

Abstract
Purpose
Several established optical imaging approaches have been applied, usually in isolation, to preclinical studies; however, truly useful in vivo imaging may require a simultaneous combination of imaging modalities to examine dynamic characteristics of cells and tissues. We developed a new multimode optical imaging system designed to be application-versatile, yielding high sensitivity, and specificity molecular imaging.
Procedures
We integrated several optical imaging technologies, including fluorescence intensity, spectral, lifetime, intravital confocal, two-photon excitation, and bioluminescence, into a single system that enables functional multiscale imaging in animal models.
Results
The approach offers a comprehensive imaging platform for kinetic, quantitative, and environmental analysis of highly relevant information, with micro-to-macroscopic resolution. Applied to small animals in vivo, this provides superior monitoring of processes of interest, represented here by chemo-/nanoconstruct therapy assessment.
Conclusions
This new system is versatile and can be optimized for various applications, of which cancer detection and targeted treatment are emphasized here.
Keywords: Multimode, Preclinical, In vivo, Spectral analysis, Fluorescence lifetime, Wide-field two-photon excitation, Chemotherapy, Nanoconstruct, Corroles
Introduction
Small animal imaging has become widely used as a non-invasive research tool since it allows the detection of primary and metastatic tumors, as well as monitoring effects of pharmacological interventions. In the past, it was necessary to—inefficiently—sacrifice the animals in order to detect tumors or monitor drug molecules inside tissues. Moreover, the excised tissues are not functional, and thus the normal physiology such as blood flow, cell–cell interactions, and metabolic activity is suspended, altered, or degraded. These limitations impede continuous/recurrent and accurate monitoring of biochemical, genetic, and pharmacological processes in vivo and in the same animal. This encouraged the development of better methods to track disease at the preclinical level, with imaging of small animals in vivo emerging as an important approach [1, 2].
Several non-invasive technologies such as ultrasound, X-rays and computed tomography, magnetic resonance imaging, positron emission tomography, single photon emission computed tomography, and optical imaging are available for small animal imaging. These were all originally developed for clinical applications and redesigned for small animal imaging. Ultrasound, computed tomography, and magnetic resonance imaging are capable of resolving the anatomy and physiology through the energy–tissue interaction. On the other hand, positron emission tomography and single photon emission computed tomography require reporter probes/contrast agents in order to monitor metabolism [1, 3]. Various optical imaging technologies that range from 2D imaging to 3D tomography of internal organs and tissues have been developed for small animal imaging [4, 5]. These methods have several advantages compared to the non-optical modalities, by offering simultaneous monitoring of multiple targets or molecular pathways, and by being less expensive and less potentially harmful [1–3].
Within optical technologies, fluorescence intensity imaging is a simple method to continuously track movements and concentrations of labeled molecules in vivo, based on the spatial distribution of intensity at a single detected wavelength. This approach allows monitoring the progression of diseases, the effects of drug candidates on the target pathology, the pharmacokinetic behavior of drugs, and the treatment outcomes. It is based on the linear dependence of fluorescence intensity on the accumulated concentration of labeled molecules [2, 3, 6–10]. However, in the presence of confounding signals from entities such as endogenous fluorophores, it is very difficult to discriminate and quantify the fluorescence signal from labeled molecules of interest. Thus, for more accurate imaging in the presence of such “noise,” it is necessary to use more advanced imaging methods [2, 9]. As a more quantitative optical technology, spectral imaging provides a high-resolution spectral signature at every pixel of an image, allowing one to identify, separate, and remove the contribution of background in analyzed fluorescence images. The entire process can be completed in seconds [9, 11, 12]; additional information may be obtained by comparing spectra acquired at different excitation wavelengths. On the other hand, fluorescence lifetime imaging (FLIM) is less dependent on the concentration of fluorophores but can be highly sensitive to tissue environment such as intra/extracellular pH distribution, blood flow, tissue oxygenation, and temperature. Unlike intensity or spectral measurements, FLIM can show the energy transfer rate from the excited state of fluorophores to their surrounding environment [13–17], and can be used for localizing diseased tissues such as tumors based on physiology [18, 19].
Two-photon excitation and intravital confocal imaging methods have been widely utilized for deep tissue exploration, as they can provide highly resolved information in vivo. In particular, two-photon excited fluorescence imaging has sectioning capability at the deeper locations than one-photon excited confocal fluorescence imaging, while exhibiting less overall photobleaching; therefore, scanning two-photon excitation has been widely used as a non-invasive imaging method in tissue samples and living animals [20–22]. Intravital confocal imaging also facilitates high-resolution studies of cellular and molecular events in vivo [23]. In particular, an optical fiber bundle, properly end-scanned, constitutes a high-resolution endoscope and can be inserted into a small animal, enabling observation of microanatomy inside a small animal, with cellular resolution. This technology has already been broadly utilized, for imaging in vivo, e.g., subcutaneous tumors, peripheral nerves, and angiogenesis [24, 25]. Finally, bioluminescence imaging enables the non-invasive study of biological processes without excitation light sources in small animals [14], based on some advantages over fluorescence imaging: (1) background from excitation and/or tissue autofluorescence is eliminated, and (2) as a consequence, good signal-to-noise allows detection of light from an enzymatic reaction at few centimeters depth [10, 26].
All of the optical imaging technologies mentioned have their inherent advantages in providing particular information when used in isolation. Thus, the use of the specific imaging technologies depends on the information required in pre-clinical studies. Many studies may benefit from using multiple imaging modes simultaneously since such a combination can offer complementary and even synergetic information in detecting diseased tissues or identifying the characteristics of molecules of interest in vivo. However, most commercial small animal optical imaging systems are single-mode, thus lacking in versatility. Although these systems perform well in the tasks they have been designed for, they do not have enhanced capabilities such as the use of multiple laser excitation sources, or advanced features such as fluorescence lifetime and two-photon excited fluorescence imaging. These limitations, and the need we see for more flexible, advanced in vivo imaging, prompted us to develop a new multimode optical imaging system that can distinguish multiple targets, provide quantification of fluorochrome biodistribution, and allow simple switching from macro- to microscopic resolution and between multiple imaging modes: fluorescence intensity, spectral, lifetime, intravital confocal, two-photon excited fluorescence, and bioluminescence imaging.
In this work, we demonstrate a new multimode optical imaging system that enables simultaneous use of these powerful multiscale optical imaging technologies for dynamic, quantitative, and functional monitoring of multiple fluorophores, discrimination between them, and the acquisition of highly resolved information in vivo. In addition, after evaluating each imaging mode by using a biological specimen of choice (including ex vivo and in vivo), we present their focused use in multimode optical imaging for chemotherapy research: We have chosen two drug molecules of interest, nanoconstructs [27, 28], and the complex of a breast cancer-targeted cell penetration protein (HerPBK10) and a sulfonated gallium corrole (S2Ga), which is denoted HerGa [29, 30]. We show here continuous fluorescence intensity/spectral imaging for dynamic monitoring and clearance examination of nanoconstructs in vivo, and the feasibility of multimode optical imaging for better chemotherapy assessment of HerGa.
