

ABSTRACT
Central to NF-κB signaling pathways is IKKγ/NEMO, a regulatory subunit of the cytoplasmic IκB kinase (IKK) complex, which undergoes various posttranslational modifications, specifically phosphorylation, to regulate its function. Furthermore, Kaposi’s sarcoma-associated herpesvirus (KSHV) FADD-like interleukin-1β (IL-1β) converting enzyme (FLICE) inhibitory protein (vFLIP) activates the NF-κB signaling pathway by directly interacting with IKKγ/NEMO. However, the exact functions of IKKγ/NEMO phosphorylation and its KvFLIP interaction in NF-κB activation remain elusive. Here, we report two novel phosphorylation sites of IKKγ/NEMO and their negative effect on the IKKγ/NEMO-mediated NF-κB signaling pathway. First, the Src family protein tyrosine kinases (SF-PTKs), including Src, Fyn, Lyn, and Fgr, interact with and phosphorylate tyrosine residue 374 (Y374) of IKKγ/NEMO. Mutation of the Y374 residue to phenylalanine (Y374F) specifically abolished SF-PTK-mediated tyrosine phosphorylation, leading to increased tumor necrosis factor alpha (TNF-α)-induced NF-κB activity. Moreover, our mass spectrometry analysis found that the serine 377 residue (S377) of IKKγ/NEMO underwent robust phosphorylation upon KvFLIP expression. Replacement of the IKKγ/NEMO S377 residue by alanine (S377A) or glutamic acid (S377E) resulted in a significant increase or decrease of NF-κB activity and TNF-α-mediated IL-6 cytokine production, respectively. Our study thus demonstrates that the Y374 or S377 residue located at the C-terminal proline-rich domain of human IKKγ/NEMO undergoes phosphorylation upon TNF-α treatment or KvFLIP expression, respectively, resulting in the suppression of IKKγ/NEMO activity to induce NF-κB activation. This study suggests the potential phosphorylation-mediated feedback negative regulation of IKKγ/NEMO activity in the NF-κB signaling pathway.
IMPORTANCE
Since unchecked regulation of NF-κB has been linked to uncontrolled proliferation and cell death, the downregulation of the NF-κB signaling pathway is as important as its activation. Specifically, the phosphorylation-mediated modification of IKKγ/NEMO is a critical regulatory mechanism of NF-κB activity. Here, we report two novel phosphorylations of IKKγ/NEMO and their negative effects on the NF-κB signaling pathway. First, the Src family protein tyrosine kinase interacts with and phosphorylates tyrosine residue 374 of IKKγ/NEMO, suppressing tumor necrosis factor alpha (TNF-α)-induced NF-κB activity. Additionally, Kaposi’s sarcoma-associated herpesvirus (KSHV) FADD-like interleukin-1β (IL-1β) converting enzyme (FLICE) inhibitory protein (KvFLIP) expression induces a robust phosphorylation of the serine 377 residue of IKKγ/NEMO, resulting in a significant decrease of NF-κB activity. Our study thus demonstrates that the Y374 or S377 residue of IKKγ/NEMO undergoes phosphorylation upon TNF-α treatment or KvFLIP expression, respectively, resulting in the suppression of IKKγ/NEMO activity to induce NF-κB activation. This also suggests the potential phosphorylation-mediated feedback negative regulation of IKKγ/NEMO activity in the NF-κB signaling pathway.
Introduction
Members of the NF-κB family are transcription factors involved in the regulation of diverse cellular processes, including inflammation, innate/adaptive immunity, apoptosis, and tumorigenesis (1). Under normal conditions, the NF-κB complex is primarily sequestered in the cytoplasm by forming a complex with a family of inhibitory proteins or IκBs (2). A number of biological stimuli, including proinflammatory cytokines (tumor necrosis factor alpha [TNF-α] and interleukin-1β [IL-1β]), microbial infection, growth factors, and ligands for antigen receptors, converge at the activation of the IκB kinase (IKK) complex, which is composed of two structurally related catalytic subunits, IKKα and IKKβ, and a noncatalytic regulatory subunit, IKKγ/NF-κB essential modulator (NEMO) (3). Activation of the IKK complex subsequently leads to phosphorylation and ubiquitination of IκBs for proteasome-mediated degradation. Released NF-κB translocates into the nucleus and induces transcription of numerous target genes involved in cell proliferation, survival, differentiation, immune responses, and inflammatory responses (2).
Inappropriate activation of the NF-κB signaling pathway, however, is linked with several human diseases (4, 5). Thus, under normal conditions, the NF-κB signaling pathway in response to a variety of stimuli has to be transient and tightly regulated by both positive and negative regulatory mechanisms. While a precise regulatory mechanism underlying IKK complex activation still remains unclear, various posttranslational modifications on the different subunits of the IKK complex appear to be clearly involved in NF-κB activation (6, 7). IKKγ/NEMO has been reported to undergo phosphorylation, sumoylation, and ubiquitination by a number of stimuli (3, 6, 7). IKKγ/NEMO is composed of a helical domain (HLX1), coiled-coil domain 1 (CC1), a second helical domain (HLX2), another coiled-coil domain (CC2) containing a minimum oligomerization domain, a NEMO ubiquitin (Ub) binding (NUB) domain, a leucine zipper (LZ) domain, a proline-rich domain, and a zinc finger (ZF) domain (8). Extensive characterization has found that IKKγ/NEMO undergoes K63-linked polyubiquitination at the lysine residues 285 and 399 upon TNF-α and T-cell receptor (TCR) stimulation, allowing the IKK complex to be recruited to TAK1, a kinase known to phosphorylate IKKβ for its activation (9, 10, 11, 12). Sumoylation, followed by the monoubiquitination of IKKγ/NEMO, has been reported to play an important role in the activation of NF-κB upon genotoxic stress (13, 14, 15). Phosphorylation of IKKγ/NEMO at multiple serine residues has been suggested to regulate NF-κB activity in response to various stimuli (13, 16, 17, 18). IKKγ/NEMO phosphorylation at serine residues 85 and 141 by protein kinase C (PKC) is involved in the IKKβ-mediated phosphorylation of IκB (16). Phosphorylation of IKKγ/NEMO at serine residue 85 has been identified as a crucial site by ATM for IKK activation upon genotoxic stress (13). Several additional serine residues at positions 31 and 43 in the N terminus and at positions 376 and 377 in the proline-rich domain of the C terminus of IKKγ/NEMO have been identified to undergo phosphorylations upon stimulation with inflammatory cytokines (TNF-α and IL-1), by overexpression of TNF receptor I (TNFRI), or by Tax, an oncoprotein of the human T cell lymphotrophic virus type 1 (HTLV-1) (18). Mutation of the S369 residue in mouse IKKγ/NEMO (equivalent to S376 in human IKKγ/NEMO) to alanine (S369A) has been reported to increase its ability to stimulate NF-κB activity (17). In addition, phosphorylation of IKKα and IKKβ at two serine residues of their activation T loop has been shown to activate their kinase activity upon cytokine treatments (19, 20, 21, 22). These studies strongly indicate that the posttranslational modifications of each subunit of the IKK complex can play either a positive or a negative role in activation of the NF-κB signaling pathway.
