|---|---|
| EAE + PBS (n = 10) | 100 | 12.3 ± 0.2 | 3.3 ± 0.2 | 20.4 ± 1.1 | 18.9 ± 1.1 |
| EAE + IL-17 (n = 10) | 100 | 12.0 ± 0.4 | 3.6 ± 0.1 | 22.9 ± 0.9 * | 21.6 ± 0.9 * |
| EAE + MOG i.v. (n = 10) | 50 | 16.8 ± 1.1 | 0.4 ± 0.1 | 2.4 ± 0.9 | 2.2 ± 0.8 |
| EAE +MOG i.v. + IL-17 (n = 10) | 90 | 15.1 ± 0.9 | 1.2 ± 0.2 # | 5.2 ± 1.0 # | 4.7 ± 1.0 # |
EAE and i.v. tolerance induction and IL-17 administration was described in Fig. 2 and in Materials and Methods section.
Disease incidence is defined as the percentage of mice that displayed any clinical signs of disease.
Mean day of disease onset is defined as the first day of 2 consecutive days with a clinical score of 0.5 or more.
Mean cumulative score is defined as the mean of the sum of daily clinical scorers observed between days 12 and 20.
* refers to comparison between EAE + PBS and EAE + IL-17; # refers to comparison between EAE + MOG i.v. and EAE + MOG i.v. + IL-17. * and #, p<0.05.
MOG i.v. inhibits phosphorylation of STAT4, STAT1, ERK1/2 and NF-κB
It has been shown that STAT4 and NF-κB regulate the production of a range of cytokines in autoimmune responses and promote auto-reactive T cell differentiation and activation of APCs [21-22]. To determine which intracellular signaling pathways are modulated in MOG i.v.-induced immune tolerance, we analyzed the phosphorylation profiles in splenocytes and spinal cords by flow cytometry and immunohistochemical staining. We found basal tyrosine phosphorylation of STAT1 (5.4 ± 0.3%), STAT4 (8.3 ± 1.0%), ERK1/2 (7.8 ± 0.1%) and NF-κBp65 (7.9 ± 0.3%) in splenocytes of naïve control mice; however, phosphorylation of STAT1, STAT4, ERK1/2 and NF-κBp65 in spleen was significantly elevated to 22.5 ± 1.9%, 26.4 ± 1.8%, 25.6 ± 0.3% and 23.8 ± 0.8%, respectively, at the peak of EAE. In contrast, in comparison to spleen of control EAE mice, phosphorylation of STAT1, STAT4, ERK1/2, and NF-κBp65 was significantly decreased to 14.6 ± 0.2 %, 15.9 ± 0.9%, 17.8 ± 0.7% and 10.7 ± 0.4%, respectively, after MOG i.v. treatment (Fig. 3A, B). Consistent with our findings in splenocytes, proportions of cells that stained positive for intracellular phosphorylated STAT1 and STAT4 were significantly enhanced in spinal cords of non-tolerized EAE mice compared to naïve mice. In tolerized mice, the proportions of these cells were significantly reduced compared to non-tolerized mice (Fig. 4A and B; p<0.05).
Fig. 3. MOG35 i.v. inhibited tyrosine phosphorylation of STAT4 and NF-κB in splenocytes.
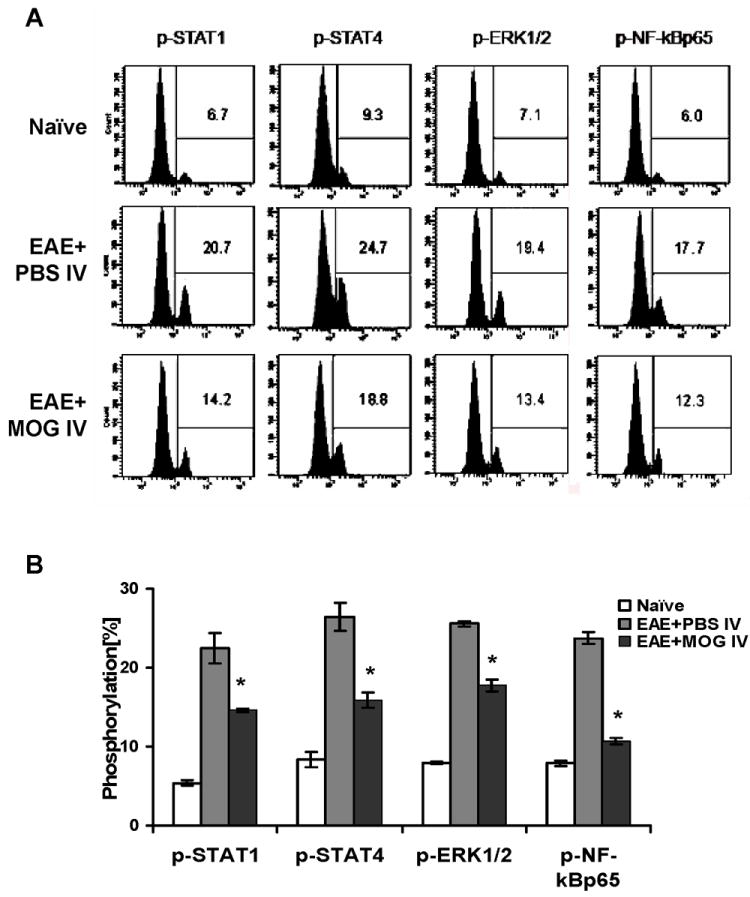
2-3 × 106 spleen cells from mice at day 21 p.i. were stimulated in vitro with 10 μg/ml MOG35-55 for 12 hrs. Cells were harvested and incubated with antibodies against p-STAT1, p-STAT4, p-ERK1/2, p-NF-κBp65 for flow cytometric analysis. One representative data set of 3 experiments is shown in (A). (B) Statistical analysis of phosphorylation in each group. Data were pooled from two independent experiments and presented as mean percentage of positive populations in splenocytes per group ± s.e.m. (n =10 each group). * p<0.05 as comparison between EAE + PBS i.v. and EAE + MOG i.v. mice.