Materials and Methods
Multimode Optical Imaging System
A schematic diagram of the instrument we designed and built is shown in Fig. 1a. It consists of a light-tight enclosure within which various optical imaging modes are implemented as follows: fluorescence intensity, spectral, lifetime, intravital confocal, scanning/wide-field two-photon excited fluorescence (WTEF), which has been developed outside but will be incorporated into the system, and bioluminescence imaging. A wide selection of light sources is available, including a tunable femtosecond (fs) pulsed laser (MaiTai, SpectraPhysics) (710–990 nm), a picosecond pulsed laser (Tsunami, SpectraPhysics; 700–1,080 nm), and continuous wave (CW) lasers [405 and 670 nm solid-state, HeCd (442 nm), argon–krypton (488, 514, and 647 nm), and HeNe laser (633 nm)]. The light sources are delivered via free space, or through a fiber-based delivery system allowing flexible delivery of various lasers’ output inside a light-tight enclosure. The light is deflected/divided by a 50:50 dichroic mirror and delivered onto specimens through diffusers and mirrors. Here, uniform laser excitation of the specimen is realized by scattering the laser light onto the 90%+ transmission broadband diffusers (“ThorLabs, 1” round 20° circle tophat diffuser). Light from the specimen is collected by a telecentric lens (Melles Griot, Invaritar™ 59LGL428 and 59LGG950, NA: 0.24).
Fig. 1.

Multimode optical imaging system. a System schematic. This system is capable of several imaging modes, including fluorescence intensity, spectral, life time, intravital confocal, and bioluminescence imaging. Also, for 3D fluorescence imaging, optical components and a control program are installed. Furthermore, scanning/WTEF imaging mode will be incorporated with the system for deep tissue imaging at high resolution. b Photographic image of the multimode optical imaging system. MFW motorized filter wheel, RL relay lens, TCL telecentric lens, ER mode exchange rail, GM galvo mirror, TL tube lens, F1 band-pass filter, F2short-pass filter, DM dichroic mirror, L doublet lens, HP heating pad, D diffuser, RS rotational stage, FR faraday rotator, R retarder, RF rotational filters, BS beam sampler, ET external trigger for FLIM.
The collected light passes through either (1) standard interference filters (Chroma Technology) installed in a motorized filter wheel, (2) an Acousto-Optical Tunable Filter (AOTF) (ChromoDynamics), or (3) a FLIM module with a time-gated intensifier (LaVision PicoStar HR), before arriving onto a cooled charge-coupled device (CCD) camera (Princeton Instruments, PIXIS 400) located on top of the light-tight imaging chamber. Every component is modular, and switching between modes is fast (~seconds, if necessary). The AOTF and FLIM modules can be flexibly exchanged by using a sliding rail. In addition, for high-resolution endoscopic imaging, an optical device and its probe (Mauna Kea Technologies) have been incorporated for intravital confocal imaging in the system. For 3D fluorescence imaging, the telecentric lens and CCD camera can be attached onto the enclosure’s sidewall. The synchronization between a rotational stage and CCD camera for image acquisition with different angle views is controlled by a program we developed. Furthermore, a two-photon imaging module was developed for high-resolution deep tissue imaging outside the system and can be integrated within it.
The experimental animal (mouse or rat) is placed inside the light-tight enclosure on a moving stage that has a spatial resolution of 5 μm in the x and y-axes, 100 μm in the z-axis, and 2.16 arcsec in the rotational axis. The sample stage can move along x-, y-, z-axis, and rotate clockwise and counterclockwise to vary the field of view and a sample position. The stages are actuated by a stepper and a servo motor using software we developed (using CVI/National Instruments). In order to prevent animals from moving during image acquisition, a gated anesthesia system we developed (capable of stopping breathing for the duration of image acquisition alone) that uses a mixture of oxygen and isoflurane is attached to the imaging box.
Imaging Modes
Fluorescence Intensity Imaging Mode
In this mode, a laser source can be selected by flipping mirrors for the excitation of a variety of fluorophores. Light from the selected source is delivered onto a specimen through mirrors, filters, and diffusers. Before a fluorescence image from the specimen is acquired, a background image including a spatial profile of an excitation light source is recorded for flat-field correction. Then, fluorescence collected by a telecentric lens is recorded in a high sensitive cooled CCD camera, after being selected by an emission filter in a computer-controlled filter wheel. Finally, the fluorescence image is corrected by the background image in order to reduce the artifacts due to the profile of an excitation light source. Meanwhile, a photographic-equivalent image is taken with LED illumination for overlaying the fluorescence image. Fig. 2a shows the schematic of the fluorescence imaging mode.
Fig. 2.

Experimental setup of each mode. a Fluorescence imaging mode. Selected light sources divided by a 50:50 dichroic mirror in a light-tight enclosure are delivered into a specimen through two diffusers. Then, fluorescence collected by a telecentric lens from a specimen is recorded in CCD through a selected filter. b Spectral imaging mode. An excitation method procedure is identical with the method in fluorescence intensity imaging. AOTF is employed between a filter and CCD for band-sequential spectral selection. c mosaic FLIM using fs pulsed laser light. fs pulsed laser light was used for the excitation of the molecules of interest. A specimen was scanned by x–y translation motorized stage for LFOV controlled by the developed program, which is connected with the actuation motor control program through a TCP/IP internet connection. d Intravital confocal imaging mode. S-probe with a diameter, 0.65 mm is utilized for intravital confocal imaging. The probe has 0 μm working distance, 5 μm lateral resolution, and 15 μm axial resolution. The image obtained using the probe has 599×500 μm field of view and 450×384 pixels. e Scanning/WTEF imaging modes. L1 is a doublet lens that enables wider excitation and is removed in scanning two-photon imaging mode. F1 and F2 are shortpass filter (<700 nm) and interference filter, respectively. L2 is a tube lens. f Bioluminescence imaging modes. Bioluminescence signal is collected by a telecentric lens, and then recorded in the cooled CCD. After bioluminescence imaging is completed, a photographic image is acquired with LED illumination for overlaying with the bioluminescence image.
Spectral Imaging Mode
In this mode (Fig. 2b), the laser delivery for excitation of specimen is the same as in fluorescence intensity imaging. In detection, the emitted fluorescence from a specimen is collected by a telecentric lens and passes through a long-pass filter, which rejects the excitation light source. Then, the light is delivered onto a high-sensitivity, low-noise CCD camera cooled to −70°C (Princeton Instruments, PIXIS 400) for imaging through an imaging AOTF system (ChromoDynamics, Inc.) which can rapidly and (random-access) sequentially select a narrow bandwidth (1.5–4.0 nm). After the sequential images within the certain range of wavelength are acquired, the spectral signatures on each pixel are generated by our custom-developed program [12]. The image is then classified based on predefined spectral signatures. In addition, spectral unmixing is performed in order to reject autofluorescence from fluorescence signals of interest using a module we incorporated into ImageJ [31].