Several viruses, including HIV-1, HTLV-1, Kaposi’s sarcoma-associated herpesvirus (KSHV; human herpesvirus 8 [HHV-8]), and Epstein-Barr virus (EBV), are known to induce the activation of the NF-κB signaling pathway (23). Among them, KSHV/HHV-8, which is a main causative agent of Kaposi’s sarcoma (KS) and associated with rare lymphoproliferative diseases such as primary effusion lymphoma (PEL) and multicentric Castleman’s disease (MCD), expresses a protein called K13 during the latent stage of the viral life cycle (24). K13, encoded by ORF71 of the KSHV viral genome, is a viral homolog of cellular FLICE (FADD-like IL-1β converting enzyme) inhibitory protein (FLIP) and is thus referred to as KSHV viral FLIP (KvFLIP). Similarly to cellular FLIPs (cFLIPs) and several vFLIPs encoded by herpesvirus saimiri (HVS), equine herpesvirus, and poxvirus, KvFLIP contains two tandem death effector domains (DEDs) (25, 26, 27). KvFLIP has been shown to directly interact with IKKγ/NEMO, leading to the constitutive activation of the NF-κB signaling pathway and ultimately contributing to transformation and tumorigenesis (28, 29, 30, 31). However, the exact function of KvFLIP interaction with IKKγ/NEMO in the constitutive activation of NF-κB remains elusive.
Here, we report that Fyn, a member of the Src family protein tyrosine kinases (SF-PTKs), interacts with and phosphorylates IKKγ/NEMO at the tyrosine 374 (Y374) residue, leading to the negative effect on the TNF-α-mediated induction of the NF-κB signaling pathway. Moreover, KvFLIP expression resulted in the substantial increase of S377 phosphorylation of human IKKγ/NEMO, regulating the activation of TNF-α-mediated NF-κB activation. Thus, our study demonstrates that novel posttranslational modifications in the proline-rich domain of human IKKγ/NEMO have important negative regulatory effects on the activation of the NF-κB signaling pathway induced by TNF-α or KvFLIP.
RESULTS
Effect of the phosphorylations of the IKKγ/NEMO proline-rich domain on TNF-α-mediated NF-κB activation.
To identify posttranslational modifications of IKKγ/NEMO by KvFLIP, IKKγ/NEMO was purified from HEK 293T cells transiently transfected with KvFLIP, followed by mass spectrometry analysis. The peptides derived from human IKKγ/NEMO revealed that IKKγ/NEMO was phosphorylated at serine residue 377 located at the C-terminal proline-rich domain. Furthermore, a large-scale proteomics analysis of the human tyrosine-phosphorylated proteins has suggested that IKKγ/NEMO undergoes tyrosine phosphorylation at residue 374 (Y374) in the same C-terminal proline-rich domain upon T cell receptor signal transduction (32). To examine if the phosphorylation sites of IKKγ/NEMO at the C-terminal proline-rich domain affect NF-κB activation upon stimulation, amino acid residues of human IKKγ/NEMO at Y374 and S377 were individually mutated to either phosphoinhibitory phenylalanine (F), alanine (A), phosphomimetic aspartate (D), or glutamate (E) as depicted in Fig. 1A (upper panel). IKKγ/NEMO-knockout mouse embryonic fibroblasts (IKKγ/NEMO−/− MEFs) were then stably reconstituted with the hemagglutinin (HA)-tagged IKKγ/NEMO wild type (WT) or each mutant, and their expressions were confirmed by immunoblotting with anti-HA antibody (Fig. 1A, lower panel). Interestingly, the phosphoinhibitory forms of IKKγ/NEMO mutants migrated faster than did WT and phosphomimetic forms of IKKγ/NEMO. Next, we examined whether these phosphorylation residue changes at the proline-rich domain of IKKγ/NEMO affected the inflammatory cytokine-induced NF-κB signaling pathway. To do this, IKKγ/NEMO−/− MEFs reconstituted with WT or each mutant of IKKγ/NEMO were stimulated with TNF-α and the levels of IκBα phosphorylation were examined by immunoblotting. As shown in Fig. 1B, IKKγ/NEMO−/− MEFs reconstituted with the Y374F or S377A mutant showed faster kinetics of IκBα phosphorylation upon TNF-α stimulation than did those reconstituted with WT. In contrast, IKKγ/NEMO−/− MEFs reconstituted with the Y374D or S77E mutant showed delayed or reduced IκBα phosphorylation, respectively, compared with those reconstituted with WT (Fig. 1B). These results suggest that the Y374 and S377 phosphorylations at the proline-rich domain of IKKγ/NEMO appear to negatively regulate its function in TNF-α-mediated NF-κB activation.
FIG 1.
