

Background: Binding of Dab2 to sulfatides results in platelet aggregation inhibition.
Results: The structure of a Dab2-derived peptide (SBM) embedded in dodecylphosphocholine micelles, characterization of its minimal functional sulfatide-binding site, and its inhibitory platelet aggregation activity were determined.
Conclusion: An amphipathic helical region of Dab2 SBM binds sulfatides, leading to platelet aggregation inhibition.
Significance: Dab2 SBM may lead to the design of novel aggregatory inhibitors.
Keywords: Membrane Proteins, NMR, Peptide Interactions, Sphingolipid, Surface Plasmon Resonance (SPR), Disabled-2, Platelet Aggregation, Sulfatides
Abstract
Disabled-2 (Dab2) targets membranes and triggers a wide range of biological events, including endocytosis and platelet aggregation. Dab2, through its phosphotyrosine-binding (PTB) domain, inhibits platelet aggregation by competing with fibrinogen for αIIbβ3 integrin receptor binding. We have recently shown that the N-terminal region, including the PTB domain (N-PTB), drives Dab2 to the platelet membrane surface by binding to sulfatides through two sulfatide-binding motifs, modulating the extent of platelet aggregation. The three-dimensional structure of a Dab2-derived peptide encompassing the sulfatide-binding motifs has been determined in dodecylphosphocholine micelles using NMR spectroscopy. Dab2 sulfatide-binding motif contains two helices when embedded in micelles, reversibly binds to sulfatides with moderate affinity, lies parallel to the micelle surface, and when added to a platelet mixture, reduces the number and size of sulfatide-induced aggregates. Overall, our findings identify and structurally characterize a minimal region in Dab2 that modulates platelet homotypic interactions, all of which provide the foundation for rational design of a new generation of anti-aggregatory low-molecular mass molecules for therapeutic purposes.
Introduction
Disabled-2 (Dab2)2 is a cargo-specific adaptor protein involved in endocytosis, cell surface receptor turnover, and cell signaling (1–4). Expression of Dab2 is frequently lost in about 90% of all breast and ovarian cancers, and its homozygous deletion has been identified in numerous tumors, pointing to a role for this protein in tumor suppression (5, 6). Two isoforms of Dab2 have been identified: a long isoform (known as p96 or p82) that is localized at the plasma membrane (2–4) and a shorter form of the protein (known as p67 or p59) that exhibits a diffuse distribution in the cell with remarkable nuclear localization (7). The ability of Dab2 to display a wide range of functions is centered in its modular architecture. The N-terminal region of Dab2 includes a phosphotyrosine-binding domain (PTB) followed by two Asp-Pro-Phe (DPF) and five Asn-Pro-Phe (NPF) motifs and a C-terminal proline-rich SH3 domain (8).
The PTB domain has been involved in a variety of cellular functions ranging from signal transduction, membrane trafficking, and cytoskeletal assembly (9). Despite its initial characterization as a tyrosine-phosphorylated Asn-Pro-any residue-Tyr (NPXY) motif-binding module, the PTB domain exhibits no preference for phosphorylation in tyrosine residues or for nonphosphorylated NPX(Y/F)-containing proteins (10). The crystal structures of the Dab1 and Dab2 PTB domain exhibit seven central β-strands, which form two orthogonal β-sheets followed by a C-terminal α-helix (11, 12). In addition to recognizing short motifs, the PTB domain binds to phosphoinositides (9). The Dab1 and Dab2 PTB domains bind to both phosphatidylinositol 4-phosphate and phosphatidylinositol (4,5)-bisphosphate (PtdIns(4,5)P2), at a lipid-binding site that faces opposite of the NPXY motif (11–13). Moreover, the N-terminal region of Dab2, including its PTB domain (N-PTB), has been recently reported to bind the αIIbβ3 integrin receptor to negatively regulate platelet-fibrinogen interactions (14, 15). To exert this function, Dab2 binds to platelet surface sulfatides through two conserved sulfatide-binding motifs within N-PTB. This association is proposed to partition Dab2 into two extracellular pools, the sulfatide- and the integrin-bound complexes, modulating the extent of platelet aggregation (15). Sulfatide binding by Dab2 is required to efficiently compete, among other receptor molecules, with P-selectin, a sulfatide-binding pro-aggregatory protein, whose surface expression is consequently reduced (16).
Recently, we characterized the molecular mechanism by which Dab2 N-PTB recognizes its lipid ligands, PtdIns(4,5)P2 and sulfatides, using multiple biophysical approaches (17). First, PtdIns(4,5)P2 and sulfatides compete with each other for Dab2 N-PTB binding, consistent with earlier findings that both lipids require the conserved residue Lys-53 (15). Although sulfatides contribute to Dab2 membrane penetration on the platelet surface, PtdIns(4,5)P2 likely allows the protein to contact the inner leaflet of the plasma membrane to trigger clathrin-mediated endocytosis (17).
