

Summary
The 2′ OH of the peptidyl-tRNA substrate is thought to be important for catalysis of both peptide bond formation and peptide release in the ribosomal active site. The release reaction also specifically depends on a release factor protein (RF) to hydrolyze the ester linkage of the peptidyl-tRNA upon recognition of stop codons in the A site. Here we demonstrate that certain amino acid substitutions (in particular those containing hydroxyl or thiol groups) in the conserved GGQ glutamine of release factor RF1 can rescue defects in the release reaction associated with peptidyl-tRNA substrates lacking a 2′ OH. We explored this rescue effect through biochemical and computational approaches that support a model where the 2′-OH of the P-site substrate is critical for orienting the nucleophile in a hydrogen bonding network productive for catalysis.
Introduction
Termination of translation is signaled when a stop codon in the mRNA is positioned in the A site of the ribosome decoding center. In bacteria, stop codons are recognized by two class 1 release factors (RF1 and RF2) with overlapping specificity for the three codons, UAG, UGA and UAA. Upon recognition of stop codons, release factors engage the active site of the large ribosomal subunit where they catalyze the nucleophilic attack of water on the ester linkage of the peptidyl-tRNA. The reaction is expected to proceed through a tetrahedral intermediate that breaks down to form the free peptide and deacylated tRNA, now protonated at the 3′ OH leaving group.
While peptidyl transfer and peptide release involve distinct chemical reactions (aminolysis vs. hydrolysis, respectively), their core mechanisms are thought to share a number of features in common. Conformational rearrangements stabilized by A-site substrate binding have been documented by biochemical (Beringer and Rodnina, 2007; Brunelle et al., 2006; Caskey et al., 1971; Shaw and Green, 2007) and structural (Laurberg et al., 2008; Schmeing et al., 2005b) studies. Moreover, ribosomal active site nucleotides are not believed to play a direct role in chemical catalysis as mutations in these nucleotides generally had little effect on either reaction, with one exception being the identity of nucleotide A2602 for peptide release (Polacek et al., 2003; Youngman et al., 2004). Interestingly, abasic substitutions at this position were active for release (Amort et al., 2007) and subsequent structural work on release complexes indicated that A2602 is occluded from the catalytic center by the release factor protein, making even this nucleotide unlikely to be directly involved in catalysis (Laurberg et al., 2008).
The class 1 release factors in bacteria and eukaryotes have a universally conserved GGQ motif that is positioned near the site of catalysis (Frolova et al., 1999; Mora et al., 2003; Rawat et al., 2003; Song et al., 2000). Mutation of the GGQ glycines in the bacterial release factors decreased the rate constant of the release reaction by up to four orders of magnitude (Shaw and Green; Zavialov et al., 2002), while mutation of the glutamine showed more modest defects (Mora et al., 2003; Seit-Nebi et al., 2001; Zavialov et al., 2002). The glycines are likely important for the conformation of the active site as it was found that on the ribosome the GGQ tripeptide backbone takes on an orientation not allowed by any other amino acid (Laurberg et al., 2008). Biochemical studies found that the identity of the glutamine side-chain affects the nucleophile specificity of the reaction (Shaw and Green, 2007), consistent with structural data placing the GGQ glutamine in a position to interact with the nucleophile of the reaction or other functionally critical moieties (like the 3′ OH leaving group or oxyanionic intermediate) (Korostelev et al., 2008; Laurberg et al., 2008; Trobro and Aqvist, 2009; Weixlbaumer et al., 2008). These studies together suggest that the glutamine may play a more important role in the reaction than initially suggested by relatively modest kinetic effects.
The A76 2′ OH of the peptidyl-tRNA substrate is the only moiety proposed to be critical for both peptidyl transfer and peptide release. For both reactions, when the 2′ OH (of A76) of the peptidyl-tRNA substrate is substituted with deoxy (dA76) or fluoro (fA76) groups, the rate of the reaction is substantially diminished (Brunelle et al., 2008; Hecht et al., 1974; Weinger et al., 2004). While substitution of the 2′ OH is expected to affect the inherent chemical reactivity of the substrates (Herschlag et al., 1993; Hiller et al., 2010), the observed rate defects on the ribosome appear to surpass the expected losses in chemical reactivity. Further experiments in an in vitro translation system have called the importance of the 2′ OH into question, as a modified 2′ dA76 tRNASer was able to incorporate a catalytic serine into an active esterase 2 enzyme (Huang and Sprinzl, 2011; Koch et al., 2008). The discrepancy between the results appears to be explained in part by a role for the 2′ OH in preventing inactivating conformational rearrangements in the active site, though there remain substantial contributions from the 2′ OH to catalysis (Zaher et al., 2011).
There are multiple models for how the 2′ OH of the peptidyl-tRNA substrate could contribute to catalysis. In one model, the 2′ OH is simply important for the conformation of the active site and positioning of the substrates, likely facilitated by a network of hydrogen bonding interactions. While structural studies detected little difference in the orientation of deoxy-substituted substrate and transition state analogs bound to the P-site, the binding affinity of the deoxy-substituted substrate was somewhat affected, consistent with a perturbation of the hydrogen bonding network (Schmeing et al., 2005a). In another model, the 2′ OH plays an active role in proton transfer, acting as a general base to abstract a proton from the nucleophile or as a general acid to protonate the leaving group. Such a model would require the 2′ OH to assume a net positive or negative charge (i.e. to have a shifted pKa), though crystallographic evidence fails to identify nearby groups capable of causing such a pKa shift (Schmeing et al., 2005a). Moreover, pH studies of the PT reaction found that the only titratable group affecting the reaction was the amine nucleophile (Brunelle et al., 2006; Katunin et al., 2002).
A third and favored model invokes a proton-shuttle mechanism wherein the critical 2′ OH simultaneously accepts a proton from the attacking nucleophile and donates a proton to the 3′ OH leaving group, through a concerted transfer (Chamberlin et al., 2002; Das et al., 1999; Dorner et al., 2002; Griffin and Reese, 1964; Weinger and Strobel, 2006). This model fits with much of the experimental literature including mutational (Brunelle et al., 2008; Dorner et al., 2003; Quiggle et al., 1981; Weinger et al., 2004) and thermodynamic (Sievers et al., 2004) studies. Structural and molecular modeling studies (Hansen et al., 2002; Schmeing et al., 2005a; Trobro and Aqvist, 2005) provide further support for such a model since the nucleophilic water in the ribosomal active site appears to be wholly inaccessible to solvent, thus making proton movement through the 2′OH of the peptidyl-tRNA plausible. A model invoking concerted proton transfer was not supported by recent proton inventory studies which found only one proton transferred in the rate-limiting transition state, however a number of other models involving hydroxide catalysis or step-wise proton transfer through the 2′OH have been proposed (Kuhlenkotter et al., 2011). Here we report that certain amino acid substitutions of the Q of the GGQ (those that provide a group capable of proton donation) can rescue the defects associated with loss of the P-site substrate 2′ OH. These results are consistent with the observation that the glutamine side-chain is positioned to affect catalysis of peptide release and support a model where the P-site substrate 2′ OH is a critical component of a pre-organized hydrogen bonding network essential to peptide release catalysis.