Fluorescence Lifetime Imaging (FLIM) Mode
For FLIM in multi-mode optical imaging system, we developed a mosaic FLIM system with fs pulsed laser, for a large field of view (LFOV) described elsewhere. In this method, fs pulsed laser light is tuned to 400~480 nm, a repetition rate of 80 MHz, generated by the second harmonic of fs tunable pulsed laser with 800~960 nm in Barium–Borate (BBO) crystal, and used for the excitation of molecules inside a specimen. An ultra-fast time-gated camera (LaVision, PicoStar HR) was utilized for FLIM. Fig. 2c shows the schematic of the mosaic FLIM setup. The fs pulsed laser is delivered through a small opening in a light-tight enclosure; then, the light passes through two mirrors and a diffuser in order to excite a specimen. The beam size of the light is controlled by distance between a specimen and the diffuser that induces a 20° divergence of light. The fluorescence from a specimen is collected by the telecentric lens. The fluorescence light passes through a selected emission filter, and then is delivered onto the CCD connected with time-gated intensifier (TGI), synchronized with the pulsed light via a delay unit that connects with the external trigger. The scanning for LFOV is performed sequentially after each image acquisition by x–y translation via the motorized stage under the control of the program that we developed using National Instrument CVI. It can control scanning width, height, and step size. Also, the program can be connected with a Joystick program that allows control of the motorized stages and scanning features at different locations through TCP/IP connection [9].
Intravital Confocal Imaging Mode
Intravital confocal imaging enables in vivo endoscopic imaging with high resolution, using the proven CellVizio instrument from Mauna Kea Technologies, used here (Fig. 2d) as a new mode added to our system. In this mode, we constructed and used a holder for positioning a fiber bundle probe in order to reduce the artifacts due to operators’ hand movements. The fiber bundle probe is directly connected with a confocal scanner including a scanning unit, an excitation laser source, an emission filter, and a detection module (an avalanche photodiode).
Multiphoton (Scanning/WTEF) Imaging Mode
The scanning/WTEF imaging mode has been developed in order to examine details at deeper locations in multimode optical imaging in vivo; we first built this capability outside our black box system. For wide-field non-linear excitation, a polarized fs pulsed laser with 780~990 nm, 50–300 mW average power, and 80 MHz repetition rate was utilized. The fs pulsed laser beam passes through a Faraday rotator, several mirrors, and galvo mirrors. Then, it is focused on the back focal plane of an objective (Nikon 40×, 1.3 NA, oil, Nikon 40×, 0.75 NA, air, or Nikon 60×, 0.75 NA) through a doublet lens (L1; Melles Griot, FL200) in order to obtain a quasi-parallel beam which makes a larger spot size at the sample. The fluorescence from a specimen is collected by the same objective, and then passes through short/band-pass filters and a tube lens (L2) before being recorded on a cooled CCD. In addition, in order to compensate a non-uniform excitation generated by the Gaussian profile of the beam, flat-field correction of a WTEF image was performed by a normalized and inverted Gaussian mask [32]. The Gaussian mask has been constructed through the convolution of the original image with a Gaussian function (radius, over 40) at a focal plane using ImageJ. The corrected image, R, is obtained by the entry-by-entry product of the original image O and the Gaussian mask M (R=O×M). Moreover, for two-photon excited fluorescence imaging of in vitro specimens, a scanning unit is incorporated into the system. In the scanning two-photon imaging mode, the CCD is replaced with a photomultiplier tube (PMT; Hamamatsu, H6780-20), and the doublet lens (L1) is removed from an optical beam path. Here, galvo mirrors and PMT output signals are synchronized by the program we developed. Fig. 2e shows the experimental setup.
Bioluminescence Imaging Mode
A bioluminescence imaging mode was developed in order to detect ATP and enzymatic activity in engineered nude mice. The experimental setup is shown in Fig. 2f. A photographic image of the mouse was also recorded, using two light-emitting diodes. Bioluminescence was collected by a telecentric lens (Melles-Griot, Invaritar™) and imaged onto a cooled CCD (Princeton Instruments, PIXIS 400). The bioluminescence image was thresholded and overlapped with the photographic image.
Sample Preparations
Four nude mice and one rat were prepared for the evaluation of each imaging mode. Three of the mice (provided by the labs of Drs. Lali K. Medina-Kauwe and Julia Ljubimova) bore bilateral flank tumor xenografts, one mouse has implanted brain tumors, and one is an engineered mutant mouse. Also, a rat was prepared for intravital confocal imaging. Before intravital confocal imaging of rat spine is performed, the rat was euthanized, and then the spine of the rat was exposed. For evaluation of fluorescence intensity imaging, 100 μl of nanoconstruct solution at a concentration of 2.5 mg/kg was prepared for the tail vein injection. On the other hand, 10 μM fluorescein solution was diluted by phosphate buffered saline solution (pH 7.4) for spectral and FLIM. For evaluation of bioluminescence imaging, we prepared a stock solution of a luciferin enzyme substrate (Xenogen) at 15 mg/ml in Dulbecco’s phosphate-buffered saline solution, and then injected 100 μl of luciferin into an abdominal cavity of the engineered mouse. For WTEF imaging, a 16-μm cryostat section of mouse intestine (specifically, the filamentous actin prevalent in the brush border) stained with Alexafluor 568 phalloidin (FluoCells slide #4 (F-24631), Invitrogen) was prepared. Finally, an extracted mouse liver was stained nonspecifically with 500 μM fluorescein solution for evaluation of scanning two-photon excited fluorescence imaging mode.
In nanoconstruct experiments, epidermal growth factor receptor (EGFR)-positive MDA-MB 468 human breast tumor was implanted into the right posterior mid-dorsum of nude mice [Tac: Cr:(MCr)-Foxnnu]. After anesthetizing the mouse using the gated anesthesia machine we built, the 100 μl of nanoconstruct solution was intravenously injected via the tail vein at a concentration of 2.5 mg/kg [27, 28].
Mice with implanted MDA-MB-435 human breast tumor with over 30mm2 were prepared for assessment of HerGa in chemotherapy. 46 nmol of HerGa was injected (IV) into the tail of one mouse for imaging tumor regions using FLIM. In addition, specific organs and tumors were extracted from the same mouse at 4 days after intravenous injection of HerGa, and the organs and tumors were washed with PBS in order to examine the drug accumulation. In addition, after injection of HerGa into two tumor and non-tumor regions, we euthanized the mouse as provided by our IACUC protocol. Multimodal imaging of the mouse was performed to test HerGa capacity for tumor detection and delineation.
Results
Evaluation of Individual Modes in a Multimode Optical Imaging System
Fluorescence Intensity Imaging
Fluorescence intensity imaging has been performed in order to identify the targeting capability of a nanoconstruct drug molecule [27, 28], which is conjugated with Alexafluor 680, for brain tumors. For excitation, laser light at 647 nm was utilized. An emission filter with a central wavelength of 705 nm and bandwidth of 20 nm was utilized for the detection of fluorescence from the drug molecules. After the drug was injected into the tail vein of the mouse, the fluorescence of Alexafluor 680 was detected in 24 h. Fig. 3a illustrates a typical pseudocolor image obtained through the fluorescence intensity imaging mode, clearly showing that the drug molecules preferentially accumulate into the brain tumors, as indicated by an arrow.