Fig. 4. Detection of phosphorylation of STAT1 and STAT4 protein in spinal cords.
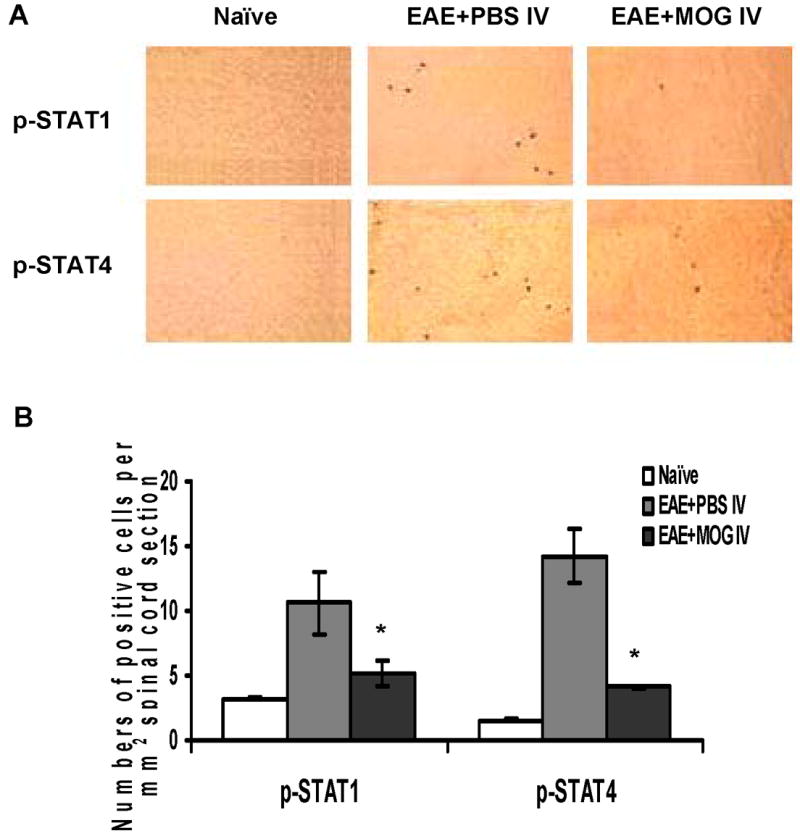
Cryosections of lumbar spinal cord from mice collected at day 21 p.i. were incubated with rabbit anti-p-STAT1 or goat anti-p-STAT4 antibodies followed by incubation with HRP-conjugated secondary antibody. Sections were developed in DAB and visualized by light microscopy. (A) One representative immunostaining for p-STAT1 and p-STAT4 in each group is shown, magnification (× 200). (B) Positive cells were quantitatively assessed under high-power microscopy (× 400) using a gridded eyepiece. Results were pooled from two independent experiments and expressed as numbers of positive cells per mm2 of spinal cord section (n =10 each group). * p<0.05, as comparison between EAE +PBS i.v. and EAE + MOG i.v. mice.
IL-17 activates STAT4 in CD11b+ cells in vitro
To address whether IL-17 affected the JAK/STAT4 signaling pathway in tolerized and non-tolerized EAE mice, splenocytes of these mice were treated with different doses of recombinant mouse IL-17, and analyzed by flow cytometry. We found that IL-17 enhanced tyrosine phosphorylation of STAT4 in splenocytes from EAE control and EAE + MOG i.v. mice (Fig. 5A). Similar results were obtained for STAT1 and NF-κB, whereas IL-17 had no effect on ERK1/2 activation in vitro (data not shown). To identify cells with elevated phosphorylation of STAT4 we stained splenocytes from EAE mice for a range of cell surface markers including CD4, CD8, CD11b and CD11c, in conjunction with intracellular phosphorylated STAT4 staining. Phosphorylation of STAT4 was almost completely confined to CD11b+ splenocytes (19.3%), with a small proportion staining positive for CD4 (0.5%), CD8 (0.9%) and CD11c (0.6%) (Fig. 5B; representative plot). A similar cellular profile of p-STAT4 expressing cells was observed in splenocytes of both PBS-i.v. and MOG-i.v. mice, pre- and post-IL-17 stimulation (data not shown). Together, these results indicate that i.v. tolerance-induced regulation of STAT4 and other IL-23/IL-17 pathways mostly occurs in CD11b+ cells and a small number of CD4+, CD8+ T cells and DCs.
Fig. 5. IL-17 enhanced phosphorylation of STAT4 in CD11b+ splenocytes in vitro.