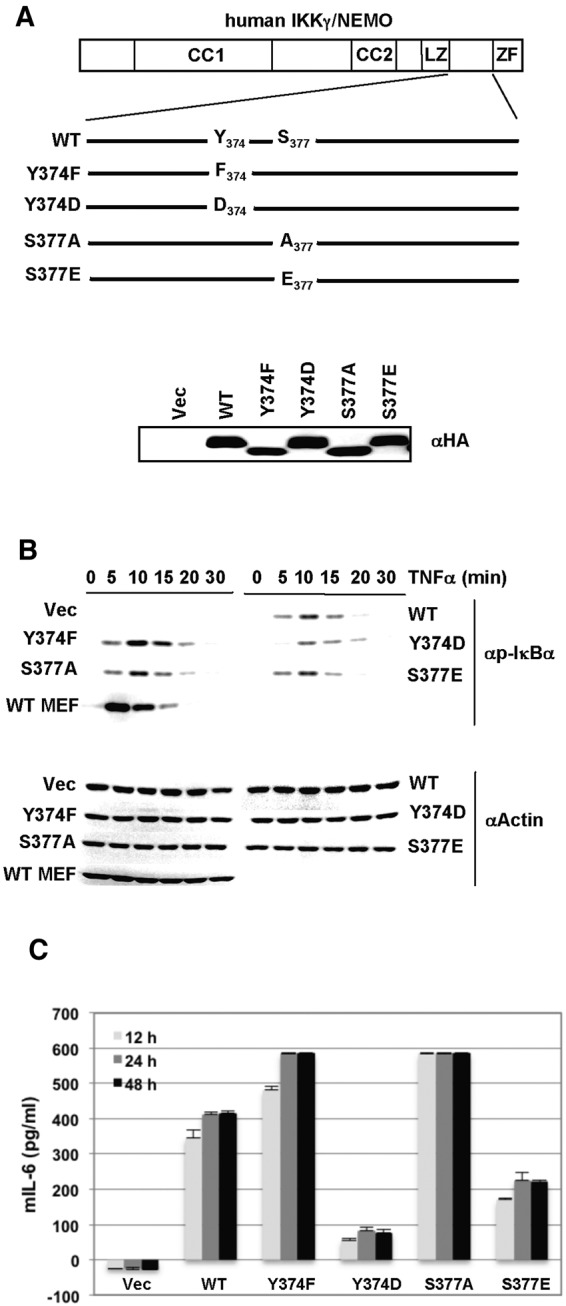
The Y374 or S377 phosphorylation of IKKγ/NEMO exerts negative effects on TNF-α-mediated NF-κB activation. (A) A schematic representation of the location of Y374 and S377 in the C-terminal proline-rich domain of human IKKγ/NEMO. Individual mutations were generated by site-directed mutagenesis. IKKγ/NEMO-knockout (IKKγ/NEMO−/−) mouse embryo fibroblasts (MEFs) were transfected with puroIRES vector carrying the HA-tagged IKKγ/NEMO WT or mutant gene. IKKγ/NEMO−/− MEFs stably expressing the IKKγ/NEMO WT or mutant were then established by puromycin resistance (2 µg/ml). Expression of the IKKγ/NEMO WT or mutant was confirmed by immunoblotting with anti-HA antibody. (B) IKKγ/NEMO−/− MEFs stably expressing either HA-tagged IKKγ/NEMO WT or mutant were stimulated with mouse TNF-α (10 ng/ml) for indicated times. Whole-cell lysates (WCLs) were then analyzed by immunoblotting with anti-phospho-IκBα (p-IκBα) antibody. Immunoblotting with antiactin antibody served as a loading control. (C) IKKγ/NEMO−/− MEFs stably expressing either HA-tagged IKKγ/NEMO WT or mutant were stimulated with mouse TNF-α (10 ng/ml) for the indicated times. Supernatants were analyzed by ELISA for IL-6 cytokine production. Experiments were repeated twice, and the average of triplicate samples from one experiment is shown with the error bars denoting the standard error of the mean.
Since the NF-κB signaling pathway induces the production of a number of inflammatory cytokines, we next measured the IL-6 levels from IKKγ/NEMO−/− MEFs reconstituted with WT or each mutant of IKKγ/NEMO upon TNF-α stimulation. As shown in Fig. 1C, all cells, except for vector-reconstituted IKKγ/NEMO−/− MEFs, produced IL-6 cytokine following TNF-α treatment. Interestingly, IKKγ/NEMO−/− MEFs reconstituted with Y374F or S377A produced higher IL-6 levels upon TNF-α stimulation than did those reconstituted with WT (Fig. 1C). In contrast, IL-6 production was significantly attenuated in IKKγ/NEMO−/− MEFs reconstituted with the Y374D or S377E mutant compared to that in WT-reconstituted IKKγ/NEMO−/− MEFs; specifically, IKKγ/NEMO−/− MEFs reconstituted with the Y374D mutant showed the most severe attenuation of IL-6 production, consistent with the delayed and reduced phosphorylation of IκBα as shown in Fig. 1B. Taken together, these results indicate that the phosphorylations of Y374 and S377 residues negatively affect IKKγ/NEMO activity.
Roles of the IKKγ/NEMO Y374 and S377 phosphorylations in IKK complex formation and IKKγ/NEMO ubiquitination.
Previous studies have demonstrated that in vivo the IKK complex predominantly forms a large complex at least 700 to 900 kDa in size containing IKKα (85 kDa), IKKβ (87 kDa), and IKKγ/NEMO (52 kDa) (33, 34). The interaction of IKKγ/NEMO with IKKα and IKKβ, which bind at the beginning of CC1 of IKKγ/NEMO (amino acids 50 to 100) (6, 35, 36), is critical for the assembly of this high-molecular-mass IKK complex, leading to the IKKβ stimulation and IκB recruitment (1). Thus, we examined whether the Y374 or S377 phosphorylation at the proline-rich domain of IKKγ/NEMO affects its ability to assemble with IKKα and IKKβ. To do this, whole-cell lysates (WCLs) of IKKγ/NEMO−/− MEFs reconstituted with the IKKγ/NEMO WT or each mutant were subjected to immunoprecipitation of IKKγ/NEMO, followed by immunoblotting for IKKα or IKKβ. As shown in Fig. 2A, either IKKγ/NEMO WT or its mutant bound endogenous IKKα or IKKβ at similar levels. It has been also suggested that the self-assembly activity of IKKγ/NEMO plays a critical role for activation of the IKK complex (1, 37, 38). In order to examine whether the Y374 or S377 phosphorylation at the proline-rich domain of IKKγ/NEMO influenced its oligomerization, HEK 293T cells were cotransfected with FLAG-tagged IKKγ/NEMO WT and HA-tagged IKKγ/NEMO WT or mutant, and whole-cell lysates were immunoprecipitated with anti-FLAG antibody, followed by immunoblotting with anti-HA antibody. This showed that IKKγ/NEMO WT and mutant showed similar levels of self-assembly activity (Fig. 2B). Our data collectively indicate that the Y374 or S377 phosphorylation at the proline-rich domain of IKKγ/NEMO does not affect its activity to assemble with IKKα and IKKβ, nor to self-assemble.
FIG 2.
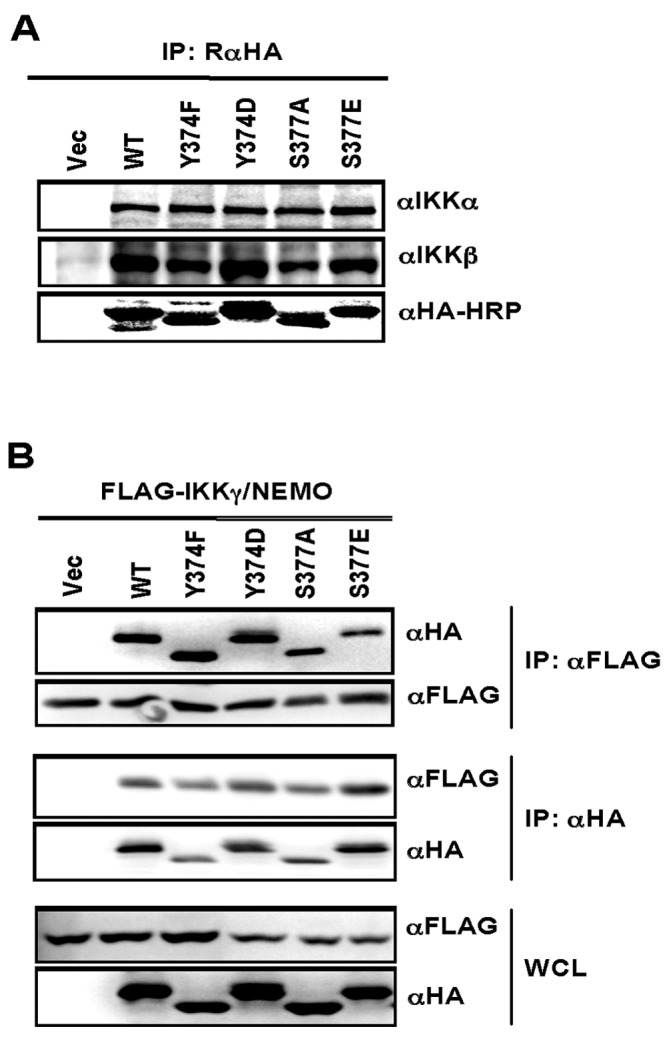
Mutation of the Y374 or S377 residue of human IKKγ/NEMO shows no effect on IKK complex formation or self-oligomerization. (A) IKKγ/NEMO−/− MEFs stably expressing either HA-tagged IKKγ/NEMO WT or mutant were subjected to immunoprecipitation (IP) with rabbit anti-HA antibody, followed by immunoblotting with antibodies against IKKα and IKKβ. (B) HEK 293T cells were transfected with HA-tagged IKKγ/NEMO WT or mutants together with FLAG-tagged WT IKKγ/NEMO. WCL was immunoprecipitated (IP) with anti-FLAG or anti-HA antibody, followed by immunoblotting with anti-HA or anti-FLAG antibody. WCL was analyzed by immunoblotting with the indicated antibodies to confirm the expression of IKKγ/NEMO.