Fig. 3.

Images obtained by using each mode of multimode optical imaging system. a Fluorescence intensity image of the drug molecules preferentially accumulating into implanted brain tumors in the mouse. The arrow indicates a brain tumor. b Spectral classification image of a nude mouse with injection of fluorescein into implanted tumors and an engineered mouse pup with GFP expression. While red pseudocolor represents fluorescence of fluorescein (the left image) and GFP (the right image), green pseudocolor represents autofluorescence. c Fluorescence lifetime image obtained by mFLIM. The mouse was scanned by the system step-by-step for LFOV. Two sets of images were merged into a single image. For each fluorescence lifetime image, a total of 39 images have been acquired within 0 to 7,800 ps with a time step, 200 ps. d Intravital confocal image of a rat spine. The image (skeletal muscles) was extracted from the video file recorder for 8 min. e WTEF images (scale bar, 50 μm) of a mouse intestine and scanning two-photon excited fluorescence image (scale bar, 100 μm) of a mouse liver stained with fluorescein. A WTEF image (upper) is acquired with 795 nm excitation pulsed laser and an emission filter (620±60 nm), and then a flat-field corrected image was generated by a constructed Gaussian mask. For the scanning two-photon excitation of the liver, fs pulsed laser light at 780 nm was here used, and a shortpass filter (<700 nm) and a bandpass filter (540±40 nm) were utilized for the emission light selection. A Nikon 40× (NA: 0.75) and a Nikon 20× objective (NA: 0.50) were used for WTEF and scanning two-photon excited fluorescence imaging, respectively. f Bioluminescence image from engineered mice (normalized by highest value).
Spectral Imaging
We have extensive experience in pre-clinical and clinical spectral imaging [33, 34]. The feasibility of spectral imaging in our new multimode system has been investigated after 10 μM fluorescein, diluted with PBS (pH 7.4), was injected into breast tumors implanted in the back of a nude mouse. 488nm laser light was utilized for the excitation of fluorescein inside the mouse, and then a total of 12 images were recorded within the spectral range of 500 to 720 nm with a step size of 20 nm. The images were analyzed using software we developed. Spectral classification based on a least mean square distances method was performed for separating fluorescein fluorescence from autofluorescence. Fig. 3b shows the interpreted image obtained through spectral classification: the fluorescence of fluorescein (red pseudocolor) is clearly discriminated from autofluorescence (green pseudo-color).
Mosaic Fluorescence Lifetime Imaging Mosaic (field of view scanning)
FLIM of the same mouse (used in the previous spectral imaging) was performed for the evaluation of this imaging mode. Fig. 3c shows the fluorescence lifetime image displayed in pseudocolor. While the fluorescence lifetimes from a mouse skin cluster around 3~3.8 ns, the fluorescence lifetimes of fluorescein injected into tumor regions show values of 3.8~4.5 ns. In the image, the tumor regions with fluorescein are clearly distinguished from other regions by the lifetime differences exhibited. Also, the lifetime values of fluorescein are in good agreement with the those in literature [13].
Intravital Confocal Imaging
The feasibility of intravital confocal imaging of a rat spine has been evaluated. After euthanizing the rat, its spine was exposed. Then, autofluorescence images of the rat spine were recorded for 8 min by using an MKT S-probe (0 μm working distance). The typical frame rate is 12 fps. Fig. 3d shows the results obtained by using it. The image in Fig. 3d clearly shows skeletal muscle fibers and vessels around spine regions, with high resolution.
Multiphoton (Scanning/WTEF) Imaging
Fig. 3e shows the results obtained by WTEF imaging of a mouse intestine stained with Alexa 568 Phalloidin and scanning two-photon excited fluorescence imaging of a mouse liver stained with fluorescein nonspecifically. In the WTEF image, the filamentous actins are clearly shown like one-photon excited fluorescence image we do not show here. The image size is approximately 80×65 μm. Moreover, the WTEF image (Fig. 3e, upper) corrected by the constructed mask shows uniform baseline compared to that of the uncorrected image [32, 35]. Also, liver cell-like structures stained with fluorescein are clearly shown in the scanning two-photon excited fluorescence image (Fig. 3e, down).
Bioluminescence Imaging
Using bioluminescence mode in this system, we imaged the presence of ATP and enzymatic activity in nude mice. After luciferin was injected into their abdominal cavity, we took a photographic image and then recorded the bioluminescence signal for 2 min. Fig. 3f shows the overlay of the photographic and bioluminescence image. The result demonstrates the capability to record bioluminescence signals with good signal-to-noise and versatility, opening up the possibility for further studies using this modality.
Contrast Comparisons
In order to quantitatively evaluate the quality of the fluorescence images recorded, we compared the contrast of tumor areas on the images of a mouse obtained with a commercial system and our system. Here, we injected tumor targeting drug molecules into intra-vein of the mouse, and then the images were acquired after 24 h. We compared the contrasts as calculated with average intensities around tumor and non-tumor regions. The contrast values calculated based on Michelson contrast [It(tumor) − Ib(background)/(Ib+Ib)] for the (best) commercial system and our system are 0.33 and 0.66, respectively [28]. Our system’s roughly two-fold improvement is not our highest ratio obtained, only a representative one.
Multi-mode Optical Imaging in Chemo- and Nanoconstruct Therapy Research for Quantitative, Dynamic, and Functional Monitoring
Fluorescence Imaging of the Tumor Targeting Capability and Accumulation Kinetics of Nanoconstructs
We monitored a nanoconstruct distribution in a nude mouse with breast tumors after injection of the nanoconstructs conjugated with Alexafluor 680 into a tail vein in order to examine the tumor targeting capability and the accumulation kinetics of the nanoconstructs. In the study [28], we could clearly distinguish the preferential nanoconstruct accumulation in the tumor region in 3 h after its injection as well as observe that the nanoconstructs were still retained in the tumor region after 24 h and perfusion. This indicates that fluorescence intensity imaging enables dynamic monitoring of the distributions of nanoconstructs in the tumors, thus allowing determination of their targeting capability and accumulation kinetics.
Spectral Imaging for Quantitative Examination of Clearance of the Nanoconstructs In Vivo and the Extent and Topology of HerGa Distribution into Specific Organs and the Tumor
In chemotherapy assessment, drug clearance is critical in the determination of dosage. We utilized here spectral imaging for monitoring nanoconstruct temporal clearance from the whole mouse. For classification, the spectral signatures of autofluorescence and Alexafluor 680 fluorescence were acquired before and after nanoconstruct injection respectively. Fig. 4a shows the spectral signatures. Then, the images before injection and at 10 min, 3h, and 24 h after injection were analyzed with the spectral signatures. Fig. 4b shows the spectral classification images at those time points. Before injection (0 h), autofluorescence was seen over the whole mouse. However, in 10 min after injection of nanoconstructs, the nanoconstructs (red pseudocolor) were distributed over the overall mouse. The nano-constructs still remained over the whole area of the mouse at 24 h. This result shows that spectral imaging and analysis provides better sensitivity and specificity in the determination of the clearance of nanoconstructs, allowing detection of minute drug amounts and of their changes.
