

SUMMARY
We sought to explain seasonality and other aspects of Campylobacter jejuni epidemiology by integrating population genetic and epidemiological analysis in a large 3-year longitudinal, two-centre, population-based study. Epidemiological information was collected for 1505 isolates, which were multilocus sequence-typed. Analyses compared pathogen population structure between areas, over time, and between clinical presentations. Pooled analysis was performed with published international datasets. Subtype association with virulence was not observed. UK sites had nearly identical C. jejuni populations. A clade formed by ST45 and ST283 clonal complexes showed a summer peak. This clade was common in a Finnish dataset but not in New Zealand and Australian collections, countries with less marked seasonality. The UK, New Zealand and Australian collections were otherwise similar. These findings map to known in-vitro differences of this clade. This identifies a target for studies to elucidate the drivers of the summer peak in human C. jejuni infection.
Key words: Campylobacter, epidemiology, genetics, gastrointestinal infections
INTRODUCTION
Campylobacter is the most common bacterial cause of gastroenteritis in industrialized and non-industrialized countries [1]. Between 40 000 and 60 000 laboratory-confirmed cases in England and Wales annually [2] are estimated to represent over 300 000 infections [3], similar to other industrialized countries [1]. About 93% of laboratory-confirmed cases of campylobacteriosis in England and Wales are due to Campylobacter jejuni [4]. Phenotypic subtyping has made limited impact in understanding this infection with weak prediction of genetic relatedness by assayed phenotypes [5]. Multilocus sequence typing (MLST) has been applied to human C. jejuni isolate collections from many countries [6–12] and to non-human animal isolates [6, 9, 13–19]. This has allowed genetic attribution of human infections to source [13, 14, 18], which indicates that MLST may also support investigation of basic patterns of epidemiology such as temporal and spatial distribution. The explanation for many of these basic epidemiological features remains elusive.
A summer peak in Campylobacter is consistently reported from temperate countries [1, 20–22] often doubling the weekly incidence at the same time each year [22] with later peaks in colder countries [21, 22]. Exceptions include Estonia and Germany with less distinct or repeatable summer peaks [21, 22]. Seasonality may be driven by exposure to differing sources or varying Campylobacter prevalence in sources of infection with a lack of empirical evidence to differentiate between these. Prevalence of different multilocus sequence types varies between reservoirs [13, 14, 18]. A 1-year UK human study noted a peak of ST45 complex in C. jejuni isolates during that summer [7]. The temporal pattern of C. jejuni subtypes infecting humans may thus help to explain seasonality.
Reported Campylobacter population structures have varied among countries. These differences could represent geographical structuring, seasonal patterns, or transient expansions of particular clones [8, 12]. A common-source local outbreak is suggested as the source of a sequence type (ST) common in a New Zealand collection [12]. The same main clonal complexes contribute to human disease in much of the world, which finding is less affected by sampling concerns than observed differences. Substantial published multilocus sequence-typed human isolate collections from Australia [11], New Zealand [12] and Finland [10] allow direct comparison of infecting subtypes. Joint analysis may clarify similarities and differences in international epidemiology.
Incidence rates of C. jejuni vary by age group [23]. Higher incidence in males than in females is usual [24–26]. Variation in subtype with age and gender has not been studied apart from one study reporting increased serotype diversity with older age [27].
Several open questions thus persist in the basic epidemiology of C. jejuni. The integration of MLST in epidemiological analysis of structured longitudinal samples and joint analysis of data from separate studies may address some of these questions. This study applied MLST to a two-site longitudinal sample of C. jejuni in humans and extended this by joint analysis with published international datasets. The UK sample was a 3-year population-based isolate collection with accompanying descriptive epidemiological information. The international collections used were those with fully published isolate details. The main questions addressed were whether population structure changed systematically over a 3-year period; varied seasonally; correlated with demography or symptoms; was distinguishable in geographically separate UK populations; and varied among countries.