Results
Release activity with 2′ deoxy A76 peptidyl-tRNA substrate is restored with RF1 Q235 mutants
To further explore the molecular basis of the overall diminished reactivity of the 2′ dA76 peptidyl-tRNA substrates, we evaluated release activity for a series of RF1 enzymes carrying a variety of side-chains substituted at position Q235 (of the conserved GGQ). Ribosome “termination” complexes with ribose or deoxy-substituted A76 dipeptidyl-tRNA (f-Met-Lys-tRNALys) in the P site (rA76 or dA76, respectively) and a UAG stop codon poised in the A site were prepared (Brunelle et al., 2008; Weinger et al., 2004). The complexes were then reacted with a saturating amount of the RF1 variants and the rate of dipeptide release was measured as previously described (Shaw and Green, 2007). Consistent with earlier studies, the control complexes with rA76 tRNA substrate reacted efficiently with release factor RF1 with a rate constant of ~0.1 s−1 while the dA76 substrate was severely deficient in the rate of peptide release with a near background rate constant of 6.8 × 10−6 s−1 (Fig. 1A, Table 1) (Brunelle et al., 2008). We next tested several of the RF1 variants, Q235G and Q235A, that had been shown to be highly active on rA76 substrates but which exhibited substantial losses in nucleophile specificity (Shaw and Green, 2007). Both the glycine and alanine side-chains are relatively small and we reasoned might permit increased solvent accessibility with the dA76 peptidyl-tRNA substrate, as previously suggested for the standard rA76 reaction (Trobro and Aqvist, 2007). While the Q235A showed no increase in reactivity with the 2′ dA76 substrate, we observed a substantial increase (9-fold over wild type RF1) with the Q235G variant on 2′ dA76 (and a modest decrease, 3-fold, in activity on 2′ rA76) (Fig. 1A, Table 1).
Figure 1. Peptide release from deoxy tRNA substrate with RF1 mutants.

(A) Log-scale bar graph of rate constants for peptide release on P-site 2′ rA76 (white) or dA76 (dark grey) programmed ribosomes with RF1 wild type or variants. Reactions were carried out at saturating concentration of RF1 (5 μM) and error bars represent standard error from at least two repeated measurements. (B) Endpoint assay testing reactivity of RF1 serine variants with same 2′ dA76-programmed ribosomes. Time point for GGS variant was 1 min while others were taken after 2 hrs. (C) Plot of the rate enhancement relative to the background rate for RF1 and unacylated tRNA stimulated release with both rA76 and 2′ dA76 substrates.
Table 1.
Rate constants for peptide release with RF1 mutants on rA76 or dA76 tRNA substrates
| rA76
|
dA76
|
|||
|---|---|---|---|---|
| kobs (s−1) | Relative to wild type (-Fold) | kobs (s−1) | Relative to Wild type (-Fold) | |
| RF1 | 9.8 × 10−2 ± 0.4 × 10−2 | 6.8 × 10−6 | ||
|
| ||||
| GGG | 2.8 × 10−2 | 0.29 | 6.1 × 10−5 | 8.9 |
|
| ||||
| GGA | 1.9 × 10−1 | 1.9 | 6.2 × 10−6 | 0.90 |
|
| ||||
| GGS | 4.0 × 10−1 | 4.1 | 2.7 × 10−2 ± 0.1 × 10−2 | 4000 |
|
| ||||
| GGC | 7.1 × 10−3 | 0.073 | 1.7 × 10−4 ± 0.3 × 10−4 | 25 |
|
| ||||
| GGT | 1.0 × 10−1 | 1.0 | 1.2 × 10−2 ± 0.1 × 10−2 | 1800 |
|
| ||||
| GGM | 3.7 × 10−2 | 0.37 | n.a. | |
|
| ||||
| GGL | 5.3 × 10−3 | 0.054 | 9.8 × 10−7 | 0.14 |
|
| ||||
| GGN | 7.0 × 10−4 | 0.0072 | 3.8 × 10−6 | 0.55 |
|
| ||||
| GGD | 2.4 × 10−4 | 0.0025 | 1.9 × 10−6 | 0.28 |
|
| ||||
| GGE | 6.6 × 10−3 | 0.068 | 8.6 × 10−7 | 0.13 |
|
| ||||
| GGH | 2.3 × 10−4 | 0.0024 | n.a. | |
|
| ||||
| GGK | 1.0 × 10−2 | 0.10 | 4.5 × 10−6 | 0.66 |
|
| ||||
| GGW | 7.6 × 10−3 | 0.078 | 6.4 × 10−7 | 0.094 |
|
| ||||
| GAQ | 3.1 × 10−5 | 0.00031 | 6.1 × 10−6 | 0.89 |
|
| ||||
| AGQ | 4.0 × 10−5 | 0.00041 | 1.3 × 10−5 | 2.0 |
|
| ||||
| RF- | 3.3 × 10−5 ± 1.1 × 10−5 | 0.00034 | 9.6 × 10−7 ± 0.9 × 10−7 | 0.14 |
n.a. = no activity observed
Most interestingly, we observed dramatic increases in release activity on the 2′ dA76 substrate with either the Q235S or Q235T mutants (4000- and 1800-fold faster than the wild type RF1 on dA76, respectively) (Fig. 1A). These variants (Q235S and Q235T) catalyzed the reaction with rate constants of 2.8 × 10−2 s−1 and 1.2 × 10−2 s−1, respectively, only about 4- to 8-fold slower than the rate with the normal rA76 substrate and wild type RF1 (Fig. 1A, Table 1). The only other RF1 variant that exhibited notable increases of dA76 substrate reactivity was Q235C with a rate constant of 1.7 × 10−4 s−1, 25-fold faster than the wild type RF1 on dA76 (Table 1). Thus, while many RF1 variants retained activity on rA76 substrates (Frolova et al., 1999; Mora et al., 2003; Shaw and Green, 2007), only certain amino acid substitutions rescued the defect of the dA76 substrate.
Release activity with 2′ dA76 tRNA substrate critically depends on the position of the rescuing amino acid substitution
Next we set out to determine whether the observed dA76 rescue effect was specific to the Q235 position of RF1. To test this we made serine substitutions in several positions in RF1 immediately surrounding Q235 including G233, G234 and H236. These variant RFs were reacted with ribosome complexes containing the 2′ dA76 peptidyl-tRNA substrate and release activity was evaluated. After 1 minute, the GGSH (Q235S) variant reaction had reacted to completion while with the other three variants (GGQH (WT), SGQH (G233S), GSQH (G234S) and GGQS (H236S)) essentially no reaction had occurred after 2 hours (Fig. 1B). Based on these results it appears that placement of a hydroxyl group specifically at the Q235 position is critical for the rescue of peptide release activity with the dA76 substrate.
Stimulation of peptide release from 2′ dA76 tRNA substrate with unacylated A-site tRNA
Early biochemical experiments demonstrated that addition of an unacylated tRNA, or simply a CCA trinucleotide, to the ribosomal A site could stimulate peptide release (Caskey et al., 1971; Zavialov et al., 2002). This release activity is thought to be triggered by conformational changes induced by A-site substrate engaging the A loop of the ribosomal RNA (Brunelle et al., 2006; Schmeing et al., 2005b; Youngman et al., 2004) and by interaction of the 3′ OH of the A-site substrate with an active site water that is then oriented for nucleophilic attack (Simonovic and Steitz, 2008; Trobro and Aqvist, 2007). We asked whether unacylated tRNA could similarly stimulate peptide release from the 2′ dA76 dipeptidyl-tRNA substrate by adding near-saturating levels of unacylated tRNAPhe (20 μM) to release complexes programmed with the corresponding UUU sense codon in the A site. The unacylated tRNA stimulated release from the rA76 substrate by 60-fold (as previously reported) and on the dA76 substrate by about 24-fold over background (Fig. 1C). The equivalence of these values is striking when compared with the very different reactivity of the rA76 and dA76 dipeptidyl-tRNA substrate with wild type RF1.
Activity of release complexes after long incubations
In order to provide some estimate of the contribution of the 2′ OH moiety to catalysis in both the PT and peptide release reactions, we wanted to assess whether the P-site tRNA remained not only bound to the ribosome, but bound in a ribosome complex that is competent for catalysis. We took advantage of the ability of the GGS RF1 variant to catalyze release with either rA76 or dA76 substrates to test the activity of the dipeptidyl-tRNA termination complexes after long periods of incubation. Complexes were incubated for the indicated time points, and then an aliquot was removed and reacted with RF1 GGS for an additional 10 minutes to test complex reactivity. For complexes bearing rA76 substrate (with no RF present), the rate constant for background hydrolysis was measured at 3.3 ×10−5 s−1, consistent with previous reports (Brunelle et al., 2008; Zavialov et al., 2002), and appeared to derive from substrate that is bound to active ribosome complexes as indicated by the high degree of reactivity with RF1 GGS throughout the time course (Fig. 2A).