Fig. 4.
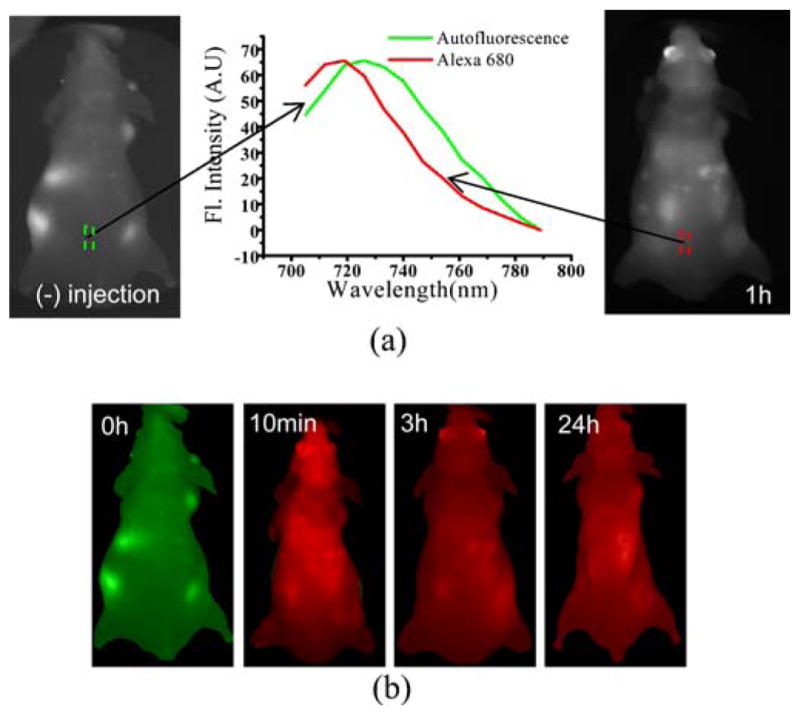
Clearance examination of the drug nanoconstruct using spectral imaging. a Spectral signatures of autofluorescence and fluorescence of Alexafluor 680. The spectral signature of autofluorescence and Alexafluor 680 fluorescence is obtained from a spectral image cube (700 to 790 nm) acquired before and after intravenous injection of the nanoconstruct respectively. b Spectral classification images at different time points. A total of 13 images were recorded within the spectral range of 705 to 789 nm with a step size 7 nm at different time points (0 h, 10 min, 3 h, and 24 h). Then, those images were analyzed with the previously acquired signatures using our developed program. Here, while green pseudocolor represents autofluorescence, red pseudocolor represents Alexafluor 680.
Investigation of the Extent and Topology of HerGa Distribution into Specific Organs and the Tumor Using Multimode Optical Imaging Ex Vivo
We reported that HerGa is very effective for tumor detection and delineation in HER2+ breast cancer animal models. However, for the successful translation of HerGa chemotherapy into the clinic, we need to do a better assessment, in vivo. Thus, we here evaluated the extent and topology of HerGa distribution into specific organs and the tumor itself using multimode optical imaging in order to examine the tumor targeting capability and tumor retention of HerGa as well as drug effects on the other organs. Fig. 5a shows the fluorescence intensity image at 620 nm. In the figure, HerGa is preferentially accumulated in tumors compared to other organs, and the fluorescence signal from tumors is significantly higher than that from liver. This figure clearly indicates that HerGa specifically and preferentially targets the tumor and is better retained there. Fig. 5b shows the spectral classification image analyzed with the predefined spectral signatures. In the image, while tumor and liver are clearly classified as HerGa fluorescence, most areas of other organs are classified as autofluorescence. The spectral imaging here offers better quantitative discrimination between HerGa and autofluorescence than fluorescence intensity imaging. Fig. 5c shows fluorescence lifetime images of the tumors and the liver. Here, other organs except for the tumors and the liver are not considered for analysis since the signal from them is too low. Fluorescence lifetime values of HerGa accumulated in tumors are significantly higher than those in liver. In addition, the histograms of fluorescence lifetime in tumors and liver were constructed for detailed examination (Fig. 5c), underscoring the higher HerGa lifetimes in tumors vs. liver. This result may suggest the environmental information around tumor region as well as the potential of HerGa in tumor detection and delineation. Finally, we performed two-photon excited fluorescence imaging of tumors in order to examine them in detail with high resolution. Fig. 5d shows the two-photon excited fluorescence image of tumor, clearly showing the bright cellular structures due to HerGa accumulations. Taken together, these results indicate that multimode optical imaging can provide complementary and highly resolved information on tumor retention/accumulation of HerGa (intensity), quantitative concentration (spectral), and tumor environment (lifetime), simultaneously, thus improving the assessment of this chemotherapy molecule’s action.
Fig. 5.

Specific organs and tumor images obtained using multi-mode optical imaging. a Fluorescence intensity image of organs and tumors at 620 nm. b Spectral classified image. A total of ten images were recorded within the spectral range of 500 to 680 nm with a step size of 20 nm. Then, the images were analyzed using software we developed. While the red color represents HerGa fluorescence, the green color represents autofluorescence. Spectral signatures of autofluorescence (green) and HerGa (red) were extracted from a pre-constructed signature library. c Fluorescence lifetime images of tumors and liver (the upper image) and the fluorescence lifetime histograms (the lower image). d Two-photon excited fluorescence image of tumors. The image of tumor was acquired using fs pulsed laser at 848 nm, an emission filter with 620±60 nm, Nikon 60× objective.
Feasibility of Multimodal Optical Imaging of HerGa for Tumor Detection and Delineation In Vivo
As shown previously, the multimodal optical imaging ex vivo provides synergetic information in the assessment of HerGa, suggesting its potential for tumor detection and delineation in vivo. Thus, we performed multimodal optical imaging (fluorescence intensity, spectral, lifetime, and two-photon excited fluorescence imaging) of HerGa for tumor detection and delineation, after the injection of HerGa into tumors and non-tumor regions. Fig. 6 shows the images obtained by using multimodal optical imaging. The fluorescence intensity and the spectral classification image (Fig. 6a, b) show the HerGa-injected regions. In the fluorescence intensity image, we can observe HerGa relative concentration in the injected regions (Fig. 6b). On the other hand, in the spectral classification image, we can clearly distinguish HerGa localization more quantitatively and specifically (Fig. 6a). Here, the injected regions are classified as red and blue pseudocolor, indicating highly concentrated accumulation of HerGa compared to autofluorescence (A•F) [36]. In addition, we performed two-photon excited fluorescence imaging of a tumor region (region 1) of the mouse shown in Fig. 6d. The two-photon excited fluorescence images provide highly resolved information around the tumor regions where HerGa accumulates. Most importantly, the fluorescence lifetimes (1.9–2.2 ns) of HerGa in tumors (region 1 and 2) are higher than those (1.2–1.5 ns) in muscles (region 3). Thus, we can discriminate between the tumors and normal regions by the fluorescence lifetime difference. This shows the potential of HerGa for the tumor detection and delineation, with additional potential for assessing micro-environmental status in the tumors such as pH and oxygenation.