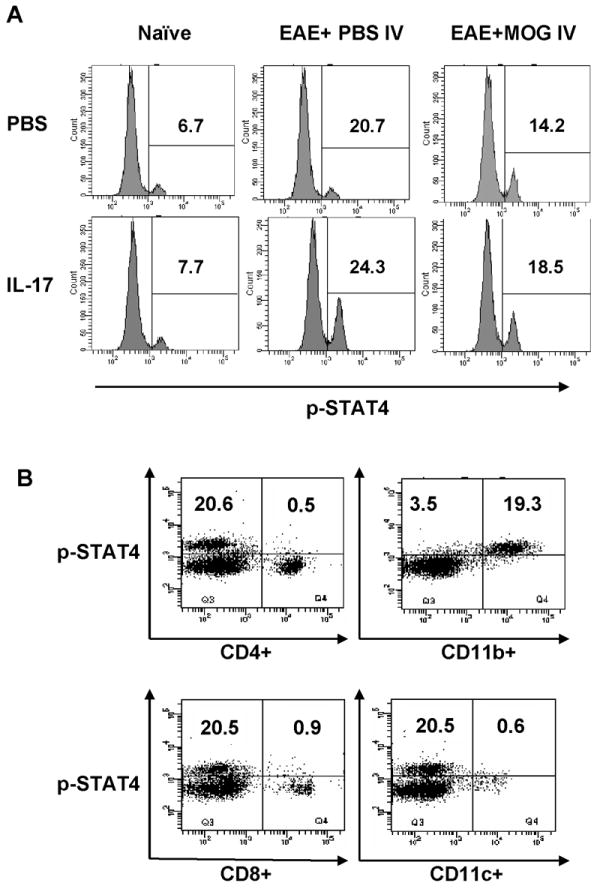
(A) 2-3 × 106 splenocytes from naïve mice, EAE + MOG i.v. mice or EAE +PBS i.v. mice were treated with or without 1 ng/ml IL-17 for 60 min, then incubated with fluorescent antibodies against p-STAT4 for flow cytometric analysis. Expression of p-STAT4+ splenocytes of these groups. (B) Identification of the cellular origin of gated p-STAT4+ cells. Splenocytes were treated as described in (A) and p-STAT4+ cells were gated for expression of CD4, CD8, CD11c, and CD11b. The flow cytometry plots shown in (B) are for cells from EAE + PBS i.v. mice before IL-17 stimulation. These data serve as an example, a similar cell origin pattern having been observed in both PBS-i.v. and MOG-i.v. mice, both pre- and post-IL-17 stimulation (data not shown).
Adoptive transfer of CD11b+ cells of tolerized mice ameliorated EAE
To confirm the biological significance of inhibition of STAT4 phosphorylation in macrophages induced by i.v. tolerance and to determine whether these cells are tolerogenic in recipient mice, we purified CD11b+ splenocytes from MOG i.v. tolerized mice at day 10 after the last MOG i.v. injection by FACS. These cells were transferred to recipient mice that had been immunized to develop EAE 10 days p.i., at which time myelin-reactive T cells had been activated. We found significantly reduced clinical severity in mice that received CD11b+ cells from tolerized mice compared to those that received these cells from naïve or EAE mice (Fig. 6A). There was an approximately 1.8-2-fold decrease in p-STAT4 and p-ERK1/2 phosphorylation observed in splenocytes of mice that received CD11b+ cells from tolerized mice (Fig. 6B). These results suggest that suppression of JAK/STAT and NF-κB signaling in CD11b+ cells may be involved in i.v. tolerance induction.
Fig. 6. Adoptive transfer of tolerized mouse-derived CD11b+ cells ameliorates EAE.
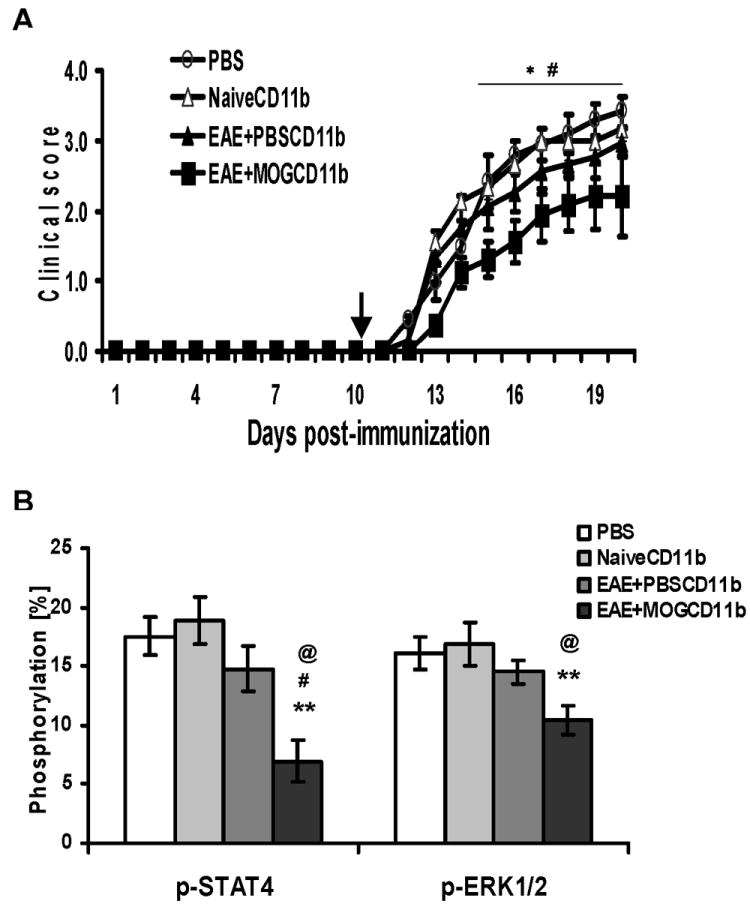
Splenocytes from MOG i.v. or PBS i.v.-treated mice at day 10 p.i. or from untreated naïve B6 mice as negative control were stimulated in vitro with 10 μg/ml MOG35-55 for 3 days, and CD11b+ cells were purified by FACS (purity >97%). 2 × 106 purified cells or the same volume of PBS as a negative control were injected i.v. into B6 recipient mice at day 10 p.i. (A) Mean daily clinical scores ± s.e.m. of each group (n = 10 each group; pooled from two independent experiments). * refers to comparison between EAE mice that received CD11b+ cells of MOG i.v. mice (EAE + MOGCD11b) and naïve mice (NaïveCD11b); # refers to comparison between EAE mice that received CD11b+ cells from MOG i.v. mice (EAE + MOGCD11b) and EAE + PBS i.v. mice (EAE + PBSCD11b). (B) Mice were sacrificed at week 3 p.i. Activation of STAT4 and ERK1/2 in recipient total splenocytes was analyzed by flow cytometry and showed suppressed phosphorylation of STAT4 and ERK1/2 in recipient mice after adoptive transfer. *, comparison between EAE mice that received CD11b+ cells of MOG i.v. mice (EAE + MOGCD11b) and naïve mice (NaïveCD11b); #, comparison between EAE mice that received CD11b+ cells from MOG i.v. mice (EAE + MOGCD11b) and EAE + PBS i.v. mice (EAE + PBSCD11b); @, comparison between EAE mice that received CD11b+ cells from MOG i.v. mice (EAE + MOGCD11b) and PBS only. # and @, P<0.05; **, P<0.01.