To determine whether IKKγ/NEMO ubiquitination is affected by mutations of the Y374 and S377 phosphoacceptor residues, HEK 293T cells were transiently transfected with FLAG-tagged ubiquitin (Ub) and HA-tagged WT or each mutant of IKKγ/NEMO and subjected to pulldown with agarose beads conjugated with mouse anti-HA antibody, followed by immunoblotting with rabbit anti-FLAG antibody. The experiment showed that the WT and each mutant of IKKγ/NEMO carried similar levels of ubiquitination (Fig. 3A). These results were further confirmed in IKKγ/NEMO−/− MEFs reconstituted with the WT or mutant form of IKKγ/NEMO (Fig. 3B). These results imply that mutations of the Y374 and S377 phosphoacceptor residues of IKKγ/NEMO show little or no effect on its TNF-α-induced ubiquitinations.
FIG 3.

Mutation of the Y374 or S377 residue of human IKKγ/NEMO does not affect its ubiquitination. (A) HEK 293T cells were transfected with HA-tagged IKKγ/NEMO WT or mutants together with FLAG-tagged ubiquitin (Ub). WCL was used for the purification of the IKKγ/NEMO complex using agarose beads conjugated with mouse anti-HA antibody, followed by immunoblotting with rabbit anti-FLAG or rabbit anti-HA antibody. WCL was analyzed by immunoblotting with the indicated antibodies to confirm expression of IKKγ/NEMO. (B) IKKγ/NEMO−/− MEFs stably expressing either HA-tagged IKKγ/NEMO WT or mutant were treated with TNF-α for 10 min, followed by immunoprecipitation with agarose beads conjugated with mouse anti-HA antibody and immunoblotting with rabbit anti-HA antibody (upper panel). A longer exposure of the blot marked with an asterisk is shown in the lower panel (both panels are marked with an asterisk). WCL was analyzed by immunoblotting with anti-HA antibody to confirm the IKKγ/NEMO expression.
SF-PTKs interact with and phosphorylate IKKγ/NEMO.
A large-scale proteomics analysis of the human tyrosine-phosphorylated proteins has previously suggested that IKKγ/NEMO undergoes tyrosine phosphorylation at residue 374 (Y374) upon T cell receptor signal transduction (32, 39). To examine whether members of the SF-PTKs phosphorylate IKKγ/NEMO, 293T cells were cotransfected with WT IKKγ/NEMO and various SF-PTKs, including Fyn, Lck, Fgr, Src, and Lyn. Whole-cell lysates were used for immunoblotting with phosphotyrosine (pY)-specific antibody. Besides the auto-tyrosine-phosphorylated Fyn (58 kDa), Lck (53 kDa), Fgr (55 kDa), Src (55 kDa), and Lyn (55 kDa), an additional tyrosine-phosphorylated 50-kDa protein, at a molecular mass equivalent to that of IKKγ/NEMO, was detected in cells expressing IKKγ/NEMO WT with Fyn, Fgr, Src, or Lyn, but not with Lck (Fig. 4A). The 50-kDa tyrosine-phosphorylated protein was not detected in cells expressing the IKKγ/NEMO Y374F mutant (Fig. 4A). Furthermore, the anti-pY immunoblotting of immunopurified IKKγ/NEMO showed that the WT, but not the Y374F mutant, underwent tyrosine phosphorylation upon coexpression with Fyn, Fgr, Src, or Lyn (Fig. 4B). It should be noted that the levels of IKKγ/NEMO tyrosine phosphorylation were highest upon expression of Fyn, followed by Src, Fgr, and Lyn, whereas Lck expression did not induce the tyrosine phosphorylation of IKKγ/NEMO (Fig. 4B). Consistently, IKKγ/NEMO WT readily interacted with Fyn, Fgr, Src, or Lyn, but not with Lck, whereas the Y374F mutant failed to interact with any of these SF-PTKs (Fig. 4C). These results suggest that Fyn, Fgr, Src, and Lyn interact with and phosphorylate IKKγ/NEMO at its Y374 residue.
FIG 4.