Sulfatides are well characterized lipids that contribute to hemostasis (18). Their surface local concentration increases upon platelet activation (19), leading to the recruitment of extracellular pro-aggregatory (i.e. von Willebrand factor, P-selectin) (20, 21) and anti-aggregatory proteins (Dab2) (14, 15). Given the critical role of membrane sulfatides in platelet function and the presence of multiple ligand-binding sites in Dab2 N-PTB, we have generated a Dab2-derived peptide that contains the two sulfatide-binding motifs (SBM). Here, we report the solution structure of Dab2 SBM embedded in dodecylphosphocholine (DPC) micelles. Using NMR spectroscopy, we have investigated the nature of Dab2 SBM-sulfatide interaction and established that the majority of the interacting residues map on the second sulfatide-binding motif. The orientation of Dab2 SBM relative to a sulfatide-enriched DPC micellar surface was probed using paramagnetic agents on the NMR resonances of Dab2 SBM. We demonstrate that Dab2 SBM exhibits a parallel orientation below the micellar surface but does not cross to its center. Importantly, we found that Dab2 SBM exhibits an anti-aggregatory activity comparable with that of the fibrinogen-derived peptide, Arg-Gly-Asp-Ser (RGDS), when evaluated under hemodynamic conditions that mimic those of the vasculature.
EXPERIMENTAL PROCEDURES
Peptide Synthesis
A peptide corresponding to human Dab2 SBM (24SKKEKKKGPEKTDEYLLARFKGDGVKYKAKLIGID58) was chemically synthesized (Biopeptide Co., Inc.)
Peptide Expression and Purification
cDNA representing the human Dab2 SBM peptide (residues 24–58) was cloned into pGEX6P1 (GE Healthcare) and the glutathione S-transferase (GST) fusion protein expressed in Escherichia coli (Rosetta; Stratagene). Bacterial cells were grown in either Luria-Bertani or minimal media at 37 °C until they reached an optical density of ∼0.8. Induction of GST fusion protein resulted from the addition of 1 mm isopropyl β-d-thiogalactopyranoside (Research Products International) followed by incubation for 4 h at 25 °C. The fusion protein, bound to glutathione beads (GE Healthcare), was cleaved by incubation with PreScission protease (GE Healthcare) for 3 h at room temperature. Free Dab2 SBM was concentrated to dryness using a centrifugal evaporator (Savant SpeedVac). The dried peptide film was then suspended in 6 m urea for 30 min at room temperature and subjected to size-exclusion chromatography using a Superdex 30 column, equilibrated with 50 mm Tris-HCl (pH 8), 250 mm NaCl, and 3 m urea, driven by an FPLC system (GE Healthcare). Dab2 SBM fractions were pooled and desalted on a C18 column (Waters Sep-Pak) and then dried again as described above and stored at −80 °C until use. The purity of the peptide was higher than 95% as estimated by mass spectrometry analysis.
NMR Spectroscopy
NMR samples containing 0.2–1 mm of uniformly unlabeled, 15N labeled-, and 15N, 13C-labeled Dab2 SBM were prepared in 90% H2O, 10% 2H2O and 10 mm d4-citrate (pH 5), 40 mm KCl, and 1 mm NaN3 (NMR buffer) with either 40 mm or 200 mm d38-DPC. Sulfatide (brain source), dioleoyl phosphatidylserine (PtdSer), dioleoyl phosphatidylethanolamine, or dioleoyl phosphatidylcholine (Avanti Lipids) were added into the 15N-labeled DPC-embedded Dab2 SBM (0.2 mm) at 1:8 peptide:lipid ratio and analyzed by 1H-15N heteronuclear single-quantum coherence (HSQC) experiments. Peptide and lipid stock solutions were prepared in NMR buffer containing 40 mm d38-DPC. The weighted chemical shifts were calculated using the following equation (22).