METHODS
Study population
The Campylobacter Sentinel Surveillance Scheme (CSSS) ran between May 2000 and April 2003. Laboratories referred all isolates to the Health Protection Agency (HPA) reference laboratory. Demographic, clinical and exposure data was collected by postal questionnaire [28]. Two populations in the CSSS, Nottingham and Southampton and parts of their surrounding counties, were selected because they were geographically separated, 168 miles apart, and recruitment had been successful and relatively stable. Isolates linked to a questionnaire with complete postcode and no recent international travel were included. Travel-related cases were excluded to maximize the data available to study endemic disease within available resources. Post-code completeness exceeded 96% apart from the Nottingham area during the first year when it was 72%. These criteria identified 1596 isolates, 885 from Nottinghamshire and 711 from Hampshire. No Campylobacter outbreaks were identified in these populations during the study period.
Laboratory methods
The HPA reference laboratory supplied all available C. jejuni isolates. Amplification and sequencing used methods and primers from published protocols [16, 29] and the MLST website (http://pubmlst.org/). Sequence extension reactions used BigDye™ reaction mix at 1/32 of the manufacturer's recommended quantities [29]. Products were detected on an ABI Prism 3730 DNA analyser. Traces were assembled using Sequence Typing Analysis and Retrieval System software developed by Dr Man-Suen Chan and Dr Nicki Ventress [(http://sara.molbiol.ox.ac.uk/userweb/mchan/stars/adminguide.htm) © University of Oxford, 2001]. Failed sequences were assembled manually or repeated. Consensus were queried against the pubMLST Campylobacter database (http://pubmlst.org/campylobacter/) to give an allele number and sequence types using standard Campylobacter MLST nomenclature [29]. Genotypes are available from the pubMLST database.
International published human disease data
Published structured human disease datasets from defined time and place were from populations in Australia [11], New Zealand [12] and Finland [10] (Table 1). The Australian dataset was from a population-based case-control study of sporadic cases of Campylobacter in the catchment area of two laboratories in New South Wales. The New Zealand isolates were consecutive isolates form the hospital laboratories serving eight district health board populations in New Zealand excluding repeat isolates from individuals or members of the same family. The Finnish isolates were from domestically acquired human sporadic cases of gastroenteritis collected at Helsinki University Central Hospital Laboratory during separate years.
Table 1.
C. jejuni isolates by country and year assembled from published literature
| Year | Country | ||
|---|---|---|---|
| Australia | Finland | New Zealand | |
| 1999–2001 | 153 | ||
| 1996 | 92 | ||
| 2002 | 109 | ||
| 2003 | 97 | ||
| 2006 | 107 | ||
Analysis
Geographical distribution of sequence types
The two English populations were compared and then combined for comparison with the datasets from other countries. Comparison was by χ2 test for clonal complex distribution, FST fixation indices calculated by Arlequin at the nucleotide and allele level for gene pools [30] and Simpson's index for diversity [31]. Pairwise FST measures the extent to which isolates differ between populations compared to within populations. A value of 0 indicates identical population (i.e. no genetic differentiation) and a value of 1 complete genetic separation. It is generally used to assess short-term genetic distances between populations [30].
Temporal distribution of sequence types within the UK dataset
Subtype distribution was compared between peak and non-peak periods. A peak day of year was identified by Poisson regression of daily counts modelling seasonal and secular variation using date in sine and cosine terms and in a linear term [32]. The 50% of isolates closest to this day of year were considered peak and others non-peak. A χ2 test on clonal complex and FST tests were then performed comparing peak and non-peak populations. Having confirmed significant variation, a harmonic logistic regression model was used to explore seasonal patterns by clonal complex in more detail and without the assumption that seasonality was limited to the presence of absence of a summer peak. This modelled season using sine and cosine coefficients for day of year and with year modelled as both a continuous and categorical variable to assess nonlinear effects of year. Isolates from each clonal complex were compared to all other isolates. For example, to assess seasonal ST45 complex distribution compared to other isolates the model was:
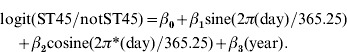 |
Age and gender distribution of genotypes within the UK dataset
Subtype distribution was compared by gender, between older (aged ⩾60 years) and younger (aged <30 years) people, and between children (aged <18 years) and adults using the same analyses as for geographical analyses.
Genotype and symptoms in the UK dataset
The association of genotype with presence of particular symptoms and with illness duration was tested using logistic and linear regression models, respectively. Bivariate and age-adjusted analyses were performed.