Figure 2. Reactivity of complexes with rA76 or dA76 substrates.

Ribosome complexes containing (A) rA76 or (B) 2′ dA76 peptidyl-tRNA substrate were incubated in reaction buffer at 37°C. At each indicated time point, aliquots were taken and reacted with (○) RF1 Q235S to test reactivity or (■) 1x buffer to measure uncatalyzed hydrolysis.
The rate constant for background hydrolysis from dA76 substrate complex was measured at 9.6 × 10−7 s−1, consistent with previous measurements (Brunelle et al., 2008), and substantially slower than the rate constant for rA76 substrate (approximately 35- to 50-fold) (Fig. 2B). Even after 120 hours of incubation at 37°C the majority of these complexes remained reactive with RF1 GGS indicating that dipeptidyl-tRNA substrate is bound to the ribosome throughout the time course of the reaction.
Computer simulations
Earlier MD simulations with the wild type (GGQ) RF1 and rA76 substrate predicted several key features of the termination reaction: (1) an interaction between the oxyanion of the transition state and both a backbone NH group of the Gln residue and a nearby water molecule, (2) an interaction between the Gln (Q) side-chain and the water that serves as the reaction nucleophile, and (3) a proton shuttle involving the 2′ OH of the peptidyl-tRNA (Trobro and Aqvist, 2007, 2009). The simulations predicted a mechanism where the nucleophilic water forms one H-bond with the RF1 Q235 side chain carbonyl group, and donates its other proton to the A76 2′ OH. This geometry yields a proper orientation both of the nucleophile for attack on the ester carbon and of the proton transfer chain (via the 2′ OH) to the leaving group (Trobro and Aqvist, 2007, 2009). This orientation of Q235 is also supported by crystal structures showing H-bonds between the Q235 nitrogen and U2506 (Jin et al., 2010; Korostelev et al., 2008; Laurberg et al., 2008).
To gain insight into the structural and energetic effects of the RF1 mutations, we carried out MD simulations of the termination reaction on the ribosome with GGQ (wt), GGS, GGT, GGC and GGL variants and the 2′ dA76 substrate, utilizing the empirical valence bond (EVB) method as described earlier (Trobro and Aqvist, 2007, 2009). The results from the present calculations are summarized in Fig. 3A. What emerged from this approach is the prediction that the GGS and GGT variants will significantly accelerate the hydrolysis reaction on the dA76 substrate (by a factor of 1000–4000 relative to the uncatalyzed reaction in water). The GGC mutant is also predicted to catalyze the reaction with the dA76 substrate, although at a slower rate than GGS or GGT (1 × 10−4 s−1 in the neutral form, 1 × 10−5 s−1 with an ionized side-chain (data not shown)). In contrast, neither the GGQ nor the GGL mutants are predicted to promote this same reaction. Thus the simulation energetics are in very good general agreement with the experimentally measured biochemical effects.
Figure 3. Predicted energetic and structural effects of RF mutations.

(A) Calculated free energy profiles (in kcal/mol) for the uncatalyzed ester hydrolysis reaction in solution and for termination on the ribosome with the GGQ, GGL, GGC (neutral), GGS and GGT RF variants with 2’dA76 substrate. R, TI and P denote reactants, tetrahedral intermediate and products, respectively. The calculated activation free energy barriers are 26, 25, 26, 23, 22 and 21 kcal/mol for the water, GGQ, GGL, GGC, GGS and GGT reactions, respectively. Average MD structures for GGQ (B), GGL (C) and GGS (D) in the reactant state and for GGS (E), GGT (F) and GGQ (G) in the transition state separating R and TI. Ribosomal RNA, the P-site peptide and the RF are shown with yellow carbons. Water molecules are depicted in red (O) and white (H) and the transition state (E–F) involves partial bonding between the attacking water (wat1) and an auxiliary water molecule (wat2, replacing the A76 2′-OH), as well as H-bonding to the O3′ of dA76. A third conserved water (wat3) is shown stabilizing the oxyanion. The corresponding transition state with a hydroxide ion taking the place of A76 2′-OH is also shown for the GGS reaction (H).
To explore the mechanistic basis for the RF1 variant activity predictions, we examined in more detail the average MD structures along the reaction trajectory. In the MD simulations of all five RF1 variants (including the wild type GGQ) with the dA76 substrate, a second water molecule (wat2), in addition to the nucleophile (wat1), is now seen beneath the dA76 ribose where it can potentially replace the function of the 2′ OH group in proton transfer (Fig. 3B–G). However, the simulations for the GGQ and GGL variants (that predict minimal catalysis) suggest that the availability of a nearby OH group per se is not sufficient for catalysis. Instead, the calculations indicate that the efficiency of the proton shuttle depends on a favorable H-bond network extending from the attacking water molecule (wat1) through the additional water (wat2) to the O3′ that is accessed by GGS, GGT, and GGC but not by GGQ and GGL.
In the GGQ case with the 2′ dA76 substrate, the Gln side-chain carbonyl group accepts an H-bond from the attacking water in the reactant state, which renders the second, bridging, water molecule sub-optimally oriented for mediating proton transfer to the 3′ OH (note the absence of H-bonding between the bridging H2O (wat2) and the 3′ OH in Fig. 3B). In the GGL reactant state, the Leu side-chain fills the remaining space adjacent to the nucleophilic water, similar to the Gln side-chain, but excluding potential polar interactions (Fig. 3C). By contrast, with the GGS and GGT mutants, the side-chain OH group can donate hydrogen bonds both to the attacking water molecule in the reactant state (Fig. 3D) and perhaps more importantly to the developing negative charge on the substrate carbonyl group in the transition state (Fig. 3E,F). Note also that the bridging water molecule (wat2) in the GGS mutant becomes properly pre-oriented for proton transfer to the 3′ OH (Fig. 3D), forming a hydrogen bond with the 3′ leaving group not present with wt-RF1 (Fig. 3B). In the transition state, the interaction between the oxyanion and serine or threonine side-chain and (Fig. 3E,F) has no equivalent interaction with the Q of the GGQ (Fig. 3G). We propose that the additional stabilizing interaction with the oxyanion is necessary with the dA76 substrate, in addition to the normal contributions from the RF backbone NH group (at position 235) and a conserved water molecule (wat3) (Korostelev et al., 2008; Laurberg et al., 2008; Schmeing et al., 2005a; Trobro and Aqvist, 2009; Wallin and Aqvist, 2010; Weixlbaumer et al., 2008). It seems that serine and threonine are the only amino acids of “correct” length, that also have a strong H-bond donor in the γ position to provide this stabilizing interaction.
Simulation of the GGC variant reaction reveals that the sulfhydryl group has a relatively weak interaction with the oxyanion and can partly rotate away from the reaction site, allowing an interaction not observed with the other RF variants between a second water near the A2451 2′ OH and the attacking water (data not shown). The resulting productive orientation of the nucleophile appears to be the main contribution to catalysis as the sulfhydryl does strongly not interact with the oxyanion, probably due to the smaller dipole moment of this group relative to hydroxyl. It seems likely that the GGG mutant acts similarly, allowing extra solvent water to orient the nucleophile and restoring some catalytic activity with dA76 tRNA substrate.
As mentioned, the GGC thiolate derivative yielded a weaker rate acceleration than a neutral cysteine side-chain in our simulation. In this case the thiolate is positioned to accept a hydrogen bond from the water nucleophile but cannot provide any stabilizing interaction with the oxyanion. However, it is possible that an ionized cysteine side-chain could facilitate the creation of a hydroxide ion in the reaction site by deprotonating one of the water molecules. Such a hydroxide could then either attack the ester carbon directly or serve as a base catalyst for neutral water attack, as recently suggested for the wild type reaction with rA76 (Kuhlenkotter et al., 2011).