Solvent accessibility of the amide backbone signals was detected using proton-deuterium exchange studies, in which the peptide was suspended in the same buffer containing 100% 2H2O. Disappearance of the amide backbone peaks as a function of time was determined using two-dimensional 1H-15N HSQC spectra. NMR experiments were acquired at 25 °C using a Bruker Avance III 600-MHz spectrometer (Virginia Tech) equipped with an inverse detected triple resonance broadband inverse probe with z axis pulse field gradients. 1H chemical shifts were referenced using sodium 4,4-dimethyl-4-silapentane-1-sulfonate (50 μm), whereas 15N and 13C chemical shifts were referenced using frequency ratios as described by Wishart et al. (23) (and see also Ref. 24). Full and unambiguous assignments of spin systems and sequential resonances were obtained from two-dimensional 1H-15N HSQC, NOESY, and total correlation spectroscopy and three-dimensional HNCO, HN(CA)CO, CBCANH, CBCA(CO)NH, and 15N HSQC-NOESY spectra. Pulse-field gradients were employed for water suppression by a WATERGATE pulse sequence (25). Spectra were processed using TopSpin (Bruker) and NMRPipe (26) and analyzed using Sparky (27).
Structure Calculations
Structure calculations were obtained using Xplor-NIH (version 2.28) (28). Distance restraints used in the structure calculations were classified as strong (1.8–2.7 Å), medium (1.8–3.3 Å), weak (1.8–5 Å), and very weak restraints (1.8–6 Å) based on NOE peak intensities. The torsion angle restraints were obtained from the TALOS program using backbone chemical shifts (29). A total of 200 structures were calculated from the structural restraints using the standard simulated annealing protocol (sa_new.inp) included in the Xplor-NIH software package and further refined using the refine.inp protocol. The structures showed no NOE violations greater than 0.5 Å, and no dihedral angle restraint violations larger than 5° were accepted using the accept.inp. Twenty structures with the lowest energy were selected as the most representative structures for further analysis. The stereochemical qualities of the structure were monitored using the PROCHECK-NMR software. The final structures were generated using MOLMOL and PyMOL. A summary of the restraints used to calculate the NMR structure of the micelle-bound Dab2 SBM and the statistics of the calculated structures are shown in Table 1.
TABLE 1.
Structural statistics of the Dab2 SBM peptide in DPC micelles at pH 5.0, 298 K
| Total number of distance NMR restraints | 619 |
| Intraresidue (|i − j| = 0) | 338 |
| Sequential (|i − j| = 1) | 221 |
| Medium (|i − j| ≤ 4) | 60 |
| Long range (|i − j| > 4) | 0 |
| Average energy (kcal mol−1) | 421.8 |
| r.m.s.d.a (Å) | |
| All residues | |
| Backbone heavy atoms | 5.62 ± 1.63 |
| All heavy atoms | 6.30 ± 1.58 |
| Residues 38–43 | |
| Backbone heavy atoms | 0.19 ± 0.10 |
| All heavy atoms | 1.05 ± 0.26 |
| Residues 48–55 | |
| Backbone heavy atoms | 0.25 ± 0.14 |
| All heavy atoms | 0.79 ± 0.16 |
| Ramachandran analysis (at the helical span) | |
| Residues in most favored region (%) | 97.1 |
| Residues in additionally allowed region (%) | 2.9 |
| Residues in generously allowed region (%) | 0 |
| Residues in disallowed region (%) | 0 |
a r.m.s.d., root mean square deviation.
Paramagnetic Studies
To determine the location of Dab2 SBM relative to the micelle surface, we used three different paramagnetic agents: 16-doxyl steric acid (16-DSA), 5-doxyl stearic acid (5-DSA) (Sigma-Aldrich), and MnCl2 (J. T. Baker). Due to their poor aqueous solubility, 5-DSA and 16-DSA were dissolved in d4-methanol. Either 16-DSA or 5-DSA (0.67 mm each) was added to the Dab2 SBM (0.2 mm) in NMR buffer containing 40 mm DPC and 1.6 mm sulfatides. The aggregation number of DPC micelles is about 60 (30); therefore, the experiments were carried out with one spin label probe per micelle. The intensity reduction of 1H-15N HSQC spectra caused by close proximity to the spin label was calculated as the ratios of the peak intensities of the spectra in the absence to the presence of the probe.
Circular Dichroism Spectroscopy
Far-UV circular dichroism (CD) spectra were recorded using a JASCO J-815 spectropolarimeter. Five accumulated scans from 260 to 190 nm were collected on 150-μl volume samples containing 20 μm Dab2 SBM in 10 mm citrate (pH 5) and 40 mm KF in the absence or presence of 10 mm DPC (Anatrace/Affymetrix) and held in a 1-mm path length cell at 25 °C. The secondary structure content of the peptide was calculated using the CDPro software.