DISCUSSION
Previous work has demonstrated that oral or i.v. administration of myelin antigens or their peptides induces tolerance and ameliorates EAE in mice [4, 25]. The mechanisms underlying tolerance induction may include physical elimination (clonal deletion) and/or functional inactivation (anergy) of autoantigen-reactive T cells [4, 25-26], and subpopulation dysregulation by the induction of Th2 immunoregulatory cells [8, 27]. However, the molecular mechanisms for induction of tolerance are not fully understood. It has been found that administration of high-dose autoantigen reduced mRNA expression of the proinflammatory molecules TNF-α and inducible NO synthase (iNOS) [10]]. Eagar et al. found that the CTLA-4 pathway is required to maintain the unresponsive state of tolerized T cells upon autoantigenic stimulation under inflammatory conditions [28]. More recently, it has been found that the Ca++-regulated calcuineurin/NFAT cascade plays a decisive role in the generation of peripheral tolerance against self-antigens [29]. In the current study we induced immune tolerance and prevented EAE in B6 mice by repetitive i.v. administration of MOG peptide after immunization. The cytokine profile and intracellular signaling transduction pathways of self-reactive T cells were investigated. We found that i.v. antigen-induced immune tolerance is associated with suppression of JAK/STATs signaling pathways and IL-17. Furthermore, increased IL-27p28 and IL-10, which are known to suppress IL-17 [17-18], were observed in tolerized mice, suggesting that these cytokines may be candidate upstream mediators of tolerance-induced suppression of IL-17.
IL-17 is expressed by a unique lineage of CD4+ T cells (Th17), which develop in response to cytokines including TGF-β, IL-6 and IL-23 [14, 30-31]. IL-17 stimulates the production and expression of other pro-inflammatory cytokines such as IL-1β and TNF-α by macrophages [32]. IL-17 production is significantly elevated in MS patients and in EAE mouse models [33-36]. Recent studies showed that IL-17 plays an important role in the development of autoimmune diseases [11] including EAE, as demonstrated by reduced EAE severity in IL-17-/- mice [13]. In this study we found decreased IL-17 production in tolerized mice, associated with suppressed phosphorylation of STAT4 and other JAK/STAT signaling members. Furthermore, addition of exogenous IL-17 partially reversed the suppression of autoantigen-induced T cell proliferative responses and clinical EAE in tolerized mice. These results provide supportive evidence for the important role of IL-17 in EAE pathogenesis, and help to elucidate a potential mechanism underlying the immunoregulatory effect of i.v. tolerance in autoimmune disorders such as EAE.
Although Th1 cells and their hallmark cytokine IFN-γ may exert suppressive effects on autoimmune disorders by an IFN-γ-NO-apoptosis pathway [37], the important role of this cell population and IFN-γ in the development of EAE has recently been re-emphasized. Kroenke et al. showed that adoptive transfer of either IL-12p70-polarized Th1 or IL-23-polarized Th17 cells into naive syngeneic mice resulted in clinically indistinguishable EAE in these two groups [16]. Th1-driven disease was characterized by macrophage-rich infiltrates and prominent NOS2 up-regulation. In contrast, neutrophils and granulocyte-colony-stimulating factor (CSF) were prominent in Th17-driven lesions. Differential chemokine profiles have also been observed in infiltrating Th1 and Th17 cells [16]. Stromnes et al. [15] demonstrate that while both Th1 and Th17 cells are required for EAE pathogenesis, the Th17/Th1 ratio of infiltrating T cells determines where inflammation occurs in the CNS. IL-17-/- mice show a partially reduced severity of EAE, indicating the involvement of other pathogenic factor(s), one of which may be IFN-γ [13]. Suppression of EAE by tolerance induction correlates with the inhibition of Th1 cells and IFN-γ production [38]. There is growing evidence that myelin-specific Th17 cells and/or IL-17/IFN-γ double-producing cells are critical effectors in EAE induced by the active immunization of C57BL/6 mice with MOG35–55 [16, 39-40]. It has also been found that IFN-γ producing Th1 cells are the major source of IL-17 in EAE [41]. Further, a supportive role for IFN-γ in migration and function of Th17 cells has recently been reported [42-43]. The finding in the present study that both IFN-γ and IL-17 are increased in EAE and significantly suppressed in tolerized mice suggests a pathogenic role of both Th1 and Th17 cytokines in EAE.
STAT4 is an important member of the JAK/STAT pathway, which is critically involved in initiating immune responses in inflammatory conditions [19-20]. The key role of STAT4 in initiating EAE is apparent in STAT4 knock-out mice, which do not develop EAE [23]. In addition to STAT4, we analyzed other important members of the signaling transduction pathway, including NF-κB, which is known to be critically involved in dendritic cell maturation, T cell activation and initiation of inflammatory immune responses [44]. In this study, we observed that NF-κB activity was markedly enhanced in EAE, but was significantly suppressed in MOG i.v. tolerized mice, demonstrating that NF-κB activation was differentially modulated in EAE progression and suppression.
Furthermore, we investigated STAT1 and ERK1/2, which are associated with Th1/Th2 polarization and regulation of cytokine production in T cells and APCs [45-46]. Similar to STAT4 and NF-κB, we observed enhanced activation of STAT1 and ERK1/2 in spleen cells of EAE mice, but activation of these molecules was suppressed in tolerized mice, consistent with findings in mice treated with 1,25 Dihydroxyvitamin-D3 [47]. Of note is that STAT1, STAT5 and ERK1/2 signaling were reported important for both pro-inflammatory and anti-inflammatory cytokine production, demonstrating both pro-inflammatory and regulatory functions under physiological and pathological conditions. For instance, ERK1/2 knock-out mice are more susceptible to EAE and display a shift from Th2 toward Th1 responses [46]. The contributions of STAT1 and ERK1/2 to immune tolerance will be further investigated in future studies.