Based on these ideas, we modeled a hydroxide catalyzed reaction for all of the RF variants with dA76. These simulations do not reveal any obvious stable binding site for a hydroxide ion but indicate that it could transiently occupy the position of the water molecule substituting for the A76 2′ OH (see above). With this placement, we ran reaction simulations where one proton is removed from the system, i.e. where the 2′ OH group of rA76 is effectively replaced by a hydroxide ion rather than by a water molecule. Unfortunately, it is not possible to computationally estimate the absolute free energy of a hydroxide (i.e., the local pKa of water) in the peptidyl transferase center (PTC), and as such the absolute hydrolysis rates cannot be predicted. However, we can calculate the relative rates for this type of mechanism and these turn out to be very similar to those obtained for the neutral water mechanism. Hence, the simulations yield the following rate-acceleration relative to the wild type GGQ, GGS: 14000-fold (−5.7 kcal/mol), GGT: 7000-fold (−5.3 kcal/mol), GGL: 0.06-fold (+1.7 kcal/mol) and GGC: 30-fold (−2.1 kcal/mol), where the free energy of the rate limiting barrier relative to GGQ is given within parentheses. The rate limiting transition state for the hydroxide catalyzed reaction with the GGS variant and the dA76 substrate is shown in Fig. 3H where the structural similarity to the neutral reaction can be seen.
The effects of pH on 2′ dA76 tRNA substrate reactivity with RF1 Q235T and Q235C
To further address the mechanism by which RF1 Q235 mutants can restore peptide release activity to 2′ dA76 P-site substrates, we tested the pH dependence of the reactions. Measurement of the rate constant for these reactions as a function of pH can help distinguish between different possible mechanisms. We first tested the pH-dependence of the release reaction with wild type RF1 and rA76 substrate. Analysis of the plot of log(kobs) versus pH revealed a linear increase in reactivity with pH with a slope of 0.57 ± 0.03 and no plateau (Fig. 4). Thus, the group (or groups) being titrated does not appear to have a pKa in the physiological range.
Figure 4. pH dependence of release kinetics.

pH-dependence of release kinetics for wild type RF1 with 2′ rA76 Met-Lys-tRNALys ribosome complex (▲) or RF1 GGT (■) or GGC (●) with 2′ dA76 ribosome complex. For wild type RF1 and GGT the data is fit to a linear equation, while the GGC data is fit to a model with one ionizing group. Error bars represent standard error of at least two measurements.
We next examined the rates of peptide release from 2′ dA76 tRNA substrates with RF1 Q235S, Q235T and Q235C enzymes over a range of pH values from 6.1 to 8.6. While each reaction is pH dependent, the shapes of the curves differ (Fig. 4). The pH-rate profiles observed with the RF1 Q235S and Q235T mutants (GGS and GGT) increased linearly with pH over the range tested with a slope of 0.52 ± 0.06 and 0.66 ± 0.07 respectively, similar to that observed for the wild type reaction. The pH dependence of the reaction of dA76 with RF1 Q235C is somewhat different and fit to a model with a single protonation event with a pKa of about 7.5. The reaction with the cysteine mutant is also significantly slower than the wild type or threonine reaction (plateauing with a rate constant of 7.8 × 10−4), raising the possibility that this reaction is rate limited by a different step.
Solvent isotope effects on peptide release with Q235 mutants and 2′ dA76 substrate
We next measured the kinetic solvent isotope effects (KSIEs) of peptide release with RF1 and several Q235 variants. If hydrogen bonds with the water nucleophile or other exchangeable hydrogen atoms are formed or broken in the rate-limiting step, the reaction rate is predicted to be affected by isotopic substitution with deuterium. Release complexes were prepared as previously, resuspended in either H2O or D2O buffer, and reacted with RF1 enzyme in the corresponding buffer. Reaction time courses in H2O and D2O were fit to single exponential curves and the kinetic solvent isotope effects (KSIE) were determined by calculating the relative rates of the reactions in the two solvents (kobs H2O/kobs D2O).
We tested isotope effects for wild type RF1 and the Q235 variants in the reaction with rA76 substrate. In the wild type reaction we observed a small solvent isotope effect of ~1.4 (Fig. 5). With the RF1 variants, the GGA variant was more affected by the heavy water isotope with a KSIE of 4.3, compared to 2.1 for the GGG variant. Similar to the wild type reaction, only a small KSIE of ~1.8 was observed with the GGS RF1 variant while both the GGT and GGC mutants had relatively larger KSIE measurements with rA76 substrate (3.9 with GGT and 4.2 with GGC). These data suggest that the different RF1 Q235 mutants alter either the rate limiting step or mechanism of the reaction even with normal rA76 tRNA substrate.
Figure 5. Solvent isotope effects on peptide release with RF1 Q235 mutants.
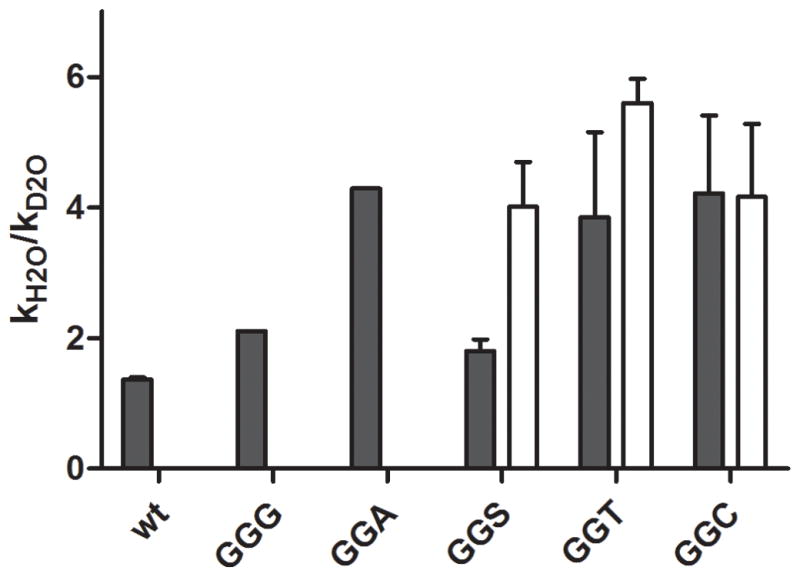
Kinetic solvent isotope effect measurements with RF1 Q235 variants and rA76- (black bar) or 2′ dA76-programmed ribosome complex (white bar). The KSIE was calculated as kobs H2O/kobs D2O. In cases where multiple measurements were available error bars represent +/− standard error.
Deuterium solvent isotope effects were also measured for the release reaction with 2′ dA76 substrate ribosome complexes catalyzed by RF1 mutants GGS, GGT and GGC (as these variants are relatively active on the dA76 substrate). The reaction of RF1 GGS with 2′ dA76 gave a KSIE of ~4.0. This is a relatively larger effect than observed in the wild type reaction or the reaction with GGS and rA76 (Fig. 5). The other dA76-reactive RF1 variants, GGT and GGC, also exhibited large measured KSIEs of 5.6 and 4.2, respectively, with dA76 substrate, similar to their effect with rA76. These results are broadly consistent with models invoking proton movement during the reaction trajectory, since the rates of the reaction are adversely affected by the additional mass of the deuterium atom.
Discussion
Our data reveal that substitutions in the GGQ glutamine side-chain of release factor RF1 can restore peptide release activity to ribosome complexes carrying 2′ dA76 peptidyl-tRNA substrate. The magnitude of the effect was surprisingly large, with variants Q235S and Q235T enhancing reactivity 28,000 and 13,000-fold over background, respectively. The resulting rate constants approach that observed for wild type RF1 and rA76 substrate (Table 1). We observed similar, albeit more modest, effects with the Q235G and Q235C variants. Rescue activity has been observed previously in other ribozymes, usually by adding free bases to an abasic site, and can be used to probe ribozyme mechanism (Peracchi et al., 1996; Peracchi et al., 1998). Interestingly, here we observe rescue of an RNA mutation by a protein moiety.