Surface Plasmon Resonance
Stocks of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine, 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine, and sulfatides (Avanti Lipids) and cholesterol (Sigma) were dissolved in organic solvents following the manufacturer's instructions. Liposomes were prepared as reported previously (16). All surface plasmon resonance (SPR) measurements were performed on a BIAcore X-100 system (GE Healthcare) at 25 °C in running buffer containing 10 mm Tris-HCl (pH 7.4) and 100 mm NaCl. Running buffer was used for the equilibration, association, and dissociation steps. The surface of an L1 sensor chip was washed by injecting 40 mm N-octyl β-d-glucopyranoside at a flow rate of 30 μl/min. Approximately 3,000 response units of liposomes were immobilized onto the surface of the sensor chip at a flow rate of 5 μl/min. Sulfatide-free liposomes were used as the control surface. Each of the sensor chip flow cells were then blocked using 0.1 mg/ml fatty acid-free BSA (Sigma Aldrich). Dab2 SBM was then injected over the liposome-coated surface of the sensor chip at a flow rate of 30 μl/min, in a range between 0 and 180 μm. In each cycle, flow cells were regenerated with 20 mm NaOH. After subtraction of nonspecific binding traces, the sensorgrams were analyzed using the BIAcore X-100 evaluation software (version 2.0).
Blood Collection and Platelet Purification
Blood samples were obtained from healthy donors by venipuncture. The procedure followed the Institutional Review Board guidelines and obtained ethical approval from the Virginia Tech Office of Research Compliance. Platelets were obtained as reported previously (16).
Microfluidic Design
The microfluidic device and the shear stress conditions used in these studies were essentially as reported (16). Briefly, the microfluidic device consisted of a simple straight channel 500 μm wide, 50 μm deep, and 3.625 cm long. Flow was driven by a microsyringe pump (Cole-Parmer), and the pump was set at 0.05 ml/h. These conditions led to an average velocity of 0.55 mm/sec in the channel, which is within the range of in vivo blood velocity (31) and produces a shear rate of 70 s−1. Channels were coated with a cell-free platelet poor plasma fraction, which was obtained by centrifugation of the sample at 13,000 × g for 10 min at room temperature. Coating was performed for 2 h at 37 °C in the presence of 5% CO2. Platelets flowing through the channel were detected using an inverted light microscope (DMI 6000B, Leica Microsystems) equipped with a digital camera (DFC420, Leica Microsystems).
Quantitative Measurements of Platelet Aggregates
Quantification of aggregate formation was carried out using MATLAB (The MathWorks, Inc.) with an in-house developed code as described (16). For this experiment, we were primarily interested in the size and number of objects that were definitely composed of more than one platelet. After stationary objects were identified and sized, the previous methodology was extended to sort objects by size into single platelets and platelet aggregates, and only the aggregates were considered for the remainder of the analysis. The threshold was chosen as >57.5 μm2 based on comparison of the size distribution of the platelet-only condition to the remaining cases. Dividing each video into four horizontal independent regions and calculating the mean and S.D. of the platelet aggregates between each segment assessed regional variability of each condition. Statistical differences in the means were tested using a balanced one-way analysis of variance test and a Tukey-Kramer honest significant difference test for multiple comparisons.
RESULTS
Resonance Assignments and Structure Constraints