Population genetic analysis in the UK dataset
A phylogeny was estimated using ClonalFrame [33] a model-based algorithm inferring phylogeny from sequence data assuming a clonal genealogy with recombination. The model assumption that distant sequence blocks are not affected by single recombination events holds for C. jejuni [34]. The assumption that recombination is from outside the dataset studied can lead to incorrect inference of relatedness in deeper branches [33]. These distant relationships are not therefore considered. Analysis used 50 000 burn-in and 50 000 subsequent iterations, sampling each 100th iteration. Three runs inferred similar structure and were combined into a 65% consensus tree.
RESULTS
Of 1596 cases, the laboratory sample was missing or contaminated for 36 (2·3%) cases. Six isolates proved to be C. coli on MLST and seven mixed cultures (with at least two alleles for MLST one locus) that could not be resolved. Of the remaining 1547 C. jejuni isolates 1505 were typed by MLST (Table 2). Profiles were incomplete for 42 where template was exhausted before completion of typing. Allele distributions and demographic characteristics of cases with complete profiles and partial profiles were similar.
Table 2.
Typed isolates by location and study year
| Year | Nottingham | Hampshire | Total |
|---|---|---|---|
| May 2000–April 2001 | 316 | 276 | 592 |
| May 2001–April 2002 | 292 | 262 | 554 |
| May 2002–April 2003 | 188 | 112 | 300 |
| Total | 796 | 650 | 1446* |
Cases reporting date of onset of illness. Of the 59 cases not recording onset date an estimated date based on laboratory data indicated onset of 17, 30 and 12 cases in years 1, 2 and 3, respectively.
Geographically separate UK populations
The clonal complex distribution in the two populations is almost identical (Fig. 1) with no statistical support for variation between them at clonal complex (χ2, P = 0·79) nucleotide (FST = 0, P = 0·4), or allele (FST = 0, P = 0·6) levels. Simpson's Index for sequence type was 0·95 in each population. The two sentinel site populations are combined as English C. jejuni isolates in comparisons below with other countries.
Fig. 1.

[colour online]. Clonal complex distribution of 1505 isolates in two geographically distinct UK populations, May 2000–April 2003 (likelihood ratio χ2 test, P = 0·79). n.a., Not assigned.
The FST values are highest between Finland and each of the other three countries and show that the Finnish C. jejuni population is distinct from the others (Table 3). There was also substantial variation of the C. jejuni clonal complex distribution among countries (Fig. 2; χ2, P < 0·0001). Some clonal complexes were widely distributed with ST21 complex the most common in England, second most common in Finland and New Zealand and fourth in Australia. ST48 complex was the most abundant in Australia and New Zealand, fourth in England and fifth in Finland. However, in Finland 46% of isolates were ST45 complex, ranging between 40% and 53% in the 3 years studied, which was fivefold higher than England, and tenfold higher than Australia or New Zealand. Within the ST45 complex, ST45, the central genotype, comprised 62% of isolates and ST137 12%, with no marked differences among countries in these proportions. The ST677 complex comprising 16/92 isolates in 1996 and 21/206 in 2002–2003 in Finland was otherwise rare with eight (0·5%) isolates in England and none from Australia or New Zealand. By contrast, ST257 complex was rare in Finland (one isolate) but comprised 14%, 13% and 5% of isolates in Australia, England, and New Zealand, respectively. There were 50 ST574 isolates in England with this sequence type absent from other countries, and just one ST574 complex isolate seen elsewhere, in Finland.
Table 3.
FST values for C. jejuni isolates from human infections in four countries, allele-based FST values above diagonal and sequence-based FST values below. All differences are statistically significant with P < 0·0001
Fig. 2.