While IL-17 has been found to play an important role in initiating EAE and IL-10 is a major cytokine in EAE suppression [48-49], we and others have recently shown a potent inhibitory effect of IL-27 in both actively induced and adoptively transferred EAE [18, 50-52]. Significantly elevated production of IL-10 and IL-27 in MOG i.v. tolerized mice suggests a regulatory role for these cytokines in the induction of MOG i.v. immune tolerance. Further, upregulation of IL-10 is considered to be a crucial mechanism underlying IL-27-induced suppression of IL-17 and adoptively transferred EAE [18]. Thus, we speculate that IL-10 and IL-27 may directly or indirectly participate in immune modulation by inhibiting IL-17 production in MOG i.v. induced immune tolerance.
Macrophages and dendritic cells (DCs) are the two most important types of professional APCs, with similar functions and cytokine profiles [53-54]. However, it has been found that, upon similar stimulation, such as with LPS + IFN-γ, DCs produced a low level of IL-10, while macrophages produced high levels of this anti-inflammatory cytokine. This indicates that macrophages may have a bias towards tolerance induction [55]. In the current study, we found increased phosphorylation of STAT4 and other intracellular signaling molecules in EAE. The phosphorylation of intracellular signaling molecules is observed primarily in CD11b+ cells, most of which are macrophages, and in small proportions of DCs, CD4+ and CD8+ T cells. After i.v. tolerance, phosphorylation of these signaling molecules was significantly inhibited. More importantly, adoptive transfer of CD11b+ splenocytes derived from tolerized mice significantly inhibited EAE in recipient mice. These results suggest that, in addition to the primary role of T cells in tolerance induction [8-9], CD11b+ cells also play an important part in the induction of i.v. tolerance, and diminished phosphorylation of STAT4 and other JAK/STAT signaling members after i.v. antigen may be one of the mechanisms underlying this process.
In summary, we conclude that i.v. administration of MOG peptide suppressed IL-17 production and JAK/STAT and NF-κB signaling, leading to immune tolerance and attenuation of EAE. These findings identify IL-17 and JAK/STAT signaling as targets in regulation of EAE by i.v. tolerance and may lead to strategies for modulating IL-17 signaling in EAE/MS treatment.
MATERIALS AND METHODS
Animals
Female C57BL/6 mice (8-10 week-old) were purchased from The Jackson Laboratory (Bar Harbor, ME). All studies were approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University.
Induction and treatment of EAE
Mice were subcutaneously (s.c.) injected with 150 μg MOG35-55 (Invitrogen, Carlsbad, CA) emulsified in CFA (Difco Lab, Detroit, MI) containing 4 mg/ml Mycobacterium tuberculosis H37Ra (Difco, Detroit, MI) at two sites on the back. A total of 200 ng pertussis toxin (List Biological Lab, Epsom, England) was given intraperitoneally (i.p.) on days 0 and 2 p.i. To induce tolerance, 150 μg MOG35-55/mouse in 100 μl of PBS was i.v. injected via the tail vein on days 0, 3 and 6 p.i. The same volume of PBS was injected into a parallel group of mice as control. For treatment of mice with exogenous cytokine, IL-17 (200 ng per mouse) was injected i.p. on days 0, 3, 6 and 9 p.i. Animals were observed daily for clinical signs and scored as follows [56]: 1, limp tail or waddling gait with tail tonicity; 2, waddling gait with limp tail (ataxia); 2.5, ataxia with partial limb paralysis; 3, full paralysis of one limb; 3.5, full paralysis of one limb with partial paralysis of a second limb; 4, full paralysis of two limbs; 4.5, moribund; and 5, death.
T cell proliferation assay
Triplicate aliquots of 5 ×105 splenocytes in a 200 μl volume were cultured in 96-well round-bottom microtiter plates (Nunc, Naperville, IL), and stimulated with 10 μg/ml MOG35-55. To determine whether IL-17 would reverse suppressed T cell proliferative responses after i.v. tolerance, exogenous murine IL-17 (1 ng/ml) was added to the culture in parallel. This concentration of IL-17 was chosen based on our optimization studies (data not shown). After 60 hrs of incubation, cells were pulsed with 1 μCi 3H thymidine/well (Amersham, Little Chalont, UK) for 18 hrs; the cells were then harvested on fiberglass filters, and thymidine incorporation was measured using a beta-1205 scintillation counter (Pharmacia Biotech).
Cytokine profiles
Splenocytes were prepared from harvested spleens by forcing the tissue through a sterile 70-μm nylon cell strainer (Falcon 2350, Lincoln Park, NJ) to generate a single-cell suspension. 5 ×106 splenocytes in duplicate were cultured in RPMI1640 with 10% FBS in 24-well plates and either left unstimulated or stimulated with 10 μg/ml MOG35-55 for 3 days. Cell-free supernatants were collected and analyzed for the production of IL-17, IFN-γ, IL-27p28 and IL-10 by ELISA. Anti-mouse IFN-γ, IL-10 antibodies were purchased from BD Bioscience, Rockville, MD. Mouse IL-27p28 and IL-17 ELISA kits were purchased from R&D Systems Inc, Minneapolis, MN.