A peptide containing the membrane-associated sulfatide-binding motifs of Dab2 (Fig. 1A) was synthetically generated and exhibited a disordered conformation based on CD (Fig. 1B) and NMR (data not shown) analyses. DPC was chosen to investigate Dab2 SBM membrane binding properties because this zwitterionic detergent contains a phosphocholine head group and, therefore, mimics the anisotropy milieu of a lipid cell membrane. DPC forms stable micelles with a molecular mass of about 20 kDa, which results in a reasonable correlation time that enables NMR studies of hydrophobic or amphipathic helical regions (32). Dab2 SBM displayed a helical conformation in DPC micelles evidenced by the presence of a minimum at 205 nm, a shoulder at 222 nm, and a maximum at 195 nm (Fig. 1B). Dab2 SBM is quite stable in DPC micelles and, therefore, is suitable for NMR measurements to determine its structure and topology in a membrane-like environment. The NMR spectra of Dab2 SBM were recorded in the presence of d38-DPC micelles at a relative stoichiometry of ∼3:1 (micelle to peptide). The HSQC spectrum of Dab2 SBM displayed well resolved 1H-15N chemical shifts, indicating that the peptide exhibited a defined structure in micelles (Fig. 1C). The Gly-31, Glu-33, and Lys-34 residues displayed two sets of cross-peaks with unequal intensities in the 1H-15N HSQC spectrum of the peptide. Because all of the chemical shift heterogeneities are around Pro-32, we hypothesize that this is due to the cis/trans isomerization of X-Pro bonds commonly observed in proteins. The NOE connectivities between Hδ of proline and HN and/or Hα of the preceding residue are diagnostic for a trans peptide bond, whereas the NOE connectivities between the Hα of proline and the HN and/or Hα of the preceding residue are characteristic of a cis conformation (33, 34). In Dab2 SBM, the Pro-32 residue of the major species was in a trans conformation as indicated by the presence of strong NOEs between Hα of Gly-31 and Hδ of Pro-32. Furthermore, in two-dimensional 1H-1H NOESY and three-dimensional NOESY-HSQC spectra, only a small number of NOE connectivities were observed from Ser-24 to Arg-42, indicating a random coil conformation of the N terminus of Dab2 SBM, whereas a large number of NOE connectivities, such as dαN(i, i + 3), dαN(i, i + 4), and dαβ(i, i + 3), appeared from Phe-43 to Gly-56, which are indicative of α-helices (Fig. 2A). The superposition of the backbone atoms of the 20 lowest energy conformers calculated from NOE-based distance constraints and the ribbon representations of Dab2 SBM in DPC micelles are shown in Fig. 2, B and C, respectively. The peptide contains a long disordered N-terminal region followed by two α-helices spanning residues Tyr-38 to Phe-43 (helix 1) and Val-48 to Ile-55 (helix 2) (Fig. 2, B and C). Visualization of the calculated surface charge distribution of the Dab2 SBM structure suggests an amphipathic nature of the peptide with positively charged residues found on opposite faces to negatively charged residues (Fig. 2D). The structural organization of the micelle-embedded Dab2 SBM is in agreement with sequence conservation among human and mouse Dab proteins, in which the C-terminal region displays the highest sequence identity (Fig. 1A). Information on experimental restraints employed to calculate the 20 structures is given in Table 1.
FIGURE 1.
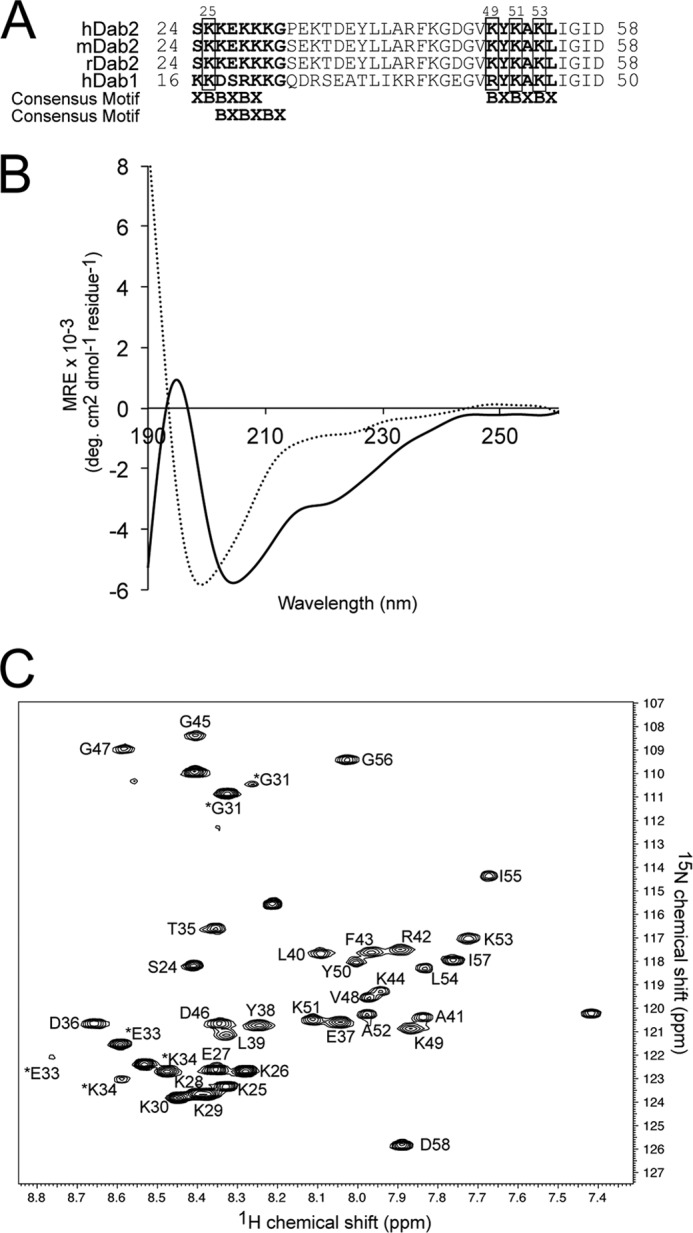
Dab2 SBM is disordered in aqueous solution but becomes structured in the presence of DPC micelles. A, sequences of the closest homologues of Dab proteins corresponding to the SBM region. The two sulfatide-binding sites identified in human Dab2 (hDab2) are bolded, and the key lysine residues are boxed. mDab2, mouse Dab2. B, far-UV CD spectra of Dab2 SBM in the absence (dotted line) and presence (solid line) of DPC micelles. deg, degree. C, two-dimensional 1H, 15N HSQC of uniformly 13C, 15N-labeled Dab2 SBM in DPC micelles. Resonances of the backbone amides are labeled according to human Dab2 protein sequence. Asterisks on Gly-31, Glu-33, and Lys-34 resonances likely are due to cis/trans isomerization of Pro-32. MRE, mean residue ellipticity.