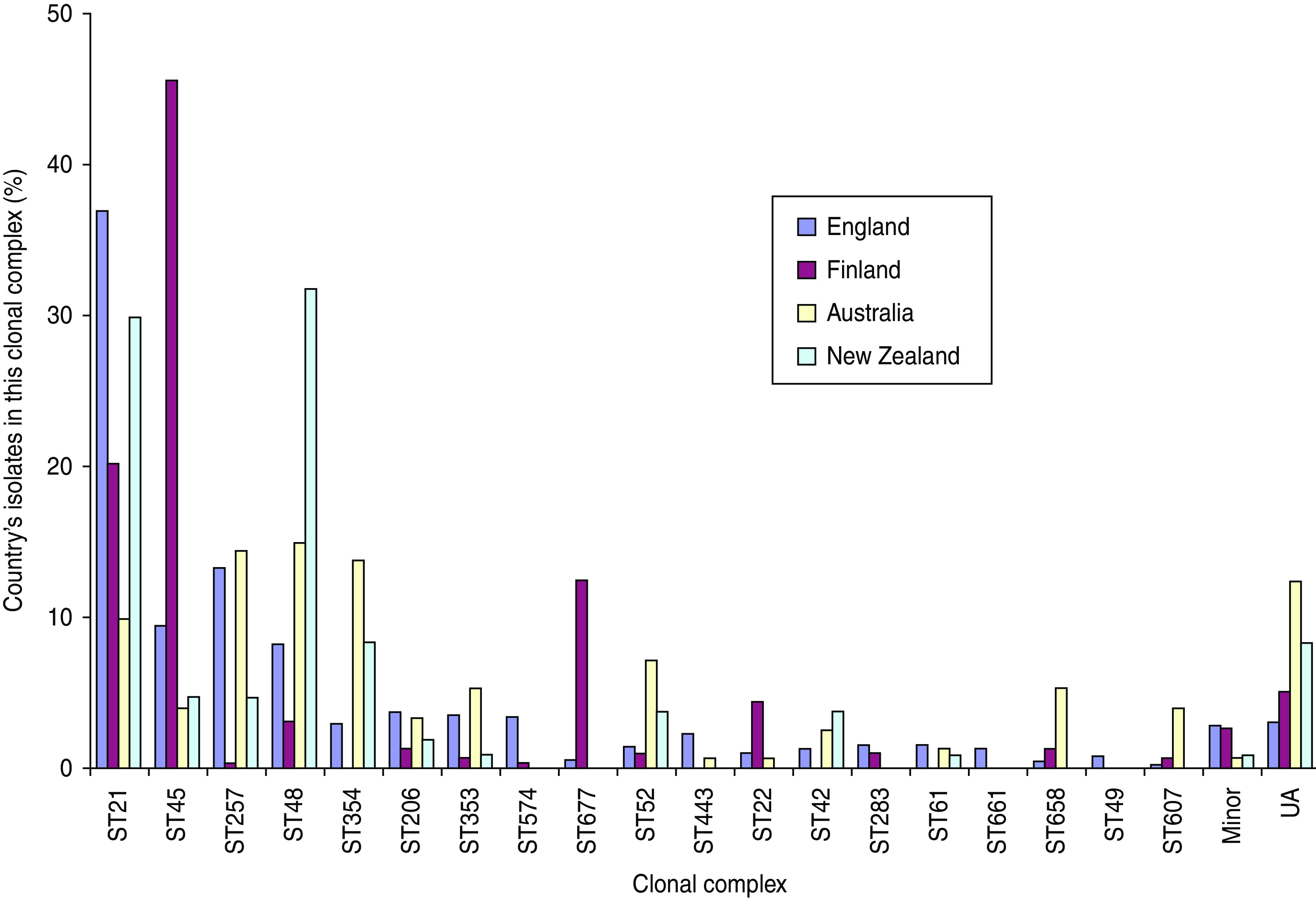
[colour online]. International comparisons of clonal complexes of C. jejuni in population-based samples. UA, Unassigned.
Temporal patterns among subtypes in the UK
There were differences in clonal complex distribution between isolates from peak and non-peak periods (Supplementary Fig.; χ2, P < 0·0001). FST differences were small but also highly significant at nucleotide (0·011, P < 0·0001) or allele (0·007, P < 0·0001) levels.
Secular and seasonal analysis for each clonal complex using harmonic logistic regression showed that the abundant ST21 complex decreased, while ST257, ST353 and ST574 complexes increased. Strong seasonal effects were evident for ST45 and ST283 complexes, both peaking during the overall seasonal peak, and for ST257 complex which did not rise with the seasonal peak but comprised a greater proportion of isolates at other times of the year. Other clonal complexes with evidence for seasonal patterns were ST354 complex peaking in the first half of the year and ST49 and ST52 complexes peaking in the latter part of the year (Table 4).