Immunohistochemical staining
Cryostat sections (7 μm) were prepared from frozen spinal cord samples, washed with PBS, and incubated with 4% paraformaldyhyde and permeabilization buffer (BD PharMingen) for 10 min, then blocked with 10% goat serum in PBS for 1 hour. Primary antibodies rabbit anti-p-STAT1 (1:100 dilution) or goat anti-p-STAT4 (1:100 dilution) (Santa Cruz) were added to sections overnight at 4°C. After being washed 3 times with PBS, sections were incubated with appropriate HRP-conjugated polyclonal second antibody (1:1500 dilution) or HRP-avidin (1:1000 dilution) (Jackson ImmunoResearch Laboratories, West Grove, PA) at room temperature for 1 hour. After washing three times, color development was performed with a nickel-cobalt-diaminobenzidine (DAB) Kit (Vector). Sections were visualized using light microscopy and scored quantitatively under 400 × magnification using a gridded eyepiece. Results are expressed as number of positive cells per mm2 spinal cord section.
Flow cytometry
To determine Th17 cells after i.v. tolerance, splenocytes of each group were pre-stimulated with MOG35-55 for three days before staining. After staining of the cell surface marker CD4, intracellular IL-17 was stained and analyzed by flow cytometry. For the analysis of STAT4 and other IL-23/IL-17 related signaling pathways, 2-3 × 106 cells from spleens were stimulated in vitro with 10 μg/ml MOG35-55 for 12 hrs, then washed with 3% FBS/PBS buffer. 0.5 ×106 cells were then incubated with mAbs (0.5 μg/sample) against cell surface proteins CD4, CD8, CD11b, and CD11c (BD Bioscience, San Diego, CA) for 30 min, washed twice, followed by fixation in PBS/4% paraformaldehyde for 10 min and permeabilized with Permeabilization Buffer (BD Bioscience) for 5 min. Cells were then incubated with rabbit anti-p-STAT1 antibody (Tyr701) (1:100 dilution), rabbit anti-phospho-ERK1/2 (Thr202/Tyr204) antibody (1:100 dilution) (Cell Signaling Technology Inc, Danvers, MA), goat anti-p-STAT4 antibody (Ser 721) (1:100 dilution) (Santa Cruz, Inc. Santa Cruz, CA), rabbit anti-p-NF-κBp65 (Ser536) (1:200 dilution) (Cell Signaling) for 30 min at 4°C, followed by the addition of secondary Cy3 conjugated anti-goat antibody (1:500 dilution) or Rhodamine conjugated anti-rabbit antibody (1:200 dilution) (Jackson ImmunoResearch Laboratories, West Grove, PA) for 20 min at 4°C. After being washed twice, the stained cells were analyzed on a FACSAria flow cytometer with CellQuest software (BD Bioscience).
Adoptive transfer of tolerized mouse-derived CD11b+ splenocytes
Splenocytes from MOG i.v.- and PBS i.v.-treated mice at day 10 p.i. or from untreated naïve B6 mice (as a negative control) were stimulated in vitro with 10 μg/ml MOG35-55 for 3 days, and CD11b+ splenocytes were purified from different groups of mice by FACS sorting (purity >97%). 2 × 106 purified cells were injected into MOG/CFA-immunized B6 recipient mice (n =10) at day 10 p.i. via the tail vein. EAE mice that received PBS only served as another control group. Clinical scores were recorded daily. At week 3 p.i., recipient mice were sacrificed and splenocytes were isolated. Activation of STAT4 and ERK1/2 in these cells was analyzed by flow cytometry.
Statistical analysis
EAE experiments were analyzed by calculating the area under the curve for each mouse over the clinical period of the experiment and performing statistical analysis on each group using these values. All experiments were tested for statistical significance using unpaired, two-tailed, Student’s t tests. Differences were considered significant at a value of p < 0.05.
Acknowledgments
This work was supported by grants from the National Institutes of Health, the National Multiple Sclerosis Society, and the Groff Foundation. We would like to thank Katherine Regan for editorial assistance.
Abbreviations
- MOG
myelin oligodendrocyte glycoprotein
- MNCs
mononuclear cells
References
- 1.Correale J, Tenembaum SN. Myelin basic protein and myelin oligodendrocyte glycoprotein T-cell repertoire in childhood and juvenile multiple sclerosis. Mult Scler. 2006;12:412–420. doi: 10.1191/135248506ms1282oa. [DOI] [PubMed] [Google Scholar]
- 2.Maier J, Kincaid C, Pagenstecher A, Campbell IL. Regulation of signal transducer and activator of transcription and suppressor of cytokine-signaling gene expression in the brain of mice with astrocyte-targeted production of interleukin-12 or experimental autoimmune encephalomyelitis. Am J Pathol. 2002;160:271–288. doi: 10.1016/S0002-9440(10)64371-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miljkovic D, Momcilovic M, Stojanovic I, Stosic-Grujicic S, Ramic Z, Mostarica-Stojkovic M. Astrocytes stimulate interleukin-17 and interferon-gamma production in vitro. J Neurosci Res. 2007;85:3598–3606. doi: 10.1002/jnr.21453. [DOI] [PubMed] [Google Scholar]
- 4.Critchfield JM, Racke MK, Zuniga-Pflucker JC, Cannella B, Raine CS, Goverman J, Lenardo MJ. T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science. 1994;263:1139–1143. doi: 10.1126/science.7509084. [DOI] [PubMed] [Google Scholar]
- 5.Whitacre CC, Gienapp IE, Orosz CG, Bitar DM. Oral tolerance in experimental autoimmune encephalomyelitis. III. Evidence for clonal anergy. J Immunol. 1991;147:2155–2163. [PubMed] [Google Scholar]
- 6.Zhang GX, Xu H, Kishi M, Calida D, Rostami A. The role of IL-12 in the induction of intravenous tolerance in experimental autoimmune encephalomyelitis. J Immunol. 2002;168:2501–2507. doi: 10.4049/jimmunol.168.5.2501. [DOI] [PubMed] [Google Scholar]