Fig. 6.
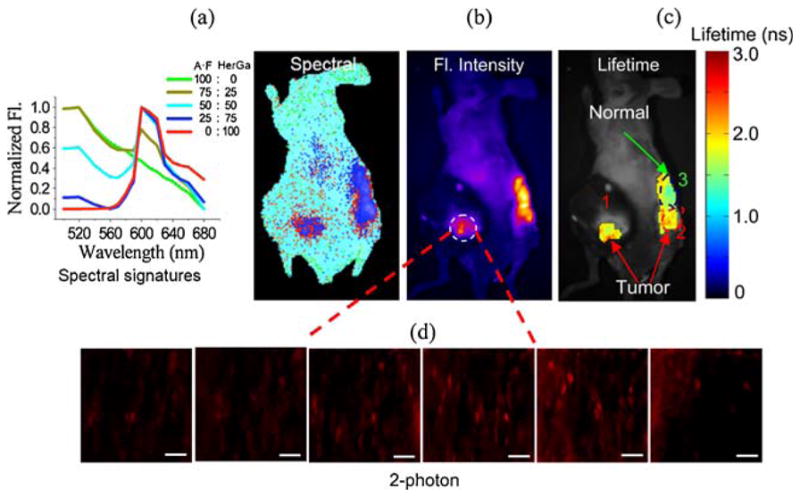
Multimodal optical imaging for cancer detection and delineation of HerGa. a Spectrally classified image. This spectral classified image was obtained through using the signatures (left side) constituted of various ratios of autofluorescence and HerGa fluorescence. b Fluorescence intensity image. c Fluorescence lifetime image. HerGa injection area: (1) tumor, (2) tumor, and (3) non-tumor. d Two-photon excited fluorescence images of tumor regions. These two-photon excited fluorescence images of tumor were acquired at the different focal planes (20, 40, 60, 80, 100, and 120 μm; ex, 848 nm; em, 620±60 nm, and Nikon 60× objective). The scale bar represents 50 μm.
Discussion and Conclusions
The work described here validates the design and functionality of our multimode imaging system and shows that it can be optimized for various applications, benefiting from the performance of each individual imaging mode, as well as the synergetic possibilities enabled by the multimode approach. In Fig. 3a, the drug molecules accumulated in the brain tumor were clearly distinguished, allowing establishment and monitoring of their brain tumor targeting capacity in vivo. In addition, this imaging mode in our system provided significantly better contrast than the leading commercial imaging systems, mostly due to versatile wavelength selection. In our multimode imaging system, the spectral capability enhances quantitation ability in small animal imaging, as shown in Fig. 3b. In the image, the regions where fluorescein is accumulated are clearly delineated by the spectral classification. This imaging capability can correctly measure corrected fluorescence intensity under severe background noise conditions (autofluorescence or other fluorophores), as we first proposed many years ago. In addition, we have incorporated FLIM capability into our system in order to obtain additional information including the environment surrounding our molecules of interest. Using this mode, we could also discriminate fluorescein from autofluorescence in a nude mouse (Fig. 3c). Furthermore, this imaging mode can be used for monitoring tissue functional status in the vicinity of fluorophores [13, 17, 37]. Intravital confocal imaging has also demonstrated its usefulness for detecting structural and functional (including pathological) information in multimode optical imaging (Fig. 3d). Microstructures such as skeletal muscle fibers or vessels were observed using this imaging mode without a biopsy. Thus, the convergence of modes allows monitoring of intact tissues inside small animals with high magnification and resolution. Scanning/WTEF imaging can further enhance this, by providing better contrast and more penetration depth than one-photon excited fluorescence imaging in our system [20, 35]. When compared to scanned two-photon excitation, the speed of WTEF imaging has the advantage of reducing any artifacts due to the movement of animals during in vivo image acquisition. Thus, it may be highly useful for in vivo imaging even though its resolution is less than that of scanning two-photon excited fluorescence imaging. In addition, bioluminescence imaging, combined with fluorescence measurements, could allow improved bioluminescence resonance transfer (BRET) studies.
We have shown that the multimode optical imaging system can be specifically useful for the assessment of chemotherapy molecules of promise since it allows simultaneous use of multiple imaging modes (fluorescence intensity, spectral, fluorescence lifetime, and two-photon excitation imaging), yielding complementary information. In this paper, the tumor targeting capability, retention, clearance, and functional characteristics of chemotherapy molecules were investigated using in the multimode advantages of our system. In a previous study [28], we could monitor the dynamic accumulation of nanoconstructs over the whole mouse in real time. The nanoconstructs were preferentially accumulated in the tumor region, and we could identify their targeting capability using the fluorescence intensity imaging mode. Here, in addition, nanoconstruct clearance from the mouse was examined using spectral imaging at different time points after nanoconstruct IV injection, yielding more accurate information than fluorescence intensity imaging at a single wavelength (Fig. 4), since spectral imaging is sensitive to molecular signatures and more quantitative. We have already reported the use of spectral imaging in the evaluation of accumulation of nanoconstructs into different organs excised from a mouse that received the nanoconstructs through IV injection [33]. In that previous report, spectral image analysis and spectral unmixing method confirmed the fact that the nanoconstructs accumulate specifically in the tumor region and excreting organs like spleen, liver, and kidney, but not in the brain, lung, and heart [33]. In this paper, we utilized spectral imaging in order to examine the extent and topology of HerGa distribution into specific organs and the tumor itself, both ex vivo and in vivo. HerGa is an intensely fluorescent macrocyclic compound that spontaneously assembles with carrier proteins to undergo cell entry. Previously, we observed that the HerGa was preferentially accumulated in breast tumors compared to S2Ga as judged by fluorescence intensity imaging [30]. We confirmed here the drug effects on specific organs using spectral imaging, which provided better quantitative information than standard fluorescence intensity imaging as shown in Fig. 5. While HerGa was clearly shown in whole tumors and in some parts of the liver, HerGa was very sparse in other organs (kidney, lung, heart, spleen, and muscle). Furthermore, FLIM has been performed in order to acquire surrounding tissue information (acidity and/or oxygenation) around tumor regions since fluorescence lifetimes of HerGa depends on pH. The fluorescence lifetimes of HerGa accumulated in the tumors are higher than in the liver (Fig. 5c), and also appear higher than in non-tumor regions in vivo (data not shown). The histogram of fluorescence lifetimes in tumor regions has a greater population at 2.0 ns than in non-tumor regions. Differences are reproducible, and probably underestimated, as the lifetime values may be affected by skin since the fluorescence lifetime values we obtained are the average values from skin, tumor, and muscle. If the skin is removed, the difference of the fluorescence lifetime values of HerGa in tumor and non-tumor regions may become larger. In order to confirm this, we have conducted FLIM measurements after HerGa injection into tumors and normal tissues. In Fig. 6c, the fluorescence lifetime difference of HerGa in the tumors and normal tissues is clearly distinguished. This result verifies that HerGa fluorescence lifetimes in tumors differ from those in normal tissues since tumor regions may be more acidic and hypoxic than normal tissues. Finally, scanning two-photon excited fluorescence imaging of tumor regions provides high magnification and resolution topological detail of the regions of interest: in Fig. 5d, we can clearly see microstructures in the tumors. Eventually, when translated to the clinic, this may help a physician’s decision-making in cancer surgical intervention and detection without the need for rapid histopathological analysis of tissue biopsy during surgery. Also, the fluorescence lifetime difference of HerGa in tumors and liver opens the possibility of HerGa being used for cancer (margin) detection and delineation.