Table 4.
Statistical evidence for secular and seasonal variation of clonal complexes in human C. jejuni infections in Hampshire and Nottinghamshire, May 2000–April 2003 (n = 1505)
| Clonal complex (CC) | Number (in CC) | Seasonal* | Year† | Linear effect (year)‡ |
|---|---|---|---|---|
| ST21 | 557 | 0·56 | 0·001 | 0·8 (0·70–0·92) |
| ST257 | 201 | 0·02 | 0·004 | 1·29 (1·06–1·55) |
| ST45 | 142 | <0·0001 | 0·40 | |
| ST48 | 125 | 0·98 | 0·02 | 0·80 (0·63–1·02) |
| ST206 | 56 | 0·07 | 0·99 | |
| ST353 | 52 | 0·52 | 0·11 | 1·48 (1·04–2·10) |
| ST574 | 50 | 0·29 | 0·003 | 1·73 (1·20–2·47) |
| ST354 | 44 | 0·01 | 0·12 | |
| ST443 | 36 | 0·92 | 0·78 | |
| ST283 | 23 | 0·002 | 0·69 | |
| ST61 | 23 | 0·55 | 0·45 | |
| ST52§ | 22 | 0·04 § | 0·11 | |
| ST42 | 21 | 0·61 | 0·33 | |
| ST661§ | 21 | 0·56 | 0·11 | |
| ST22§ | 15 | 0·23 | 0·31 | |
| ST49§ | 12 | 0·04 § | 0·23 | |
| ST573§ | 10 | 0·87 | 0·02 § | 2·38 (1·05–5·42) |
| Unassigned | 45 | 0·83 | 0·97 | |
| Minor | 50 | 0·78 | 0·12 |
Bold indicates P values <0·05.
Likelihood ratio test of a model to predict being a member of the clonal complex under study using season modelled by sine and cosine terms and year as a categorical variable.
Likelihood ratio test of a model to predict being a member of the clonal complex under study using year as a categorical variable and including season modelled by sine and cosine terms.
Likelihood ratio test of a model using year as a continuous variable and season modelled by sine and cosine terms used to perform statistical testing. Odds ratios are per one year increase. Inclusion of day instead of year gives similar results. Values in parentheses are 95% confidence intervals.
No isolates in these clonal complexes present in 2003 sample reducing sample size to 1446 for analyses with year as a categorical variable. Analysis of seasonal effect with year as a continuous variable to include the full dataset gave similar statistical support for the seasonal variation of ST52 and ST49 complexes.
Demographic characteristics and genotype
Subtypes varied with age. Comparing individuals aged ⩾60 years with those aged <30 years (χ2 test on clonal complexes, P < 0·0001; FST = 0·012, P < 0·0001 for alleles and 0·017, P < 0·0001 for nucleotides). ST257 complex (P < 0·001) and ST574 complex (P = 0·002) were more and less frequent among older cases, respectively. There was no difference between Simpson's index (0·94 for each), or proportion of infections due to uncommon clonal complexes. Children and adults also differed (χ2 test on clonal complexes P = 0·007; FST = 0·003, P = 0·05 for alleles and 0·004, P < 0·04 for nucleotides). Isolates from children comprised 11% of the dataset overall, 30% for ST283 complex and 5% for ST257 complex. Other values fell between these. Subtypes infecting men and women were similar (χ2, P = 0·41).
Clonal complex and symptoms
There was no evidence that duration of illness varied with clonal complex. Reported bloody stool (34% of cases) varied with clonal complex but vomiting, abdominal pain and fever did not. Analysis adjusting for age (using 10-year age groups) reduced the estimated association between clonal complex and bloody diarrhoea and rendered it non-significant (P = 0·09), with bloody diarrhoea more common in younger cases. ST353 (48%) and ST52 (67%) complexes were those most associated with bloody diarrhoea and ST206 complex (20%) least.
Population genetic structure
The phylogeny estimated using ClonalFrame showed clusters largely correlating with clonal complex designations [6]. However ST283 clonal complex formed a single clonal group with ST45 complex. Two abundant clonal complexes, ST574 and ST257, showed limited diversity. ST574 comprised 51 isolates, of which 50 were ST574, all from the English sample. ST257 complex, common in Australia, England and New Zealand was dominated by the ST257 sequence type (89% of isolates).