- 7.Smith CE, Miller SD. Multi-peptide coupled-cell tolerance ameliorates ongoing relapsing EAE associated with multiple pathogenic autoreactivities. J Autoimmun. 2006;27:218–231. doi: 10.1016/j.jaut.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 9.Miller A, Lider O, Roberts AB, Sporn MB, Weiner HL. Suppressor T cells generated by oral tolerization to myelin basic protein suppress both in vitro and in vivo immune responses by the release of transforming growth factor beta after antigen-specific triggering. Proc Natl Acad Sci U S A. 1992;89:421–425. doi: 10.1073/pnas.89.1.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weishaupt A, Jander S, Bruck W, Kuhlmann T, Stienekemeier M, Hartung T, Toyka KV, Stoll G, Gold R. Molecular mechanisms of high-dose antigen therapy in experimental autoimmune encephalomyelitis: rapid induction of Th1-type cytokines and inducible nitric oxide synthase. J Immunol. 2000;165:7157–7163. doi: 10.4049/jimmunol.165.12.7157. [DOI] [PubMed] [Google Scholar]
- 11.Bettelli E, Dastrange M, Oukka M. Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci U S A. 2005;102:5138–5143. doi: 10.1073/pnas.0501675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aarvak T, Chabaud M, Miossec P, Natvig JB. IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol. 1999;162:1246–1251. [PubMed] [Google Scholar]
- 13.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 14.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 15.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, Lee J, de Sauvage FJ, Ghilardi N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 18.Fitzgerald DC, Ciric B, Touil T, Harle H, Grammatikopolou J, Das Sarma J, Gran B, Zhang GX, Rostami A. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;179:3268–3275. doi: 10.4049/jimmunol.179.5.3268. [DOI] [PubMed] [Google Scholar]
- 19.Jee Y, Kim G, Tanuma N, Matsumoto Y. STAT expression and localization in the central nervous system during autoimmune encephalomyelitis in Lewis rats. J Neuroimmunol. 2001;114:40–47. doi: 10.1016/s0165-5728(00)00446-x. [DOI] [PubMed] [Google Scholar]
- 20.Nishikomori R, Usui T, Wu CY, Morinobu A, O’Shea JJ, Strober W. Activated STAT4 has an essential role in Th1 differentiation and proliferation that is independent of its role in the maintenance of IL-12R beta 2 chain expression and signaling. J Immunol. 2002;169:4388–4398. doi: 10.4049/jimmunol.169.8.4388. [DOI] [PubMed] [Google Scholar]
- 21.Subramaniam SV, Cooper RS, Adunyah SE. Evidence for the involvement of JAK/STAT pathway in the signaling mechanism of interleukin-17. Biochem Biophys Res Commun. 1999;262:14–19. doi: 10.1006/bbrc.1999.1156. [DOI] [PubMed] [Google Scholar]
- 22.Greve B, Weissert R, Hamdi N, Bettelli E, Sobel RA, Coyle A, Kuchroo VK, Rajewsky K, Schmidt-Supprian M. I kappa B kinase 2/beta deficiency controls expansion of autoreactive T cells and suppresses experimental autoimmune encephalomyelitis. J Immunol. 2007;179:179–185. doi: 10.4049/jimmunol.179.1.179. [DOI] [PubMed] [Google Scholar]
- 23.Boyton RJ, Davies S, Marden C, Fantino C, Reynolds C, Portugal K, Dewchand H, Altmann DM. Stat4-null non-obese diabetic mice: protection from diabetes and experimental allergic encephalomyelitis, but with concomitant epitope spread. Int Immunol. 2005;17:1157–1165. doi: 10.1093/intimm/dxh293. [DOI] [PubMed] [Google Scholar]
- 24.Chitnis T, Najafian N, Benou C, Salama AD, Grusby MJ, Sayegh MH, Khoury SJ. Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest. 2001;108:739–747. doi: 10.1172/JCI12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burkhart C, Liu GY, Anderton SM, Metzler B, Wraith DC. Peptide-induced T cell regulation of experimental autoimmune encephalomyelitis: a role for IL-10. Int Immunol. 1999;11:1625–1634. doi: 10.1093/intimm/11.10.1625. [DOI] [PubMed] [Google Scholar]
- 26.Ratts RB, Arredondo LR, Bittner P, Perrin PJ, Lovett-Racke AE, Racke MK. The role of CTLA-4 in tolerance induction and ttigen administration cell differentiation in experimental autoimmune encephalomyelitis: i. v. antigen administration. Int Immunol. 1999;11:1889–1896. doi: 10.1093/intimm/11.12.1889. [DOI] [PubMed] [Google Scholar]
- 27.Hilliard BA, Kamoun M, Ventura E, Rostami A. Mechanisms of suppression of experimental autoimmune encephalomyelitis by intravenous administration of myelin basic protein: role of regulatory spleen cells. Exp Mol Pathol. 2000;68:29–37. doi: 10.1006/exmp.1999.2290. [DOI] [PubMed] [Google Scholar]
- 28.Eagar TN, Karandikar NJ, Bluestone JA, Miller SD. The role of CTLA-4 in induction and maintenance of peripheral T cell tolerance. Eur J Immunol. 2002;32:972–981. doi: 10.1002/1521-4141(200204)32:4<972::AID-IMMU972>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 29.Sakaguchi S, Wing K, Miyara M. Regulatory T cells - a brief history and perspective. Eur J Immunol. 2007;37(Suppl 1):S116–123. doi: 10.1002/eji.200737593. [DOI] [PubMed] [Google Scholar]
- 30.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 31.Vanden Eijnden S, Goriely S, De Wit D, Willems F, Goldman M. IL-23 up-regulates IL-10 and induces IL-17 synthesis by polyclonally activated naive T cells in human. Eur J Immunol. 2005;35:469–475. doi: 10.1002/eji.200425677. [DOI] [PubMed] [Google Scholar]
- 32.Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998;160:3513–3521. [PubMed] [Google Scholar]