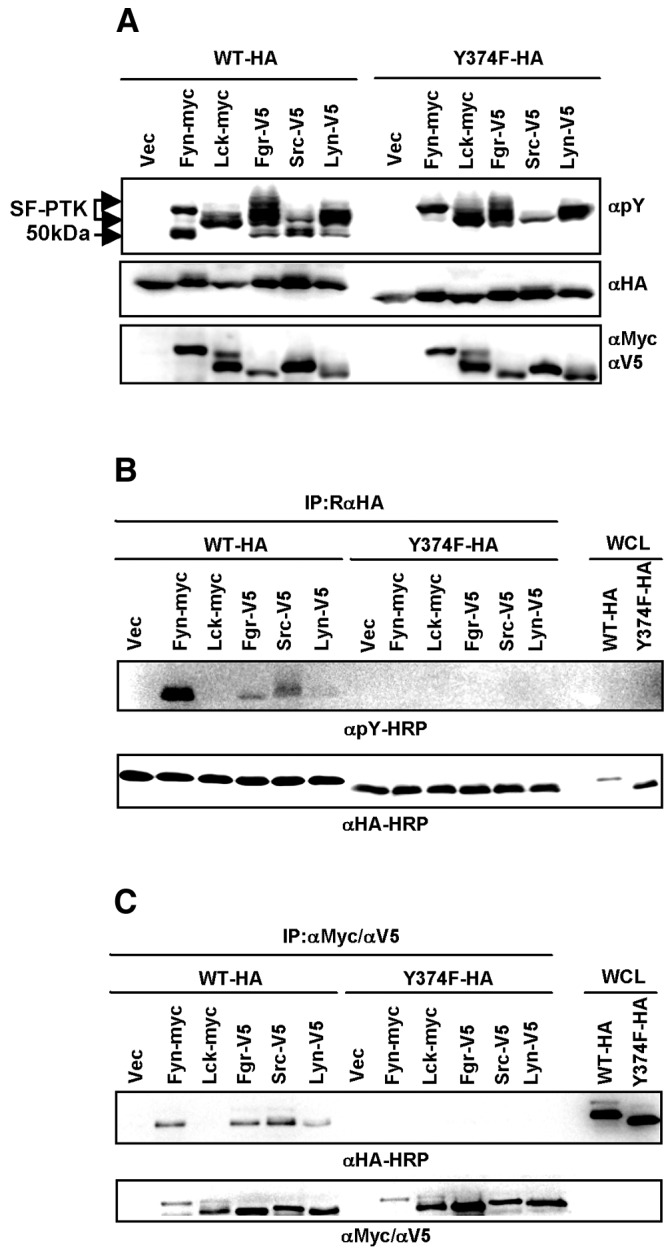
Expression of SF-PTKs leads to the Y374 phosphorylation of human IKKγ/NEMO. (A) Either IKKγ/NEMO WT or the Y374F mutant was coexpressed with each member of the Src family protein tyrosine kinases (SF-PTKs) in HEK 293T cells. WCL was then analyzed by immunoblotting with phosphotyrosine (pY)-specific antibody. HA (IKKγ/NEMO), Myc (Fyn and Lck), and V5 (Fgr, Src, and Lyn) tagging antibodies were used to demonstrate expression levels. (B) HA-tagged IKKγ/NEMO was overexpressed in HEK 293T cells along with SF-PTKs as described above and purified with rabbit anti-HA antibody in RIPA buffer, followed by immunoblotting with horseradish peroxidase (HRP)-conjugated anti-pY antibody. IP, immunoprecipitation. (C) HA-tagged WT IKKγ/NEMO was coexpressed with Myc-Fyn, Myc-Lck, V5-Fgr, V5-Src, or V5-Lyn in HEK 293T cells. WCL was subjected to immunoprecipitation with either anti-Myc or anti-V5 antibody, followed by immunoblotting with horseradish peroxidase-conjugated anti-HA antibody.
The Y374 and S377 phosphorylations of IKKγ/NEMO negatively affect TCR-mediated activation of NF-κB activity and calcium (Ca2+) influx.
To study whether the Y374 or S377 phosphorylation of IKKγ/NEMO plays a role in TCR-mediated NF-κB activation, IKKγ/NEMO-deficient Jurkat T cells were stably reconstituted with HA-tagged IKKγ/NEMO WT or Y374 or S377 mutant and subsequently stimulated with the combination of phorbol myristate acetate (PMA) and ionomycin to mimic TCR stimulation. IKKγ/NEMO-deficient Jurkat T cells stably expressing empty vector failed to induce IκBα phosphorylation upon PMA and ionomycin treatment, whereas IKKγ/NEMO-deficient Jurkat T cells stably expressing WT, Y374 or S377 mutant resulted in the robust induction of IκBα phosphorylation (Fig. 5A). The IκBα phosphorylation was detected as early as 5 min poststimulation and peaked at 20 min poststimulation in all cell lines (Fig. 5A). However, the levels of IκBα phosphorylation were stronger in IKKγ/NEMO-deficient Jurkat T cells stably expressing Y374F or S377A mutant but weaker in IKKγ/NEMO-deficient Jurkat T cells stably expressing Y374D or S377E mutant than those in IKKγ/NEMO-deficient Jurkat T cells stably expressing WT (Fig. 5A). These results further suggest that the phosphorylations of Y374 and S377 residues negatively affect IKKγ/NEMO activity to induce IκBα phosphorylation in either MEFs or Jurkat T cells.
FIG 5.
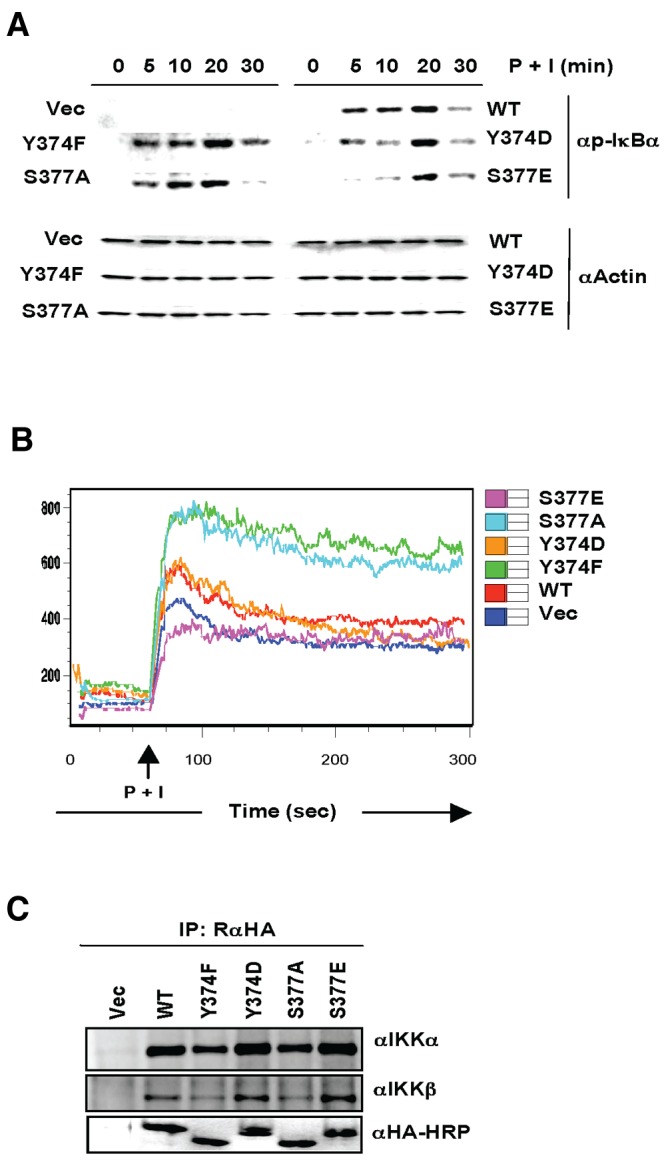
The Y374 or S377 phosphorylation of IKKγ/NEMO differentially contributes to NF-κB activation and intracellular Ca2+ influx. (A) IKKγ/NEMO-deficient Jurkat T cells stably complemented with HA-tagged IKKγ/NEMO WT or mutant were stimulated with PMA (P) and ionomycin (I) for indicated times. WCL was analyzed by immunoblotting with anti-p-IκBα antibody. Immunoblotting with antiactin antibody served as a loading control. (B) IKKγ/NEMO-deficient Jurkat T cells stably complemented with HA-tagged IKKγ/NEMO WT or mutant were loaded with Ca2+ indicator for 1 h at 37°C. Intracellular Ca2+ influx upon PMA and ionomycin (P + I) treatment was monitored for 5 min by flow cytometry analysis. (C) IKKγ/NEMO-deficient Jurkat T cells stably complemented with HA-tagged IKKγ/NEMO WT or mutant were subjected to immunoprecipitation (IP) with rabbit anti-HA antibody, followed by immunoblotting with antibodies against IKKα, IKKβ, and HA.