DISCUSSION
This study integrated population genetic analysis in the descriptive epidemiology of C. jejuni infection using a 3-year sample from two areas in England and published international datasets. The dominant clones were relatively stable in the English dataset over the 3-year study mirroring consistent patterns over time in Finland [10]. Within this stability a decrease in the abundant ST21 complex and rises in ST257 and ST574 complexes were the most substantial changes. The narrow clonal structure of the increasing ST574 and ST257 complexes, with a single sequence type dominating in each case contrasts with the prevalent, diverse, but decreasing ST21 complex. These findings suggest ST257 and ST574 as recently expanding clones and ST21 as more longstanding.
While the two English populations showed nearly identical subtype distributions of C. jejuni the international comparison showed clear differences. These large effects compared to change over time in the English and Finnish datasets indicate true geographical differences. Differentiation was not proportional to distance with the English sample being more similar to the samples from Australia and New Zealand than to the Finnish sample, which was from the Helsinki area. Geographically robust association of subtype with animal host species has been described [15]. Similarities in subtypes between England, Australia and New Zealand may therefore represent similarity of food sources of C. jejuni infection in these countries and rather different sources in Finland. One Finnish study identified that subtypes present in Finnish cases have a smaller overlap with poultry types [17] than reported in UK studies [14, 18]. The current study further supports substantial differences in the sources of C. jejuni infection in England and Finland.
Seasonally, two clonal complexes (ST45 and ST283 complexes) were particularly concentrated during the summer peak, some (e.g. ST21 and ST48 complexes) maintain a similar proportion over the year, while others (e.g. ST257 complex) contribute a greater proportion outside the seasonal peak. More complex seasonal patterns are also suggested, e.g. for ST354 and ST52 complexes which have their peaks away from the overall one. Varying seasonal patterns by subtype provides a framework for work to study whether these represent exposure of humans to different sources across the year, different dominant types of infection in these sources over the year, or seasonal variation of survival in the food chain by type. Longitudinal studies of isolates among reservoirs contributing to human infection may be particularly informative.
Complexes with lower relative prevalence during the seasonal peak were either absent (e.g. ST257, ST354, ST49) or uncommon (e.g. ST52 complex) in Finland but quite common in England, Australia and New Zealand. In contrast the ST45 complex, which contributed substantially to the summer peak in the English study, was very common in the Finland, which has a marked seasonal peak [10, 22] but less common in New Zealand which has less seasonal variation [22]. A 1-year study in northern England also reported ST45 complex as being more common during summer in humans and that this type was isolated from diverse animal species and surface water samples [35]. These authors suggested that ST45 complex might be ‘environmentally adapted’. A UK study evaluating Campylobacter carriage in broilers at depopulation over 2 years reported a summer peak in ST45 complex infection at first depopulation [19]. Laboratory studies suggest that ST45 complex has different stress responses compared to ST21 complex isolates with better survival in response to oxidative and freezing stress but poorer survival in response to heat or chilling [36]. The ST45 complex seasonal and geographical patterns in the current study and ecological and biological findings in other work suggest that identifiable biological characteristics of clonal groupings affect the dynamics of subtype distribution. Alongside ST45 the less abundant ST283 complex showed a marked summer peak in the current study. ST283 complex isolates were present in the English and Finnish samples but not the Australian or New Zealand collections. In the ClonalFrame analysis of population structure ST283 and ST45 complexes formed a single clonal group. These findings in the dimensions of time, space and genotype show that ST45 and ST283 complexes form a single clade with shared biological and ecological features.
This study identified little evidence for variation in virulence, as measured by type of symptoms and duration of illness, between clonal groups; no evidence of an association of gender and infecting subtype; limited, although clearly present, variation with age group; and marked similarity between the C. jejuni populations isolated from the two geographically separate UK study populations. The essentially identical subtype distributions across these areas sampled at the same time period confirm and extend the similar patterns of clonal complex distribution described in Oxfordshire compared to a slightly earlier study in northern England [37]. A consequence of the finding that similar types infect such separated populations is that sentinel site surveillance may be an efficient way to monitor this infection for public health purposes. Given the large number of cases annually a sentinel approach would be particularly attractive. These findings are also consistent with widely distributed foods being the main sources of infection. A caveat is the English populations in the current study comprised a city and the surrounding areas. The lack of effect does not therefore rule out geographical differences between, for example, urban and rural populations, which may have different patterns of C. jejuni epidemiology.