Several models might be invoked to explain these results. In one possible model, the RF1 variants depend on formation of an acyl-enzyme intermediate, similar to that of the serine proteases, where the modified Q235 side-chain acts as a nucleophile to attack the peptidyl-tRNA ester linkage. A mechanism such as this would likely depend on activation of the serine or threonine side-chain hydroxyl by some other active site group acting as a general base; however, we note that the pH-rate profile of the Q235S and Q235T mutants did not reveal an active site group with a physiological pKa eliminating this as a likely model.. A serine protease type mechanism would also not explain the increased activity of the Q235G mutation with the dA76 substrate as the glycine side-chain does not provide a potential nucleophile to form the acyl-enzyme intermediate. Finally, our MD simulations indicate that the Q235S and Q235T hydroxyl groups are too far away from the ester carbon for direct attack, consistent with a recent 70S-RF crystal structure in a pre-termination state (Jin et al., 2010).
In another potential model for the dA76 rescue reaction, the hydroxyl (or sulfhydryl) group provided in trans by the variant RF side-chain would replace the missing 2′ OH by transferring a proton from the nucleophile to the 3′OH. The computer simulations performed here however suggest that such a direct network is unlikely to form. Instead, proton transfer seems more likely to take place through an additional conserved water molecule (Schmeing et al., 2005a; Wallin and Aqvist, 2010). This bridging water molecule effectively replaces the missing 2′ OH and removes a proton from the nucleophile water in the transition state and subsequently donates this proton to the leaving group. Our simulations further show that a hydroxide ion in place of the water yields very similar results. The side-chain hydroxyl groups of the Q235S and Q235T variants play a key role in this type of mechanism by donating H-bonds both to the attacking water in the reactant state and to the oxyanion in the transition state (Fig. 3D–F,H). The less catalytically active Q235C mutant allows an additional solvent water to donate an H-bond to the attacking water in the reactant state, but is unable to effectively stabilize the transition state oxyanion. This model also can rationalize the increased activity of the Q235G mutant as increased space in the active site pocket would allow additional solvent to enter (Blaber et al. 1995), providing an alternative, more extended route for the proton shuttle.
This overall model, involving an intermediate water molecule (or possibly hydroxide ion) oriented by the different side-chains at position 235, is consistent with the reported kinetic solvent isotope effects, which are larger for the Q235S, Q235T, and Q235C variants with dA76 when compared with the KSIE for wild type RF1 and rA76 peptidyl-tRNA. These elevated KSIEs are consistent with the involvement of an additional solvent molecule in the transition state, and/or with a change in mechanism that involves additional hydrogen bonds with the oxyanion (Scholten et al., 1988). Interestingly, we also observed differences in the measured solvent isotope effects with RF1 Q235 variants (GGA, GGT and GGC) compared to wild type on normal rA76 tRNA substrates. These results could similarly reflect a reaction mechanism with more protons engaged in hydrogen bonding with Q235-substituted RF1 (Trobro and Aqvist, 2007) (or again with a change in rate-limiting step). We note that our measured KSIE with wild type RF1 (1.4) was smaller than the reported KSIE of 4.1 with RF2 (Kuhlenkoetter et al., 2011). Such a difference could reflect differences in the rate-limiting step of the reaction with the two enzymes or other experimental conditions such as pH (7.5 vs. 8.5) or peptidyl-tRNA substrate (f-Met-tRNAMet vs. f-Met-Lys-tRNALys). The reported proton inventory (Kuhlenkoetter et al., 2011) additionally argued that only one proton is transferred in the rate-limiting transition state. This finding is consistent with our model for the hydrolysis reaction since the transition state in all cases is early and involves only one proton in flight (Trobro and Aqvist, 2009), i.e. that of the attacking water (Fig. 3E–H).
The pH dependence of the peptidyl transfer reaction has been well studied using the minimal A-site substrates puromycin and hydroxypuromycin (Brunelle et al., 2006; Katunin et al., 2002) as well as Phelac-tRNAPhe (Bieling et al., 2006) (all substrates where chemistry is thought be rate limiting for the reaction). These studies generally concluded that peptide bond formation does not depend on acid-base catalysis mediated by ribosomal elements. We similarly found no group with a physiological pKa relevant for the release reaction with wild type RF1 and rA76 tRNA substrate and the observed pH dependence for the RF1 GGS and GGT variants with the dA76 tRNA substrate is consistent with the wild type (rA76) result. The linear increase with pH could potentially be attributed to the ionization of a reactive group with very high pKa such as the water nucleophile (Kuhlenkoetter et al., 2011). In the case of GGS, GGT or GGC, ionization of the hydroxyl or thiol side-chain groups could also contribute to the observed pH profile. Titration of either of these groups might allow them to function as a general base in the reaction, though the slopes of the curves are significantly smaller than 1 suggesting that the protonated form of these groups are also reactive. The pH-rate profile with RF1 GGC revealed a titratable group with pKa 7.5 that is tempting to ascribe to the thiol side-chain group, though our modeling of the GGC reaction with the neutral water mechanism suggests that deprotonation of this group should decrease its activity with dA76 substrate. For this reason, the possibility of a hydroxide catalyzed mechanism was addressed here and yielded very similar relative rates for the different RF1 variants compared to the neutral mechanism. Hence, it is difficult to distinguish between these two alternatives as the energetic cost of creating a hydroxide in the PTC cannot be reliably calculated. On the other hand, the involvement of a hydroxide could explain the pH dependence of RF1 GGC if the side-chain thiolate helps generate a hydroxide Another alternative is that a pH-dependent ribosome or RF conformational change that depends on titratable groups affects the rate of hydrolysis with RF1 GGC. Since both guanine and uracil ring nitrogens ionize at around pH 10 such ribosomal groups would statistically deprotonate before water under normal conditions.
The data reporting on the stimulation of dA76 release activity promoted by the unacylated tRNA in the A site are also consistent with the proposed rescue mechanism. Previous studies proposed that the 3′ OH of the unacylated tRNA might be responsible for stimulating release by positioning the nucleophilic water for catalysis (Simonovic and Steitz, 2008; Trobro and Aqvist, 2007). Our data provide biochemical support for such a model, suggesting that the 3′OH of the A-site tRNA functions like the Ser and Thr side-chains of the RF variants by orienting water molecules to promote catalysis. We cannot rule out the possibility that substrate dissociates from the P site over the time course and is blocked from re-binding by unacylated tRNA.
The results of the biochemical and computational experiments presented here support earlier models for release catalysis where a pre-organized hydrogen bonding network allows the substrate 2′ OH to facilitate catalysis through proton transfer (Schmeing et al., 2005a; Trobro and Aqvist, 2005; Wallin and Aqvist, 2010). While this model was originally supported by observed losses in reactivity with dA76 substrate (Brunelle et al., 2008), we now provide a positive read-out in the form of a robust rescue activity to validate it. Our results support the idea that loss of the 2′ OH disrupts the productive hydrogen-bonding network, resulting in mis-orientation of the nucleophile with no facile route for proton transfer (Fig. 6A,B). The Q235S and Q235T RF1 variants (and to some lesser extent Q235G and Q235C), along with additional solvent, orient the nucleophile in a productive manner and provide additional stabilization to the oxyanion in the transition state (Fig. 6C).
Figure 6. Peptide release reaction mechanisms.

(A) Proposed current model for peptide release with wild type RF1 with peptidyl-tRNA substrate. The Q235 glutamine side-chain is shown in the reactant state and the backbone amide is shown in the transition state. (B) Wild type RF1 with deoxy peptidyl-tRNA substrate. (C) RF1 mutant Q235S side chain reaction with deoxy peptidyl-tRNA substrate. Additional solvent and nucleophilic water are shown. The Q235S side chain is shown in the reactant state and both side chain and backbone amide are shown in the transition state.