Furthermore, in order to confirm the feasibility of tumor detection using multimodal optical imaging of HerGa in vivo, we used our system after HerGa injection into tumors and a non-tumor region. In Fig. 6, the tumor and non-tumor regions are clearly distinguished in the fluorescence lifetime image, but not as well in the other images. This suggests that HerGa fluorescence lifetimes in tumor certainly differ from those in normal tissues since tumor regions may be more acidic and hypoxic than normal tissues. Additionally, fluorescence intensity, spectral, and scanning two-photon excited fluorescence imaging here provide complementary information to the FLIM such as the relative injection of HerGa, HerGa localization, and HerGa distribution with high resolution and magnification at different depths.
In conclusion, we have developed and tested an advanced optical imaging system that allows functional multiscale imaging (under-the-surface whole-body or endoscopic imaging with microscopic resolution) of small animals in vivo. The multimode optical imaging system, whose concept was derived from our previous work in microscopy [38], can be utilized and optimized for various applications. Furthermore, the combination of fluorescence intensity, spectral, lifetime, and two-photon excited fluorescence imaging can provide complementary, dynamic, quantitative, functional, and highly resolved information for an assessment of nano-constructs and chemotherapy molecules. These capabilities may also enhance contrast in detecting diseased tissues or in evaluation of drug effects in chemotherapy. In this system, other imaging modes such as 3D volumetric fluorescence imaging, elastic scattering imaging, spinning disk confocal imaging, and Raman or time-gated photon propagation also can be included for more versatility and applications. Particularly, we already have constructed optical components and developed a program to enable acquisition of fluorescence images at different angles for the 3D volumetric fluorescence imaging and obtained preliminary results (data not shown). However, highly sophisticated simulations will be needed for 3D fluorescence volumetric imaging [5, 39]. This work will allow monitoring of volumetric tumor growths in vivo. Also, our scanning/wide-field two-photon excited fluorescence imaging unit needs to be better incorporated into the system in order to minimize animal disturbance during the experiment. Finally, the multimode optical imaging of HerGa suggests the possibility as a novel method for cancer detection and treatments. For clinical studies, optimization for sensitivity and specificity, and better, more time-critical decision-making in cancer management is needed. For this, a multimode optical imaging system for a human (clinical) use should be designed. For surgical detection and intervention, our multimode approach can be combined with an endoscope or surgical microscope [40–42].
Acknowledgments
We thank Dr. Mark Gaon for help developing and testing the gated anesthesia instrument. Some of this work was done in partial fulfillment of Ph.D. thesis research requirements by Dr. J.Y. Hwang, at the University of Southern California. Work at the California Institute of Technology was supported by the Arnold and Mabel Beckman Foundation. Z.G. thanks Johnson & Johnson for research support. We are grateful for the following federal support of our research: NIH (5R01CA123495-03 and 1U01CA151815-0) to JYL; NIH (1R01 CA140995 and 1R01 CA129822) and DOD W81XWH-06-1-0549 to LKMK; and US Navy Bureau of Medicine and Surgery (1435-04-04-GT-41387 and -43096), NIH (N01-CO-07119), and NSF (BESOO 79483) to DLF.
Footnotes
Conflict of Interest. The authors declare they have no conflicts of interest.
References
- 1.Weissleder R. Scaling down imaging: molecular mapping of cancer in mice. Nat Rev Cancer. 2002;2:11–18. doi: 10.1038/nrc701. [DOI] [PubMed] [Google Scholar]
- 2.Contag CH. In vivo pathology: seeing with molecular specificity and cellular resolution in the living body. Annu Rev Pathol. 2007;2:277–305. doi: 10.1146/annurev.pathol.2.010506.091930. [DOI] [PubMed] [Google Scholar]
- 3.Rice BW, Cable MD, Nelson MB. In vivo imaging of light-emitting probes. J Biomed Opt. 2001;6:432–440. doi: 10.1117/1.1413210. [DOI] [PubMed] [Google Scholar]
- 4.Deniset-Besseau A, Leveque-Fort S, Fontaine-Aupart MP, et al. Three-dimensional time-resolved fluorescence imaging by multifocal multiphoton microscopy for a photosensitizer study in living cells. Appl Opt. 2007;46:8045–8051. doi: 10.1364/ao.46.008045. [DOI] [PubMed] [Google Scholar]
- 5.Zacharakis G, Ripoll J, Weissleder R, et al. Fluorescent protein tomography scanner for small animal imaging. IEEE Trans Med Imaging. 2005;24:878–885. doi: 10.1109/tmi.2004.843254. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman RM. In vivo imaging of metastatic cancer with fluorescent proteins. Cell Death Differ. 2002;9:786–789. doi: 10.1038/sj.cdd.4401077. [DOI] [PubMed] [Google Scholar]
- 7.Paulmurugan R, Massoud TF, Huang J, et al. Molecular imaging of drug-modulated protein-protein interactions in living subjects. Cancer Res. 2004;64:2113–2119. doi: 10.1158/0008-5472.can-03-2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamauchi K, Yang M, Jiang P, et al. Real-time in vivo dual-color imaging of intracapillary cancer cell and nucleus deformation and migration. Cancer Res. 2005;65:4246–4252. doi: 10.1158/0008-5472.CAN-05-0069. [DOI] [PubMed] [Google Scholar]
- 9.Hwang JY, Agadjanian H, Medina-Kauwe LK, et al. Large field of view scanning fluorescence lifetime imaging system for multimode optical imaging of small animals. Proceeding of SPIE. 2008;6859:68590G–68598G. doi: 10.1117/12.769305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Contag CH, Bachmann MH. Advances in in vivo bioluminescence imaging of gene expression. Annu Rev Biomed Eng. 2002;4:235–260. doi: 10.1146/annurev.bioeng.4.111901.093336. [DOI] [PubMed] [Google Scholar]
- 11.Levenson RM, Lynch DT, Kobayashi H, et al. Multiplexing with multispectral imaging: from mice to microscopy. ILAR J. 2008;49:78–88. doi: 10.1093/ilar.49.1.78. [DOI] [PubMed] [Google Scholar]
- 12.Chung A, Karlan S, Lindsley E, et al. In vivo cytometry: a spectrum of possibilities. Cytometry A. 2006;69:142–146. doi: 10.1002/cyto.a.20220. [DOI] [PubMed] [Google Scholar]