This study has identified different ecologies for identifiable subtypes of C. jejuni affecting humans and the power of integrating population genetic analytical approaches in epidemiological analyses to the explanation of unresolved epidemiological features such as the seasonality of this infection. The findings of a coherent clade comprising ST45 complex and ST283 complex isolates with particular temporal, spatial, and population genetic characteristics is an area for further exploration in understanding the drivers of the summer peak in this infection. These findings also define a population structure and biological questions to guide experimental work on C. jejuni in general and this clade in particular.
Supplementary Material
Supplementary information supplied by authors.
ACKNOWLEDGEMENTS
The authors acknowledge the support of Dr Robert Owen, then Head of the Campylobacter and Helicobacter Reference Unit in this work. N.D.M. and M.C.J.M. were supported in this work by the Wellcome Trust.
Supplementary material
For supplementary material accompanying this paper visit https://doi.org/10.1017/S0950268812000192.
click here to view supplementary material
DECLARATION OF INTEREST
None.
REFERENCES
- 1.Nachamkin I, Szymanski CM, Blaser MJ. Campylobacter. ASM Press, Washington, DC, 2008, pp. 121–154. [Google Scholar]
- 2.Health Protection Agency (HPA). Laboratory reports of Campylobacter cases reported to the Health Protection Agency Centre for Infections England and Wales, 1989–2009 (http://www.hpa.org.uk/Topics/InfectiousDiseases/InfectionsAZ/Campylobacter/EpidemiologicalData/campyDataEw/) Accessed 13 October 2011.
- 3.Wheeler JG, et al. Study of infectious intestinal disease in England: rates in the community, presenting to general practice, and reported to national surveillance. The Infectious Intestinal Disease Study Executive. British Medical Journal 1999; 318: 1046–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gillespie IA, et al. A case-case comparison of Campylobacter coli and Campylobacter jejuni infection: a tool for generating hypotheses. Emerging Infectious Diseases 2002; 8: 937–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hopkins KL, et al. Fluorescent amplified fragment length polymorphism genotyping of Campylobacter jejuni and Campylobacter coli strains and its relationship with host specificity, serotyping, and phage typing. Journal of Clinical Microbiology 2004; 42: 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dingle KE, et al. Molecular characterization of Campylobacter jejuni clones: a basis for epidemiologic investigation. Emerging Infectious Diseases 2002; 8: 949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sopwith W, et al. Campylobacter jejuni multilocus sequence types in humans, northwest England, 2003–2004. Emerging Infectious Diseases 2006; 12: 1500–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duim B, et al. Molecular evidence for dissemination of unique Campylobacter jejuni clones in Curacao, Netherlands Antilles. Journal of Clinical Microbiology 2003; 41: 5593–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fitch BR, et al. Genetic diversity of Campylobacter sp. isolates from retail chicken products and humans with gastroenteritis in Central Michigan. Journal of Clinical Microbiology 2005; 43: 4221–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karenlampi R, et al. Longitudinal study of Finnish Campylobacter jejuni and C. coli isolates from humans, using multilocus sequence typing, including comparison with epidemiological data and isolates from poultry and cattle. Applied and Environmental Microbiology 2007; 73: 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mickan L, et al. Multilocus sequence typing of Campylobacter jejuni isolates from New South Wales, Australia. Journal of Applied Microbiology 2007; 102: 144–152. [DOI] [PubMed] [Google Scholar]
- 12.McTavish SM, et al. Wide geographical distribution of internationally rare Campylobacter clones within New Zealand. Epidemiology and Infection 2008; 136: 1244–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCarthy ND, et al. Host-associated genetic import in Campylobacter jejuni. Emerging Infectious Diseases 2007; 13: 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheppard SK, et al. Campylobacter genotyping to determine the source of human infection. Clinical Infectious Diseases 2009; 48: 1072–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheppard SK, et al. Host association of Campylobacter genotypes transcends geographic variation. Applied and Environmental Microbiology 2010; 76: 