It was recently reported that ribosome complexes containing a dA76 peptidyl-tRNA substrate undergo a slow inactivation (Zaher et al., 2011). Our complexes prepared according to Zaher et al. are similarly inactivated for peptidyl transfer and peptide release with WT RF1, and yet they react with the RF1 Q235S and Q235T variants. A recent study of peptide bond formation by Sprinzl and colleagues concluded that interaction of the growing polypeptide with the ribosomal peptide exit tunnel could alter the substrate conformation to allow peptidyl transfer without the 2′OH (Huang and Sprinzl, 2011). We suggest that the proposed reorientation with the Q235S mutant could mimic this “active” conformation by orienting the nucleophile, rather than the peptide substrate. Pre-steady state kinetic assays with peptide occupying the exit tunnel would be useful to address these questions in the future.
Significance
The mechanism by which the ribosome and release factor protein catalyze release of the finished polypeptide is an ongoing area of study. In one current model for catalysis of both peptidyl transfer and peptide release, the A76 2′ OH of the P-site tRNA substrate acts as a proton shuttle in a pre-organized hydrogen bonding network to remove a proton from the nucleophile and protonate the 3′ OH leaving group. Here, we demonstrate that serine or threonine mutations which place a hydroxyl at the conserved Q235 position in release factor RF1 are able to compensate for loss of the 2′OH of the P-site tRNA, restoring release activity to near wild type rates. Similar, but more modest effects were also observed with mutants Q235C and Q235G. Our results indicate that, in the absence of the P-site tRNA 2′OH, these mutants can alter the hydrogen bonding network in the active site compared to the wild type enzyme, in a way that is productive for catalysis. These results provide direct support for the idea that the 2′OH is critical for maintaining a productive hydrogen bonding network that facilitates proton transfer in the normally solvent inaccessible active site.
Materials and Methods
Preparation of RF1 mutants and modified tRNA substrates
Mutant plasmids were created using QuikChange XL (Stratagene) site-directed mutagenesis of pET15b-RF1 vector. These plasmids carrying N-terminally His-tagged RF1 were transformed into BL21 (DE3) pLysS cells, expressed and purified as described previously(Shaw and Green, 2007).
Modified tRNA substrates were prepared as described previously (Brunelle et al., 2008; Weinger et al., 2004). Briefly, the 3′ adenosine (A76) of E. coli tRNALys (Chem Block) was removed by one round of periodate-aniline treatment followed by treatment with polynucleotide kinase (PNK), and replaced by incubation with CCA-adding enzyme and adenosine triphosphate or 2′-deoxyadenosine triphosphate. Removal of A76 and subsequent addition of adenosine analogs was confirmed by observing mobility of the tRNA on a denaturing 10% (w/v) acrylamide gel. Aminoacylation of modified tRNALys (13 μM) was carried out by incubation with 27 μM [14C]-lysine (Perkin Elmer Life Sciences), 3 mM ATP, and 14 μM lysyl-tRNA synthetase at 37°C for 20 min in charging buffer (25 mM Tris-Acetate (pH 7.5), 10 mM Mg Acetate, 100 mM NH4Cl, 30 mM KCl, 1 mM DTT).
Peptide release and pH assays
Ribosomes were prepared previously described(Brunelle et al., 2008). Peptide release activity was assayed using dipeptide release complexes, formed as described previously20, which place f-[35S]Met-Lys-tRNALys in the P site and a UAG stop codon in the A site. These complexes were pelleted over sucrose cushion buffer for 2 hr at 69000 rpm, resuspended in reaction buffer C (50 mM HEPES (pH 7.5), 70 mM NH4Cl, 30 mM KCl, 7 mM MgCl2, 1 mM DTT) and stored at −80°C until use. Dipeptide release complexes (0.2 μM) were reacted with a final concentration of 5 μM RF1 wild type or mutant in buffer C at 37°C and dipeptide release was monitored as a function of time. Time points were quenched in 25% formic acid and spotted on TLC-cellulose plates to separate release products from peptide attached to tRNA by TLC electrophoresis. Rate constants for each reaction were obtained from curve fitting to single exponential equations.
For reactions using unacylated tRNAPhe as an A site substrate, ribosome complexes were formed as described, but with a cognate UUU codon in the A site. These complexes were reacted with 20 μM tRNAPhe (Sigma), determined to be a saturating concentration in earlier work (Shaw and Green, 2007), and dipeptide release was followed over time.
To determine the pH dependence of the release reaction with wild type and mutant release factors a concentrated stock of complexes (15 μM) formed as described above were diluted in buffer C at the appropriate pH. Concentrated RF1 or variant stocks (at least 700 μM) were diluted in equivalent buffer to 5 μM and reacted with 0.2 μM complex. To extend the pH range tested either 50 mM PIPES (at pH 6.1, 6.5, and 7.1) or 50 mM Bicine (at pH 7.7, 8.0, 8.25, 8.4, 8.55) were used in place of HEPES (pH 6.7, 7.3, 7.45, 7.7) in buffer C. Different buffers were used at overlapping pH’s to account for possible buffer effects on the rate of peptide release. For the GGC mutant the plot of log kobs vs pH was fit to an equation modeling catalysis dependent on one ionizing group with the equation log(kobs) = log(10−[H] Vmin + VmaxKa) − log(Ka + 10−[H]). The wild type and GGT plot were fit to a linear equation.
Complex reactivity and solvent isotope effect assays
The reactivity of release complexes over long time courses was assayed using the Q235S RF1 mutant. Both rA76 and dA76 dipeptide release complexes (0.2 μM), assembled as described, were incubated in reaction buffer C at 37°C for indicated time points. Aliquots were then mixed with 5 μM RF1 Q235S or equal volume buffer C and incubated for an additional 10 min, allowing the Q235S reaction to go to completion. The aliquot reactions were quenched in 25% formic acid, time points were spotted on TLC-cellulose and separated by TLC electrophoresis. Background release reactions carried out in buffer C only were fit to single exponential curves.
To measure the effect of heavy isotope substitution in the solvent on the release reaction, release assays were carried out in deuterium oxide. Two buffers were made by weighing out equal amounts of HEPES buffer and salts in Buffer C and dissolving the weighed out powder in equal volumes of either H2O or D2O. To remove any hydrogen donated by buffer and salts the two buffers were dried down in a speed vac and resuspended in the appropriate solvent (H2O or D2O) to give buffer and salt concentrations equal to those in a stock of 5x Buffer C (250 mM HEPES, 350 mM NH4Cl, 150 mM KCl, 35 mM MgCl2, 5 mM DTT). The pH or pD of each buffer was measured and was adjusted with appropriate base (KOH prepared in H2O or D2O) to give equal pL of 7.1 after correcting for the known isotope effect on the pH meter (pD = pH meter reading + 0.4) (Cerrone-Szakal et al., 2008; Schowen and Schowen). The pL of each buffer was measured at 37°C, the temperature at which the reactions were carried out. Release complexes were formed as described above but after pelleting, were resuspended in either 1x H2O or D2O buffers before storage. Concentrated RF1 or variant stocks (at least 700 μM) were diluted in 1x H2O or D2O buffers to 5 μM, reacted with 0.2 μM release complex in the equivalent buffer and release of dipeptide was followed over time as described. All time courses fit to single exponential curves to obtain rate constants and the solvent isotope effect was calculated as kobs H2O/kobs D2O.
Computer simulations
MD simulation systems for the termination reaction in the ribosomal PTC with dA76 substrate were prepared (Trobro and Åqvist, 2009) using rRNA and protein coordinates from the 70S T. thermophilus complex with bound RF2, PDB codes 2JL5 and 2JL6 (Weixlbaumer et al., 2008). Ion and water positions were taken from the high resolution 50S H. marismortui structure (Schmeing et al., 2005a) with PDB code 1VQN. The mutant systems were obtained by replacing the Gln235 side-chain with Ser, Thr, Cys and Leu, respectively, and all system were solvated with additional water up to 20 Å distance from the P-site A76 O3′ (Trobro and Aqvist, 2009). The MD simulations were performed as reported earlier (Trobro and Aqvist, 2009) using the program Q (Marelius et al., 1998) at 300K with the CHARMM22 force field (MacKerell et al., 1995).