To assess whether the Y374 and S377 phosphorylation of IKKγ/NEMO affected intracellular Ca2+ levels, each Jurkat T cell line was stimulated with PMA and ionomycin and the levels of intracellular Ca2+ influx were measured by flow cytometry analysis. Consistent with the IκBα phosphorylation, stimulation-induced intracellular Ca2+ influx was significantly higher in IKKγ/NEMO-deficient Jurkat T cells reconstituted with the Y374F or S377A mutant than in IKKγ/NEMO-deficient Jurkat T cells reconstituted with the WT or the Y374D mutant (Fig. 5B). Interestingly, IKKγ/NEMO-deficient Jurkat T cells reconstituted with empty vector showed levels of intracellular Ca2+ increase similar to those of T cells reconstituted with the S377E mutant upon PMA/ionomycin treatment (Fig. 5B). Finally, the interactions of IKKγ/NEMO WT or mutant with endogenous IKKα or IKKβ were not detectably altered (Fig. 5C). These results also suggest that the Y374 and S377 phosphorylations of IKKγ/NEMO negatively affect TCR-mediated Ca2+ influx.
DISCUSSION
It has been suggested that posttranslational modification of proteins can influence diverse functions of proteins, such as their activation, stability, localization, trafficking, and incorporation into higher complexes (40). A number of molecules involved in the induction of the NF-κB signaling pathway are well known to undergo posttranslational modifications in response to diverse NF-κB activators, such as TNF-α, IL-1, and microbial infection (3, 6, 7). A growing body of evidence indicates that IKKγ/NEMO is targeted for several posttranslational modifications, including ubiquitination, sumoylation, and phosphorylation. Thus, posttranslational modifications confer crucial regulatory mechanisms to IKKγ/NEMO for the activation of IKK complex under most stimulation conditions (3, 6, 7). Previous studies have demonstrated that serine residue 369 (S369) in the C terminus of mouse IKKγ/NEMO (equivalent to serine residue 376 [S376] in that of human IKKγ/NEMO) is the major site of phosphorylation by IKKβ (17). Moreover, the S376 residue of human IKKγ/NEMO was identified as the primary target for phosphorylation by IKKβ and the vicinal S377 residue was suggested to serve a compensatory role for phosphorylation upon TNF-α stimulation or expression of HTLV-1 Tax (18, 41, 42). In this study, we found that the overexpression of KvFLIP, a viral oncoprotein known to interact with IKKγ/NEMO and to constitutively activate the NF-κB signaling pathway (29, 30, 43), resulted in the phosphorylation of human IKKγ/NEMO at S377 located in its C-terminal proline-rich domain. Furthermore, we showed that mutation of the S377 residue of human IKKγ/NEMO to the phosphoinhibitory alanine (S377A) resulted in increased NF-κB activity and IL-6 cytokine production. These results are consistent with the previous study showing that the S369A mutation of mouse IKKγ/NEMO led to increased NF-κB activity (17). Most biological processes require both positive and negative regulatory mechanisms to maintain equilibrium. While the NF-κB pathway robustly induces host immune responses upon viral infection, it must slow down and ultimately stop when it reaches a late stage to avoid excessive production of inflammatory cytokines, which could lead to deleterious effects on host immunity. Thus, the S377 phosphorylation of IKKγ/NEMO may be one of these feedback inhibitory mechanisms to slow down NF-κB-mediated immune responses.
Previous genome-wide analysis of tyrosine phosphorylation in combination with mass spectrometric analysis has shown that human IKKγ/NEMO undergoes tyrosine phosphorylation at the Y374 residue (32, 39). We also found that IKKγ/NEMO interacted with members of the SF-PTKs, including Fyn, Fgr, Src, and Lyn, resulting in the Y374 phosphorylation of IKKγ/NEMO. It should be noted that murine IKKγ/NEMO does not have a tyrosine residue corresponding to Y374 of human IKKγ/NEMO, suggesting that this tyrosine phosphorylation site is specific for human IKKγ/NEMO. IKKγ/NEMO has been shown to bind itself as well as a variety of cellular and viral proteins, resulting in the modulation of IKK complex activity in either a positive or a negative manner (44, 45). Despite its apparent negative function in NF-κB activation, either the S377 or Y374 phosphorylation showed little or no effect on the ubiquitination, self-assembly, and IKKα/IKKβ interaction of IKKγ/NEMO. However, we cannot exclude the possibility that multiple serine residues and a tyrosine residue located at the C-terminal proline-rich domain of IKKγ/NEMO temporally and differentially undergo phosphorylation/dephosphorylation under diverse stimulation conditions, leading to conformational changes at a given time that subsequently alter its affinity for itself and other cellular proteins. Further detailed study is necessary to identify how the S377 and Y374 phosphorylation sites negatively regulate IKKγ/NEMO function to induce NF-κB activity.
A variety of viral oncoproteins deregulate the NF-κB signaling pathway by targeting IKKγ/NEMO (23). Earlier studies have suggested that KSHV KvFLIP is recruited to a ~700-kDa IKK complex and physically associates with IKKα, IKKβ, and IKKγ/NEMO, thereby contributing to the constitutive activation of NF-κB in KSHV-infected cells as well as in cells stably expressing KvFLIP (29, 30, 43). In addition, IKKγ/NEMO is essential for KvFLIP-mediated constitutive NF-κB activation (29), and IKKα is primarily responsible for the KvFLIP-mediated constitutive activation of NF-κB activity, whereas IKKβ contributes to maximum induction of KvFLIP-mediated activation of NF-κB activity (43, 46). A recent study showing that both KvFLIP and Tax activate the NF-κB signaling pathway without involvement of the linear polyubiquitination of IKKγ/NEMO (47) implies a distinct mechanism by which these viral proteins can induce a cellular NF-κB signaling pathway. Nevertheless, we are surprised that ectopic expression of the KvFLIP oncoprotein led to the increase of S377 phosphorylation of IKKγ/NEMO, which potentially suppresses NF-κB activation. This raises the question of why KvFLIP induces the S377 phosphorylation of IKKγ/NEMO. While it remains unclear how IKKγ/NEMO S377 phosphorylation contributes to the KvFLIP-mediated induction of NF-κB activation, it is possible that the S377 phosphorylation of IKKγ/NEMO functions as a physiological brake to keep the balance of host signaling pathways, and KvFLIP may utilize S377 phosphorylation as a decoy mechanism to deceive and evade host immune responses. Nevertheless, our study discovers novel posttranslational regulatory mechanisms to control the NF-κB signaling pathway, and future study will be dedicated to answering the questions above.