5269–5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller WG, et al. Extended multilocus sequence typing system for Campylobacter coli, C. lari, C. upsaliensis, and C. helveticus. Journal of Clinical Microbiology 2005; 43: 2315–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Haan CP, et al. Multilocus sequence types of Finnish bovine Campylobacter jejuni isolates and their attribution to human infections. BMC Microbiology. Published online: 26 July 2010. doi: 10.1186/1471-2180-10-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson DJ, et al. Tracing the source of campylobacteriosis. PLoS Genetics 2008; 4: e1000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jorgensen F, et al. Campylobacter jejuni and C. coli subtypes in housed broiler flocks reared in Great Britain: influence of season and geography. Applied and Environmental Microbiology. Published online: 1 April 2011. doi: 10.1128/AEM.02444-02410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson JS, Cahoon FE, Hodge DS. Rate of Campylobacter spp. isolation in three regions of Ontario, Canada, from 1978 to 1985. Journal of Clinical Microbiology 1986; 24: 876–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kovats RS, et al. Climate variability and Campylobacter infection: an international study. International Journal of Biometeorology 2005; 49: 207–214. [DOI] [PubMed] [Google Scholar]
- 22.Nylen G, et al. The seasonal distribution of Campylobacter infection in nine European countries and New Zealand. Epidemiology and Infection 2002; 128: 383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Health Protection Agency (HPA). Campylobacter infection rates by age, England and Wales, 2000–2009 (http://www.hpa.org.uk/Topics/InfectiousDiseases/InfectionsAZ/Campylobacter/EpidemiologicalData/campyDataEWAgeRates/). Accessed 13 October 2011.
- 24.van Hees BC, et al. Regional and seasonal differences in incidence and antibiotic resistance of Campylobacter from a nationwide surveillance study in The Netherlands: an overview of 2000–2004. Clinical Microbiology and Infection 2007; 13: 305–310. [DOI] [PubMed] [Google Scholar]
- 25.Skirrow MB. A demographic survey of Campylobacter. Salmonella and Shigella infections in England. A Public Health Laboratory Service Survey. Epidemiology and Infection 1987; 99: 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sibbald CJ, Sharp JC. Campylobacter infection in urban and rural populations in Scotland. Journal of Hygiene (London) 1985; 95: 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller G, et al. Does age acquired immunity confer selective protection to common serotypes of Campylobacter jejuni? BMC Infectious Diseases 2005; 5: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anon. Sentinel surveillance of campylobacter in England and Wales. Communicable Disease Report. CDR Weekly 2000; 10: 169–172. [PubMed] [Google Scholar]
- 29.Dingle KE, et al. Multilocus sequence typing system for Campylobacter jejuni. Journal of Clinical Microbiology 2001; 39: 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 1992; 131: 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. Journal of Clinical Microbiology 1988; 26: 2465–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naumova EN, et al. Seasonality in six enterically transmitted diseases and ambient temperature. Epidemiology and Infection 2007; 135: 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Didelot X, Falush D. Inference of bacterial microevolution using multilocus sequence data. Genetics 2007; 175: 1251–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fearnhead P, et al. Analysis of recombination in Campylobacter jejuni from MLST population data. Journal of Molecular Evolution 2005; 61: 333–340. [DOI] [PubMed] [Google Scholar]
- 35.Sopwith W, et al. Identification of potential environmentally adapted Campylobacter jejuni strain, United Kingdom. Emerging Infectious Diseases 2008; 14: 1769–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Habib I, Uyttendaele M, De Zutter L. Survival of poultry-derived Campylobacter jejuni of multilocus sequence type clonal complexes 21 and 45 under freeze, chill, oxidative, acid and heat stresses. Food Microbiology 2010; 27: 829–834. [DOI] [PubMed] [Google Scholar]
- 37.Dingle KE, et al. Extended sequence typing of Campylobacter spp., United Kingdom. Emerging Infectious Diseases 2008; 14: 1620–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information supplied by authors.
For supplementary material accompanying this paper visit https://doi.org/10.1017/S0950268812000192.
click here to view supplementary material