FIGURE 2.

NOE patterns and NMR structure of micelle-bound Dab2 SBM. A, summary of the sequential and medium range NOE connectivities for micelle-bound Dab2 SBM. The thickness of the sequential NOEs corresponds to the intensity of the NOE interaction between residues. B, the ensemble of the 20 energy-minimized conformers of micelle-bound Dab2 SBM. C, two views of a representative structure of Dab2 SBM with the two helical elements labeled in red. D, electrostatic surface representation of Dab2 SBM in two 180° orientations. Images in C and D were generated using PyMOL.
Dab2 SBM Is the Minimal Functional Unit of Dab2 N-PTB
The N-terminal region of Dab2 (N-PTB) binds sulfatides by two well conserved sulfatide-binding motifs (15). Given that Dab2 SBM was designed based on the presence of these motifs, we addressed whether the peptide was still able to associate with sulfatides using solution NMR analysis. With the Dab2 SBM structure and NMR assignments available, we were able to directly investigate sulfatide binding to the DPC-embedded peptide using HSQC titrations. The location of the sulfatide-binding site on the surface of Dab2 SBM was determined by monitoring shifts in the positions of the 1H-15N backbone amide NMR signals of the peptide induced by the presence of DPC-embedded sulfatides (1:8 peptide:sulfatide molar ratio) (Fig. 3A). The most significant chemical shift changes were observed at the C-terminal region of the peptide, including residues Arg-42, Ala-52, Leu-54, Ile-55, Gly-56, and Asp-58 (Fig. 3B, red dotted line, and C). Less significant chemical shift perturbations induced by DPC-embedded sulfatides were in residues Tyr-38, Leu-39, Leu-40, Gly-45, Val-48, Lys-49, Tyr-50, Lys-51, and Lys-53 (Fig. 3B, yellow dotted line, and C). Using site-directed mutagenesis, we have previously shown that Dab2 residues Lys-25 (located in the first sulfatide-binding motif) and Lys-49, Lys-51, and Lys-53 (located in the second sulfatide-binding motif) are critical for sulfatide binding in N-PTB (15). Thus, our functional and structural studies demonstrate that the second sulfatide-binding region in Dab2 SBM plays a major role in sulfatide recognition. Most of the N-terminal residues, including the first sulfatide-binding region, showed small or no significant perturbations (<0.04 ppm). To demonstrate that the observed spectral changes are not due to the slight change in DPC concentration and dilution of the Dab2 SBM sample, the same amount of free sulfatide d38-DPC was added to the NMR sample, and HSQC spectra were recorded. No chemical shift perturbations were detected in the spectrum (data not shown), indicating that all chemical shift perturbations are attributed to the presence of sulfatides.
FIGURE 3.

Dab2 SBM binds sulfatides. A, overlay of 1H, 15N HSQC of DPC-bound Dab2 SBM in the absence (black) and presence (red) of DPC-bound sulfatides. Perturbed chemical shifts are boxed and labeled with the corresponding residue. B, histogram representing the normalized chemical shift changes of Dab2 SBM induced by sulfatides. The colored bars represent significant perturbations: red (Δδaverage + 1 × S.D.) > yellow (Δδaverage). C, Dab2 SBM amino acids that display significant chemical shift changes are labeled and color-coded according to the scale defined in B. D, binding plot of the Dab2 SBM to sulfatide liposomes using SPR analysis. A. U., arbitrary units.
The platelet plasma membrane is similar in composition to that observed in other eukaryotic cells with phosphatidylcholine, sulfatides, and sphingomyelin found preferentially in the outer leaflet, whereas PtdSer and phosphatidylethanolamine are the most abundant lipids in the inner leaflet (35, 36). Cholesterol is abundant in both layers with a molar ratio of cholesterol to phospholipid being about 0.5 (37). Interestingly, upon stimulation of platelet aggregation, both phosphatidylethanolamine and PtdSer translocate from the inner to the outer leaflet (39). Given their transient location at the platelet surface, we therefore tested whether these lipids could also interact with Dab2 SBM. We found that although the negatively charged PtdSer interacted with Dab2 SBM (supplemental Fig. S1B), the zwitterionic phosphatidylethanolamine and phosphatidylcholine bound poorly with the peptide (supplemental Fig. S1C), suggesting that PtdSer could also be a ligand for Dab2 SBM.