- 33.Hofstetter HH, Toyka KV, Tary-Lehmann M, Lehmann PV. Kinetics and organ distribution of IL-17-producing CD4 cells in proteolipid protein 139-151 peptide-induced experimental autoimmune encephalomyelitis of SJL mice. J Immunol. 2007;178:1372–1378. doi: 10.4049/jimmunol.178.3.1372. [DOI] [PubMed] [Google Scholar]
- 34.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 37.Tarrant TK, Silver PB, Wahlsten JL, Rizzo LV, Chan CC, Wiggert B, Caspi RR. Interleukin 12 protects from a T helper type 1-mediated autoimmune disease, experimental autoimmune uveitis, through a mechanism involving interferon gamma, nitric oxide, and apoptosis. J Exp Med. 1999;189:219–230. doi: 10.1084/jem.189.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang GX, Yu S, Calida D, Zhao Z, Gran B, Kamoun M, Rostami A. Loss of the surface antigen 3G11 characterizes a distinct population of anergic/regulatory T cells in experimental autoimmune encephalomyelitis. J Immunol. 2006;176:3366–3373. doi: 10.4049/jimmunol.176.6.3366. [DOI] [PubMed] [Google Scholar]
- 39.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Natur. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 40.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suryani S, Sutton I. An interferon-gamma-producing Th1 subset is the major source of IL-17 in experimental autoimmune encephalitis. J Neuroimmunol. 2007;183:96–103. doi: 10.1016/j.jneuroim.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 42.Kryczek I, Bruce AT, Gudjonsson JE, Johnston A, Aphale A, Vatan L, Szeliga W, Wang Y, Liu Y, Welling TH, Elder JT, Zou W. Induction of IL-17+ T cell trafficking and development by IFN-gamma: mechanism and pathological relevance in psoriasis. J Immunol. 2008;181:4733–41. doi: 10.4049/jimmunol.181.7.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Connor RA, Prendergast CT, Sabatos CA, Lau CW, Leech MD, Wraith DC, Anderton SM. Cutting Edge: Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. 2008;181:3750–4. doi: 10.4049/jimmunol.181.6.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giannoukakis N, Bonham CA, Qian S, Chen Z, Peng L, Harnaha J, Li W, Thomson AW, Fung JJ, Robbins PD, Lu L. Prolongation of cardiac allograft survival using dendritic cells treated with NF-kB decoy oligodeoxyribonucleotides. Mol Ther. 2000;1:430–437. doi: 10.1006/mthe.2000.0060. [DOI] [PubMed] [Google Scholar]
- 45.Anderson P, Sundstedt A, Li L, O’Neill EJ, Li S, Wraith DC, Wang P. Differential activation of signal transducer and activator of transcription (STAT)3 and STAT5 and induction of suppressors of cytokine signalling in T(h)1 and T(h)2 cells. Int Immunol. 2003;15:1309–1317. doi: 10.1093/intimm/dxg130. [DOI] [PubMed] [Google Scholar]
- 46.Agrawal A, Dillon S, Denning TL, Pulendran B. ERK1-/- mice exhibit Th1 cell polarization and increased susceptibility to experimental autoimmune encephalomyelitis. J Immunol. 2006;176:5788–5796. doi: 10.4049/jimmunol.176.10.5788. [DOI] [PubMed] [Google Scholar]
- 47.Muthian G, Raikwar HP, Rajasingh J, Bright JJ. 1,25 Dihydroxyvitamin-D3 modulates JAK-STAT pathway in IL-12/IFNgamma axis leading to Th1 response in experimental allergic encephalomyelitis. J Neurosci Res. 2006;83:1299–1309. doi: 10.1002/jnr.20826. [DOI] [PubMed] [Google Scholar]
- 48.Slavin AJ, Maron R, Weiner HL. Mucosal administration of IL-10 enhances oral tolerance in autoimmune encephalomyelitis and diabetes. Int Immunol. 2001;13:825–833. doi: 10.1093/intimm/13.6.825. [DOI] [PubMed] [Google Scholar]
- 49.Mekala DJ, Alli RS, Geiger TL. IL-10-dependent infectious tolerance after the treatment of experimental allergic encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Proc Natl Acad Sci U S A. 2005;102:11817–11822. doi: 10.1073/pnas.0505445102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fitzgerald DC, Zhang GX, El-Behi M, Fonseca-Kelly Z, Li H, Yu S, Saris CJ, Gran B, Ciric B, Rostami A. Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat Immunol. 2007;8:1372–1379. doi: 10.1038/ni1540. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Wang G, Sun B, Li H, Mu L, Wang Q, Li G, Shi L, Jin L, Kostulas N. Interleukin-27 suppresses experimental autoimmune encephalomyelitis during bone marrow stromal cell treatment. J Autoimmun. 2008;30:222–9. doi: 10.1016/j.jaut.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 52.Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. 2008;118:1680–90. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee HK, Iwasaki A. Innate control of adaptive immunity: dendritic cells and beyond. Semin Immunol. 2007;19:48–55. doi: 10.1016/j.smim.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 54.Unanue ER. Antigen-presenting function of the macrophage. Annu Rev Immunol. 1984;2:395–428. doi: 10.1146/annurev.iy.02.040184.002143. [DOI] [PubMed] [Google Scholar]
- 55.Smith W, Feldmann M, Londei M. Human macrophages induced in vitro by macrophage colony-stimulating factor are deficient in IL-12 production. Eur J Immunol. 1998;28:2498–2507. doi: 10.1002/(SICI)1521-4141(199808)28:08<2498::AID-IMMU2498>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 56.Benson JM, Campbell KA, Guan Z, Gienapp IE, Stuckman SS, Forsthuber T, Whitacre CC. T-cell activation and receptor downmodulation precede deletion induced by mucosally administered antigen. J Clin Invest. 2000;106:1031–1038. doi: 10.1172/JCI10738. [DOI] [PMC free article] [PubMed] [Google Scholar]