- 13.Hanson KM, Behne MJ, Barry NP, et al. Two-photon fluorescence lifetime imaging of the skin stratum corneum pH gradient. Biophys J. 2002;83:1682–1690. doi: 10.1016/S0006-3495(02)73936-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng Y, Huang SH. The analysis of sinusoidal modulated method used for measuring fluorescence lifetime. Guang Pu Xue Yu Guang Pu Fen Xi. 2007;27:2523–2526. [PubMed] [Google Scholar]
- 15.Lin HJ, Herman P, Lakowicz JR. Fluorescence lifetime-resolved pH imaging of living cells. Cytometry A. 2003;52:77–89. doi: 10.1002/cyto.a.10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esposito A, Gerritsen HC, Oggier T, et al. Innovating lifetime microscopy: a compact and simple tool for life sciences, screening, and diagnostics. J Biomed Opt. 2006;11:34016. doi: 10.1117/1.2208999. [DOI] [PubMed] [Google Scholar]
- 17.Lin HJ, Szmacinski H, Lakowicz JR. Lifetime-based pH sensors: indicators for acidic environments. Anal Biochem. 1999;269:162–167. doi: 10.1006/abio.1999.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cubeddu R, Canti G, Taroni P, et al. Time-gated fluorescence imaging for the diagnosis of tumors in a murine model. Photochem Photobiol. 1993;57:480–485. doi: 10.1111/j.1751-1097.1993.tb02322.x. [DOI] [PubMed] [Google Scholar]
- 19.Cubeddu R, Canti G, Pifferi A, et al. Fluorescence lifetime imaging of experimental tumors in hematoporphyrin derivative-sensitized mice. Photochem Photobiol. 1997;66:229–236. doi: 10.1111/j.1751-1097.1997.tb08648.x. [DOI] [PubMed] [Google Scholar]
- 20.Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science (New York) 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 21.Patterson GH, Piston DW. Photobleaching in two-photon excitation microscopy. Biophys J. 2000;78:2159–2162. doi: 10.1016/S0006-3495(00)76762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chirico G, Cannone F, Baldini G, et al. Two-photon thermal bleaching of single fluorescent molecules. Biophys J. 2003;84:588–598. doi: 10.1016/S0006-3495(03)74879-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falati S, Gross P, Merrill-Skoloff G, et al. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med. 2002;8:1175–1181. doi: 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- 24.Laemmel E, Genet M, Le Goualher G, et al. Fibered confocal fluorescence microscopy (Cell-viZio) facilitates extended imaging in the field of microcirculation. A comparison with intravital microscopy. J Vasc Res. 2004;41:400–411. doi: 10.1159/000081209. [DOI] [PubMed] [Google Scholar]
- 25.St Croix CM, Leelavanichkul K, Watkins SC. Intravital fluorescence microscopy in pulmonary research. Adv Drug Deliv Rev. 2006;58:834–840. doi: 10.1016/j.addr.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Bhaumik S, Lewis XZ, Gambhir SS. Optical imaging of Renilla luciferase, synthetic Renilla luciferase, and firefly luciferase reporter gene expression in living mice. J Biomed Opt. 2004;9:578–586. doi: 10.1117/1.1647546. [DOI] [PubMed] [Google Scholar]
- 27.Lee BS, Fujita M, Khazenzon NM, et al. Polycefin, a new prototype of a multifunctional nanoconjugate based on poly(beta-L-malic acid) for drug delivery. Bioconjugate chem. 2006;17:317–326. doi: 10.1021/bc0502457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hwang JY, Moffatt-Blue C, Equils O, et al. Multimode optical imaging of small animals: development and applications. Prog Biomed Optics Imaging. 2007;8:1–10. [Google Scholar]
- 29.Agadjanian H, Weaver JJ, Mahammed A, et al. Specific delivery of corroles to cells via noncovalent conjugates with viral proteins. Pharm Res. 2006;23:367–377. doi: 10.1007/s11095-005-9225-1. [DOI] [PubMed] [Google Scholar]
- 30.Agadjanian H, Ma J, Rentsendorj A, et al. Tumor detection and elimination by a targeted gallium corrole. Proc Natl Acad Sci U S A. 2009;106:6105–6110. doi: 10.1073/pnas.0901531106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gammon ST, Leevy WM, Gross S, et al. Spectral unmixing of multicolored bioluminescence emitted from heterogeneous biological sources. Anal Chem. 2006;78:1520–1527. doi: 10.1021/ac051999h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wachsmann-Hogiu S, Hwang JY, Lindsley E, et al. Wide-field two-photon microscopy: features and advantages for biomedical applications. Prog Biomed Optics Imaging. 2007;8:1–8. [Google Scholar]
- 33.Frykman PK, Lindsley EH, Gaon M, et al. Spectral imaging for precise surgical intervention in Hirschsprung’s disease. J Biophotonics. 2008;1:97–103. doi: 10.1002/jbio.200710016. [DOI] [PubMed] [Google Scholar]
- 35.Hwang JY, Wachsmann-Hogiu S, Ramanujan VK, et al. Multimodal wide-field two-photon excitation imaging: characterization of the technique for in vivo applications. Biomed Opt Express. 2011;2:356–364. doi: 10.1364/BOE.2.000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hwang JY, Gross Z, Gray HB, et al. Ratiometric spectral imaging for fast tumor detection and chemotherapy monitoring in vivo. J Biomed Opt. 2011;16:066007. doi: 10.1117/1.3589299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kocisova E, Praus P, Rosenberg I, et al. Intracellular uptake of modified oligonucleotide studied by two fluorescence techniques. Biopolymers. 2004;74:110–114. doi: 10.1002/bip.20055. [DOI] [PubMed] [Google Scholar]
- 38.Farkas DL, Baxter G, DeBiasio RL, et al. Multimode light microscopy and the dynamics of molecules, cells, and tissues. Annu Rev Physiol. 1993;55:785–817. doi: 10.1146/annurev.ph.55.030193.004033. [DOI] [PubMed] [Google Scholar]
- 39.Soubret A, Ntziachristos V. Fluorescence molecular tomography in the presence of background fluorescence. Phys Med Biol. 2006;51:3983–4001. doi: 10.1088/0031-9155/51/16/007. [DOI] [PubMed] [Google Scholar]
- 40.Evans JA, Nishioka NS. Endoscopic confocal microscopy. Curr Opin Gastroenterol. 2005;21:578–584. doi: 10.1097/01.mog.0000174217.62214.10. [DOI] [PubMed] [Google Scholar]
- 41.Takehana S, Kaneko M, Mizuno H. Endoscopic diagnostic system using autofluorescence. Diagn ther endosc. 1999;5:59–63. doi: 10.1155/DTE.5.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ikeda N, Honda H, Katsumi T, et al. Early detection of bronchial lesions using lung imaging fluorescence endoscope. Diagn ther endosc. 1999;5:85–90. doi: 10.1155/DTE.5.85. [DOI] [PMC free article] [PubMed] [Google Scholar]