MATERIALS AND METHODS
Plasmids.
Human IKKγ/NEMO tagged with an HA epitope at the N terminus was cloned into the puroIRES vector. For the construction of phosphoinhibitory IKKγ/NEMO mutants, phenylalanine residues and alanine residues were substituted for the tyrosine residue at 374 (Y374F) and serine residues at 377 (S377A) by site-directed mutagenesis. Similarly, aspartate and glutamate residues were substituted for the tyrosine residue at 374 (Y374D) and serine residues at 377 (S377E) for the construction of phosphomimetic IKKγ/NEMO mutants. The presence of these mutations was confirmed by sequencing. Myc-tagged Fyn and Lck were previously described (48). V5-tagged Fgr, Src, and Lyn were constructed in the puroIRES vector.
Cell lines and transfection.
HEK 293T cells (ATCC) and IKKγ/NEMO-knockout MEFs (obtained from Xin Lin, University of Texas, M. D. Anderson Cancer Center) were maintained in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% fetal bovine serum (Gibco) and antibiotics (100 U/ml penicillin and 100 µg/ml streptomycin). IKKγ/NEMO-deficient Jurkat T cells (obtained from Xin Lin, University of Texas, M. D. Anderson Cancer Center) were maintained in RPMI medium (Gibco) supplemented with 10% fetal bovine serum and antibiotics. To establish stable cell lines expressing WT IKKγ/NEMO and its mutants, IKKγ/NEMO−/− MEFs were transfected with puroIRES vector carrying the WT or mutant IKKγ/NEMO gene using Lipofectamine 2000 (Invitrogen), followed by selection in the presence of 2 µg/ml of puromycin (Sigma) for 2 weeks. Similarly, IKKγ/NEMO-deficient Jurkat T cells stably expressing WT or mutant IKKγ/NEMO were established by electroporating cells with puroIRES vector carrying the WT or mutant IKKγ/NEMO gene. Transient transfection of HEK 293T cells was performed using polyethylenimine (PEI). At 36 h posttransfection, cells were harvested for immunoprecipitation.
Immunoprecipitation, immunoblotting, and reagents.
Whole-cell lysate (WCL) was prepared in 1% NP-40 lysis buffer (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 1% NP-40) containing phosphatase inhibitor cocktail (Roche) at 36 h posttransfection and then precleared for 2 h at 4°C in the presence of Sepharose beads. Precleared WCL was incubated for 2 h at 4°C with the indicated antibodies, followed by further incubation for 2 h at 4°C in the presence of protein A/G-conjugated beads. The immunoprecipitates were washed three times with 1% NP-40 lysis buffer. An SDS-PAGE gel was transferred onto a polyvinylidene difluoride (PVDF) membrane, followed by blocking in 5% nonfat milk and then immunoblotting with indicated antibodies. Antibodies against IKKα, IKKβ (L570), IκBα (L35A5), phospho-IκBα (5A5), and recombinant mouse and human TNF-α were purchased from Cell Signaling. Antibody for V5 was purchased from Invitrogen. Antibodies for HA and Myc were obtained from Covance. Antibody for FLAG, HA-conjugated beads, PMA, and ionomycin were purchased from Sigma.
Ca2+ flux assay.
IKKγ/NEMO-deficient Jurkat T cells were washed once with complete RPMI medium. Cells were then incubated with dye according to the manufacturer’s instructions (BD). Ca2+ flux was measured by flow cytometry (FACS Canto; BD). Basal fluorescence was recorded for 1 min prior to stimulation with PMA and ionomycin for an additional 5 min.
ELISA.
Enzyme-linked immunosorbent assay (ELISA) was performed using supernatants from IKKγ/NEMO−/− MEFs stimulated with mouse TNF-α (10 ng/ml) to quantify mouse IL-6 (BD OptEIA mouse IL-6 set) according to the manufacturer’s instructions.
ACKNOWLEDGMENTS
The work was supported by NIH CA082057, CA31363, and DE019085; the William Lawrence and Blanche Hughes Foundation; the Hastings Foundation; and the Fletcher Jones Foundation.
Footnotes
Citation Lee SH, et al. 2012. Novel phosphorylations of IKKγ/NEMO. mBio 3(6):e00411-12. doi:10.1128/mBio.00411-12.
REFERENCES
- 1. Häcker H, Karin M. 2006. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006:re13. http://dx.doi.org/10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 2. Vallabhapurapu S, Karin M. 2009. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 27:693–733 [DOI] [PubMed] [Google Scholar]
- 3. Israël A. 2010. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harbor Perspect. Biol. 2:a000158 http://dx.doi.org/10.1101/cshperspect.a000158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ruland J. 2011. Return to homeostasis: downregulation of NF-kappaB responses. Nat. Immunol. 12:709–714 [DOI] [PubMed] [Google Scholar]
- 5. Baldwin AS., Jr. 2001. Series introduction: the transcription factor NF-kappaB and human disease. J. Clin. Invest. 107:3–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sebban H, Yamaoka S, Courtois G. 2006. Posttranslational modifications of NEMO and its partners in NF-kappaB signaling. Trends Cell Biol. 16:569–577 [DOI] [PubMed] [Google Scholar]
- 7. Perkins ND. 2006. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 25:6717–6730 [DOI] [PubMed] [Google Scholar]
- 8. Zheng C, Yin Q, Wu H. 2011. Structural studies of NF-kappaB signaling. Cell Res. 21:183–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou H, et al. 2004. Bcl10 activates the NF-kappaB pathway through ubiquitination of NEMO. Nature 427:167–171 [DOI] [PubMed] [Google Scholar]
- 10. Yamamoto M, et al. 2006. Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat. Immunol. 7:962–970 [DOI] [PubMed] [Google Scholar]
- 11. Tang ED, Wang CY, Xiong Y, Guan KL. 2003. A role for NF-kappaB essential modifier/IkappaB kinase-gamma (NEMO/IKKgamma) ubiquitination in the activation of the IkappaB kinase complex by tumor necrosis factor-alpha. J. Biol. Chem. 278:37297–37305 [DOI] [PubMed] [Google Scholar]
- 12. Abbott DW, Wilkins A, Asara JM, Cantley LC. 2004. The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr. Biol. 14:2217–2227 [DOI] [PubMed] [Google Scholar]
- 13. Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. 2006. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science 311:1141–1146 [DOI] [PubMed] [Google Scholar]
- 14. Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. 2003. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell 115:565–576 [DOI] [PubMed] [Google Scholar]