Free energy profiles for the ester hydrolysis reaction were calculated with the empirical valence bond (EVB) method as before (Trobro and Aqvist, 2007, 2009) using the free energy perturbation (FEP) umbrella sampling approach (Aqvist and Warshel, 1993). Each free energy calculation involved 81 discrete FEP steps and a total simulation time of at least 1.4 ns for each free energy profile. Three to seven independent simulations were carried out for each of the mutant systems to evaluate the catalytic effect in ribosome. The simulation of the uncatalyzed reaction in water was performed and calibrated as previously described (Trobro and Aqvist, 2009), but with the ribose converted to a 2′ deoxy-ribose and the calibration was also verified by multiple simulations. For the hydroxide catalyzed mechanism the free energy of creating or bringing a hydroxide ion in to the PTC, which is necessary to obtain estimates of absolute rates, cannot be calculated in any reliable way and free energy profiles were in this case evaluated relative to the wt RF1 GGQ reaction with dA76 substrate.
Highlights.
Several RF1 Q235 mutations can catalyze release with normally dead dA76 substrate
The mechanism was studied using molecular modeling, pH and isotope effects
Active mutants alter hydrogen bonding, orient the nucleophile, and stabilize the oxyanion
Supports a model where tRNA 2′OH orients substrates for catalysis
Acknowledgments
We thank H. Zaher, D. Hiller and S. Strobel for helpful discussions and critical review of the manuscript. The work was supported by NIH grant R01GM059425 and salary support for R.G. was from HHMI. J.Å. acknowledges support from the Swedish Research Council and the KAW Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amort M, Wotzel B, Bakowska-Zywicka K, Erlacher MD, Micura R, Polacek N. An intact ribose moiety at A2602 of 23S rRNA is key to trigger peptidyl-tRNA hydrolysis during translation termination. Nucleic Acids Res. 2007;35:5130–5140. doi: 10.1093/nar/gkm539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aqvist J, Warshel A. Simulation of Enzyme-Reactions Using Valence-Bond Force-Fields and Other Hybrid Quantum-Classical Approaches. Chem Rev. 1993;93:2523–2544. [Google Scholar]
- Beringer M, Rodnina MV. Importance of tRNA interactions with 23S rRNA for peptide bond formation on the ribosome: studies with substrate analogs. Biol Chem. 2007;388:687–691. doi: 10.1515/BC.2007.077. [DOI] [PubMed] [Google Scholar]
- Bieling P, Beringer M, Adio S, Rodnina MV. Peptide bond formation does not involve acid-base catalysis by ribosomal residues. Nat Struct Mol Biol. 2006;13:423–428. doi: 10.1038/nsmb1091. [DOI] [PubMed] [Google Scholar]
- Blaber M, Baase WA, Gassner N, Matthews BW. Alanine scanning mutagenesis of the α-helix 115–123 of phage T4 lysozyme: Effects on structure, stability and the binding of solvent. J Mol Biol. 1995;246:317–330. doi: 10.1006/jmbi.1994.0087. [DOI] [PubMed] [Google Scholar]
- Brunelle JL, Shaw JJ, Youngman EM, Green R. Peptide release on the ribosome depends critically on the 2′ OH of the peptidyl-tRNA substrate. RNA. 2008;14:1526–1531. doi: 10.1261/rna.1057908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelle JL, Youngman EM, Sharma D, Green R. The interaction between C75 of tRNA and the A loop of the ribosome stimulates peptidyl transferase activity. RNA. 2006;12:33–39. doi: 10.1261/rna.2256706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caskey CT, Beaudet AL, Scolnick EM, Rosman M. Hydrolysis of fMet-tRNA by peptidyl transferase. Proc Natl Acad Sci U S A. 1971;68:3163–3167. doi: 10.1073/pnas.68.12.3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerrone-Szakal AL, Chadalavada DM, Golden BL, Bevilacqua PC. Mechanistic characterization of the HDV genomic ribozyme: the cleavage site base pair plays a structural role in facilitating catalysis. RNA. 2008;14:1746–1760. doi: 10.1261/rna.1140308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlin SI, Merino EJ, Weeks KM. Catalysis of amide synthesis by RNA phosphodiester and hydroxyl groups. Proc Natl Acad Sci U S A. 2002;99:14688–14693. doi: 10.1073/pnas.212527799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das GK, Bhattacharyya D, Burma DP. A possible mechanism of peptide bond formation on ribosome without mediation of peptidyl transferase. J Theor Biol. 1999;200:193–205. doi: 10.1006/jtbi.1999.0987. [DOI] [PubMed] [Google Scholar]
- Dorner S, Panuschka C, Schmid W, Barta A. Mononucleotide derivatives as ribosomal P-site substrates reveal an important contribution of the 2′-OH to activity. Nucleic Acids Res. 2003;31:6536–6542. doi: 10.1093/nar/gkg842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner S, Polacek N, Schulmeister U, Panuschka C, Barta A. Molecular aspects of the ribosomal peptidyl transferase. Biochem Soc Trans. 2002;30:1131–1136. doi: 10.1042/bst0301131. [DOI] [PubMed] [Google Scholar]
- Frolova LY, Tsivkovskii RY, Sivolobova GF, Oparina NY, Serpinsky OI, Blinov VM, Tatkov SI, Kisselev LL. Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA. 1999;5:1014–1020. doi: 10.1017/s135583829999043x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin BE, Reese CB. Some Observations on the Mechanism of the Acylation Process in Protein Synthesis. Proc Natl Acad Sci U S A. 1964;51:440–444. doi: 10.1073/pnas.51.3.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JL, Schmeing TM, Moore PB, Steitz TA. Structural insights into peptide bond formation. Proc Natl Acad Sci U S A. 2002;99:11670–11675. doi: 10.1073/pnas.172404099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht SM, Kozarich JW, Schmidt FJ. Isomeric phenylalanyl-tRNAs. Position of the aminoacyl moiety during protein biosynthesis. Proc Natl Acad Sci U S A. 1974;71:4317–4321. doi: 10.1073/pnas.71.11.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschlag D, Eckstein F, Cech TR. The importance of being ribose at the cleavage site in the Tetrahymena ribozyme reaction. Biochemistry. 1993;32:8312–8321. doi: 10.1021/bi00083a035. [DOI] [PubMed] [Google Scholar]
- Hiller DA, Zhong M, Singh V, Strobel SA. Transition states of uncatalyzed hydrolysis and aminolysis reactions of a ribosomal P-site substrate determined by kinetic isotope effects. Biochemistry. 2010;49:3868–3878. doi: 10.1021/bi901458x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Sprinzl M. Peptide bond formation on the ribosome: The role of the 2′-OH group on the terminal adenosine of peptidyl-tRNA and of the length of the nascent peptide chain. Angew Chem Int Ed. 2011 doi: 10.1002/anie.201005245. [DOI] [PubMed] [Google Scholar]
- Jin H, Kelley AC, Loakes D, Ramakrishnan V. Structure of the 70S ribosome bound to release factor 2 and a substrate analog provides insights into catalysis of peptide release. Proc Natl Acad Sci U S A. 2010;107:8593–8598. doi: 10.1073/pnas.1003995107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katunin VI, Muth GW, Strobel SA, Wintermeyer W, Rodnina MV. Important contribution to catalysis of peptide bond formation by a single ionizing group within the ribosome. Mol Cell. 2002;10:339–346. doi: 10.1016/s1097-2765(02)00566-x. [DOI] [PubMed] [Google Scholar]
- Koch M, Huang Y, Sprinzl M. Peptide-bond synthesis on the ribosome: no free vicinal hydroxy group required on the terminal ribose residue of peptidyl-tRNA. Angew Chem Int Ed Engl. 2008;47:7242–7245. doi: 10.1002/anie.200801511. [DOI] [PubMed] [Google Scholar]