We next used SPR measurements to confirm sulfatide binding of Dab2 SBM using unilamellar vesicles composed of physiologically relevant lipids found in the plasma membrane. Dab2 SBM bound to immobilized sulfatide liposomes with high association and dissociation rates (Fig. 3D). Peptides bound to lipid surfaces generally display complex biphasic traces for both association and dissociation phases because they initially bind to the lipid head groups followed by insertion into the acyl chain region of the liposome (40, 41). Initial association of Dab2 SBM to sulfatide liposomes occurs rapidly, and the process is followed by accumulation of the peptide on the lipid surface (Fig. 3D). Toward the end of Dab2 SBM injection, the resonance signal decreases rapidly early on in the dissociation phase because free and weakly bound peptides are removed by the running buffer. This phase is followed by a slow period in which the sensorgram does not return to the base line, suggesting that a population of the peptide remains bound to the lipid surface. The sulfatide liposome binding behavior of Dab2 SBM is comparable with that observed in Dab2 N-PTB (17). Dab2 SBM bound sulfatide liposomes with plots that best fitted the steady state model with a dissociation constant of 5 × 10−5 m−1.
Localization of Dab2 SBM in Sulfatide-enriched DPC Micelles
Hydrogen-deuterium exchange experiments are traditionally used to determine the location of proteins relative to membrane mimics (42). We recorded 1H, 15N HSQC spectra immediately after the addition of D2O in the lyophilized Dab2 SBM (to 0.2 mm) containing 40 mm d38-DPC. Almost all the peaks disappeared in the spectrum (data not shown) due to the extremely rapid exchange of amide protons of SBM, indicating that the peptide may remain on the micelle surface or be exposed to water. However, it is also possible that Dab2 SBM is buried but has transient access to the micelle surface.
Paramagnetic transition ions, such as Mn2+, enhance relaxation in a distance-dependent manner and, therefore, broaden NMR resonances from protein residues exposed to aqueous solvent (43). Thus, Mn2+ was used to provide the localization of Dab2 SBM in sulfatide-enriched micelles. Mn2+ leads to a reduction of the resonances of the first 16 N-terminal residues of the peptide, indicating that these residues are more solvent-exposed (Fig. 4A). This is consistent with the unstructured nature of the peptide in this region. Interestingly, the interhelical region is also broadened, whereas the two helical elements are less perturbed (Fig. 4A). In addition, we observed a series of strong cross-peaks between the HN signals of Ser-24, Lys-25, Lys-26, Lys-28–Gly-31, Thr-35, and Asp-36 residues and water molecules in NOESY spectra (data not shown), which may originate from either NOEs or exchange between water and these residues. This confirmed that the N terminus of Dab2 SBM is exposed to water. In addition to Mn2+, we also employed paramagnetic quenching with 5-DSA and 16-DSA (Fig. 4, B and C). In 5-DSA, the doxyl moiety is localized close to the polar head group of the micelle near the membrane-water interface, whereas 16-DSA contains a spin label near the end of the fatty acid chain; thus, the probe will be found at the center of the micelle. In the presence of 5-DSA, the C-terminal half of Dab2 SBM resonances was broadened (Fig. 4B), and no further significant reduction in resonance intensities was observed with 16-DSA (Fig. 4C). Altogether, these data suggest that Dab2 SBM contacts below the surface of sulfatide-enriched micelles without traversing the micelle. We propose that the two helical elements of Dab2 SBM lie parallel to the micelle surface and that the second helical element plays a role in sulfatide interactions through the second sulfatide-binding motif.
FIGURE 4.