- 15. Bergink S, Jentsch S. 2009. Principles of ubiquitin and Sumo modifications in DNA repair. Nature 458:461–467 [DOI] [PubMed] [Google Scholar]
- 16. Tarassishin L, Horwitz MS. 2001. Sites on FIP-3 (NEMO/IKKgamma) essential for its phosphorylation and NF-kappaB modulating activity. Biochem. Biophys. Res. Commun. 285:555–560 [DOI] [PubMed] [Google Scholar]
- 17. Prajapati S, Gaynor RB. 2002. Regulation of Ikappa B kinase (IKK)gamma /NEMO function by IKKbeta-mediated phosphorylation. J. Biol. Chem. 277:24331–24339 [DOI] [PubMed] [Google Scholar]
- 18. Carter RS, Pennington KN, Ungurait BJ, Ballard DW. 2003. In vivo identification of inducible phosphoacceptors in the IKKgamma/NEMO subunit of human IkappaB kinase. J. Biol. Chem. 278:19642–19648 [DOI] [PubMed] [Google Scholar]
- 19. Wang C, et al. 2001. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412:346–351 [DOI] [PubMed] [Google Scholar]
- 20. Senftleben U, et al. 2001. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 293:1495–1499 [DOI] [PubMed] [Google Scholar]
- 21. Ling L, Cao Z, Goeddel DV. 1998. NF-kappaB-inducing kinase activates IKK-alpha by phosphorylation of ser-176. Proc. Natl. Acad. Sci. U. S. A. 95:3792–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delhase M, Hayakawa M, Chen Y, Karin M. 1999. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science 284:309–313 [DOI] [PubMed] [Google Scholar]
- 23. Hiscott J, Nguyen TL, Arguello M, Nakhaei P, Paz S. 2006. Manipulation of the nuclear factor-kappaB pathway and the innate immune response by viruses. Oncogene 25:6844–6867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J. Clin. Invest. 120:939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thome M, et al. 1997. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386:517–521 [DOI] [PubMed] [Google Scholar]
- 26. Hu S, Vincenz C, Buller M, Dixit VM. 1997. A novel family of viral death effector domain-containing molecules that inhibit both CD-95- and tumor necrosis factor receptor-1-induced apoptosis. J. Biol. Chem. 272:9621–9624 [DOI] [PubMed] [Google Scholar]
- 27. Bertin J, et al. 1997. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc. Natl. Acad. Sci. U. S. A. 94:1172–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sun Q, Zachariah S, Chaudhary PM. 2003. The human herpes virus 8-encoded viral FLICE-inhibitory protein induces cellular transformation via NF-kappaB activation. J. Biol. Chem. 278:52437–52445 [DOI] [PubMed] [Google Scholar]
- 29. Liu L, et al. 2002. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J. Biol. Chem. 277:13745–13751 [DOI] [PubMed] [Google Scholar]
- 30. Field N, et al. 2003. KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell Sci. 116:3721–3728 [DOI] [PubMed] [Google Scholar]
- 31. Chaudhary PM, Jasmin A, Eby MT, Hood L. 1999. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 18:5738–5746 [DOI] [PubMed] [Google Scholar]
- 32. Oppermann FS, et al. 2009. Large-scale proteomics analysis of the human kinome. Mol. Cell. Proteomics 8:1751–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. 1997. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 91:243–252 [DOI] [PubMed] [Google Scholar]
- 34. Rothwarf DM, Zandi E, Natoli G, Karin M. 1998. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 395:297–300 [DOI] [PubMed] [Google Scholar]
- 35. Rushe M, et al. 2008. Structure of a NEMO/IKK-associating domain reveals architecture of the interaction site. Structure 16:798–808 [DOI] [PubMed] [Google Scholar]
- 36. May MJ, et al. 2000. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science 289:1550–1554 [DOI] [PubMed] [Google Scholar]
- 37. Tegethoff S, Behlke J, Scheidereit C. 2003. Tetrameric oligomerization of IkappaB kinase gamma (IKKgamma) is obligatory for IKK complex activity and NF-kappaB activation. Mol. Cell. Biol. 23:2029–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Agou F, et al. 2002. NEMO trimerizes through its coiled-coil C-terminal domain. J. Biol. Chem. 277:17464–17475 [DOI] [PubMed] [Google Scholar]
- 39. Dephoure N, et al. 2008. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 105:10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Seo J, Lee KJ. 2004. Post-translational modifications and their biological functions: proteomic analysis and systematic approaches. J. Biochem. Mol. Biol. 37:35–44 [DOI] [PubMed] [Google Scholar]
- 41. Xiao G, Sun SC. 2000. Activation of IKKalpha and IKKbeta through their fusion with HTLV-I tax protein. Oncogene 19:5198–5203 [DOI] [PubMed] [Google Scholar]
- 42. Jin DY, Giordano V, Kibler KV, Nakano H, Jeang KT. 1999. Role of adapter function in oncoprotein-mediated activation of NF-kappaB. Human T-cell leukemia virus type I tax interacts directly with IkappaB kinase gamma. J. Biol. Chem. 274:17402–17405 [DOI] [PubMed] [Google Scholar]
- 43. Matta H, Sun Q, Moses G, Chaudhary PM. 2003. Molecular genetic analysis of human herpes virus 8-encoded viral FLICE inhibitory protein-induced NF-kappaB activation. J. Biol. Chem. 278:52406–52411 [DOI] [PubMed] [Google Scholar]
- 44. Shifera AS. 2010. Protein-protein interactions involving IKKgamma (NEMO) that promote the activation of NF-kappaB. J. Cell. Physiol. 223:558–561 [DOI] [PubMed] [Google Scholar]
- 45. Shifera AS. 2010. Proteins that bind to IKKgamma (NEMO) and down-regulate the activation of NF-kappaB. Biochem. Biophys. Res. Commun. 396:585–589 [DOI] [PubMed] [Google Scholar]
- 46. Matta H, Chaudhary PM. 2004. Activation of alternative NF-kappa B pathway by human herpes virus 8-encoded Fas-associated death domain-like IL-1 beta-converting enzyme inhibitory protein (vFLIP). Proc. Natl. Acad. Sci. U. S. A. 101:9399–9404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shimizu A, et al. 2011. Kaposi’s sarcoma-associated herpesvirus vFLIP and human T cell lymphotropic virus type 1 tax oncogenic proteins activate IkappaB kinase subunit gamma by different mechanisms independent of the physiological cytokine-induced pathways. J. Virol. 85:7444–7448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Garcia MI, Kaserman J, Chung YH, Jung JU, Lee SH. 2007. Herpesvirus saimiri STP-A oncoprotein utilizes Src family protein tyrosine kinase and tumor necrosis factor receptor-associated factors to elicit cellular signal transduction. J. Virol. 81:2663–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]