- Korostelev A, Asahara H, Lancaster L, Laurberg M, Hirschi A, Zhu J, Trakhanov S, Scott WG, Noller HF. Crystal structure of a translation termination complex formed with release factor RF2. Proc Natl Acad Sci U S A. 2008;105:19684–19689. doi: 10.1073/pnas.0810953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlenkoetter S, Wintermeyer W, Rodnina MV. Different substrate-dependent transition states in the active site of the ribosome. Nature. 2011;476:351–354. doi: 10.1038/nature10247. [DOI] [PubMed] [Google Scholar]
- Laurberg M, Asahara H, Korostelev A, Zhu J, Trakhanov S, Noller HF. Structural basis for translation termination on the 70S ribosome. Nature. 2008;454:852–857. doi: 10.1038/nature07115. [DOI] [PubMed] [Google Scholar]
- MacKerell AD, Jr, Sommer MS, Karplus M. pH dependence of binding reactions from free energy simulations and macroscopic continuum electrostatic calculations: application to 2′GMP/3′GMP binding to ribonuclease T1 and implications for catalysis. J Mol Biol. 1995;247:774–807. doi: 10.1006/jmbi.1994.0180. [DOI] [PubMed] [Google Scholar]
- Marelius J, Kolmodin K, Feierberg I, Aqvist J. Q: a molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J Mol Graph Model. 1998;16:213–225. 261. doi: 10.1016/s1093-3263(98)80006-5. [DOI] [PubMed] [Google Scholar]
- Mora L, Heurgue-Hamard V, Champ S, Ehrenberg M, Kisselev LL, Buckingham RH. The essential role of the invariant GGQ motif in the function and stability in vivo of bacterial release factors RF1 and RF2. Mol Microbiol. 2003;47:267–275. doi: 10.1046/j.1365-2958.2003.03301.x. [DOI] [PubMed] [Google Scholar]
- Peracchi A, Beigelman L, Usman N, Herschlag D. Rescue of abasic hammerhead ribozymes by exogenous addition of specific bases. Proc Natl Acad Sci U S A. 1996;93:11522–11527. doi: 10.1073/pnas.93.21.11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peracchi A, Matulic-Adamic J, Wang S, Beigelman L, Herschlag D. Structure-function relationships in the hammerhead ribozyme probed by base rescue. RNA. 1998;4:1332–1346. doi: 10.1017/s1355838298980979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polacek N, Gomez MJ, Ito K, Xiong L, Nakamura Y, Mankin A. The critical role of the universally conserved A2602 of 23S ribosomal RNA in the release of the nascent peptide during translation termination. Mol Cell. 2003;11:103–112. doi: 10.1016/s1097-2765(02)00825-0. [DOI] [PubMed] [Google Scholar]
- Quiggle K, Kumar G, Ott TW, Ryu EK, Chladek S. Donor site of ribosomal peptidyltransferase: investigation of substrate specificity using 2′ (3′)-O-(N-acylaminoacyl)dinucleoside phosphates as models of the 3′ terminus of N-acylaminoacyl transfer ribonucleic acid. Biochemistry. 1981;20:3480–3485. doi: 10.1021/bi00515a027. [DOI] [PubMed] [Google Scholar]
- Rawat UB, Zavialov AV, Sengupta J, Valle M, Grassucci RA, Linde J, Vestergaard B, Ehrenberg M, Frank J. A cryo-electron microscopic study of ribosome-bound termination factor RF2. Nature. 2003;421:87–90. doi: 10.1038/nature01224. [DOI] [PubMed] [Google Scholar]
- Schmeing TM, Huang KS, Kitchen DE, Strobel SA, Steitz TA. Structural insights into the roles of water and the 2′ hydroxyl of the P site tRNA in the peptidyl transferase reaction. Mol Cell. 2005a;20:437–448. doi: 10.1016/j.molcel.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Schmeing TM, Huang KS, Strobel SA, Steitz TA. An induced-fit mechanism to promote peptide bond formation and exclude hydrolysis of peptidyl-tRNA. Nature. 2005b;438:520–524. doi: 10.1038/nature04152. [DOI] [PubMed] [Google Scholar]
- Scholten JD, Hogg JL, Raushel FM. Methyl Chymotrypsin Catalyzed Hydrolyses of Specific Substrate Esters Indicate Multiple Proton Catalysis Is Possible with a Modified Charge Relay Triad. Journal of the American Chemical Society. 1988;110:8246–8247. [Google Scholar]
- Schowen KB, Schowen RL. Solvent isotope effects of enzyme systems. Methods Enzymol. 1982;87:551–606. [PubMed] [Google Scholar]
- Seit-Nebi A, Frolova L, Justesen J, Kisselev L. Class-1 translation termination factors: invariant GGQ minidomain is essential for release activity and ribosome binding but not for stop codon recognition. Nucleic Acids Res. 2001;29:3982–3987. doi: 10.1093/nar/29.19.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JJ, Green R. Two distinct components of release factor function uncovered by nucleophile partitioning analysis. Mol Cell. 2007;28:458–467. doi: 10.1016/j.molcel.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers A, Beringer M, Rodnina MV, Wolfenden R. The ribosome as an entropy trap. Proc Natl Acad Sci U S A. 2004;101:7897–7901. doi: 10.1073/pnas.0402488101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonovic M, Steitz TA. Peptidyl-CCA deacylation on the ribosome promoted by induced fit and the O3′-hydroxyl group of A76 of the unacylated A-site tRNA. RNA. 2008;14:2372–2378. doi: 10.1261/rna.1118908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Mugnier P, Das AK, Webb HM, Evans DR, Tuite MF, Hemmings BA, Barford D. The crystal structure of human eukaryotic release factor eRF1--mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell. 2000;100:311–321. doi: 10.1016/s0092-8674(00)80667-4. [DOI] [PubMed] [Google Scholar]
- Trobro S, Aqvist J. Mechanism of peptide bond synthesis on the ribosome. Proc Natl Acad Sci U S A. 2005;102:12395–12400. doi: 10.1073/pnas.0504043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobro S, Aqvist J. A model for how ribosomal release factors induce peptidyl-tRNA cleavage in termination of protein synthesis. Mol Cell. 2007;27:758–766. doi: 10.1016/j.molcel.2007.06.032. [DOI] [PubMed] [Google Scholar]
- Trobro S, Aqvist J. Mechanism of the translation termination reaction on the ribosome. Biochemistry. 2009;48:11296–11303. doi: 10.1021/bi9017297. [DOI] [PubMed] [Google Scholar]
- Wallin G, Aqvist J. The transition state for peptide bond formation reveals the ribosome as a water trap. Proc Natl Acad Sci U S A. 2010;107:1888–1893. doi: 10.1073/pnas.0914192107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinger JS, Parnell KM, Dorner S, Green R, Strobel SA. Substrate-assisted catalysis of peptide bond formation by the ribosome. Nat Struct Mol Biol. 2004;11:1101–1106. doi: 10.1038/nsmb841. [DOI] [PubMed] [Google Scholar]
- Weinger JS, Strobel SA. Participation of the tRNA A76 hydroxyl groups throughout translation. Biochemistry. 2006;45:5939–5948. doi: 10.1021/bi060183n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weixlbaumer A, Jin H, Neubauer C, Voorhees RM, Petry S, Kelley AC, Ramakrishnan V. Insights into translational termination from the structure of RF2 bound to the ribosome. Science. 2008;322:953–956. doi: 10.1126/science.1164840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman EM, Brunelle JL, Kochaniak AB, Green R. The active site of the ribosome is composed of two layers of conserved nucleotides with distinct roles in peptide bond formation and peptide release. Cell. 2004;117:589–599. doi: 10.1016/s0092-8674(04)00411-8. [DOI] [PubMed] [Google Scholar]
- Zaher HS, Shaw JJ, Strobel SA, Green R. The 2′-OH group of the peptidyl-tRNA stabilizes an active conformation of the ribosomal PTC. EMBO J. 2011 doi: 10.1038/emboj.2011.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavialov AV, Mora L, Buckingham RH, Ehrenberg M. Release of peptide promoted by the GGQ motif of class 1 release factors regulates the GTPase activity of RF3. Mol Cell. 2002;10:789–798. doi: 10.1016/s1097-2765(02)00691-3. [DOI] [PubMed] [Google Scholar]