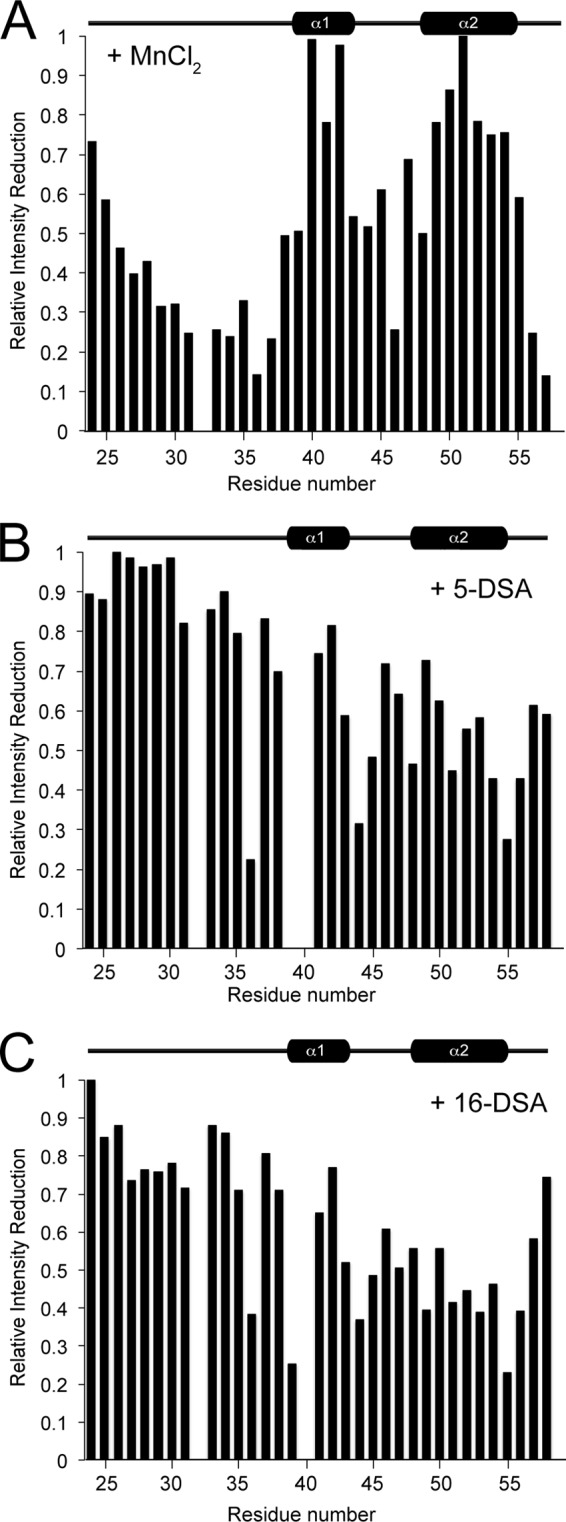
Paramagnetic quenching of Dab2 SBM in the presence of sulfatide-embedded DPC micelles. A–C, intensity retention plots of the HSQC spectrum of Dab2 SBM in the presence of MnCl2 (A), 5-DSA (B), and 16-DSA (C).
Inhibition of Platelet Aggregation by Dab2 SBM
We have previously shown that Dab2 N-PTB inhibits platelet aggregation by binding to platelet surface sulfatides under physiological shear conditions using a microfluidic system (16). For all the studies, the microchannel of the microfluidic device was coated with platelet-poor plasma, which contains adhesive proteins necessary for platelet activation. Washed platelets were flowed over the coated channel in the absence or presence of sulfatide-enriched liposomes and the indicated proteins, and the formation of platelet aggregates was followed over time. Consistent with our previous study (16), untreated platelets showed basal platelet activation and minimum platelet aggregation formation, whereas ADP-stimulated platelets incubated with sulfatide liposomes led to the formation of large aggregates (Fig. 5, A and B). Accordingly, Dab2 N-PTB inhibited platelet aggregation, whereas mutations in its sulfatide-binding motifs (Dab2 N-PTB4M) reverted the inhibitory effect (Fig. 5, C and D). Likewise, Dab2 SBM reduced platelet aggregation, albeit to a lesser degree, when compared with Dab2 N-PTB under identical experimental conditions (Fig. 5E). Remarkably, Dab2 SBM anti-aggregatory activity was comparable with that exhibited by RGDS (Fig. 5F), a low-affinity fibrinogen-derived peptide that inhibits platelet aggregation to that exhibited by acting on the αIIbβ3 integrin receptor (44). The differences found between Dab2 SBM and N-PTB regions for inhibition of platelet aggregation is believed to reflect their distinct binding affinities for the lipid (Ref. 15 and this study).
FIGURE 5.

Dab2 SBM precludes sulfatide-mediated platelet aggregation. Representative time series of images of test microchannels with platelets and aggregates under physiological flow conditions are indicated on top. Washed platelets loaded without (A) and with (B) sulfatide liposomes were preincubated with the following proteins/peptides (10 μm each): Dab2 N-PTB (C), Dab2 N-PTB4M (D), Dab2 SBM (E), and RGDS (F). Images were taken 0, 3, and 10 min after the start of flow. The last column on the right illustrates how unadhered objects (green), isolated platelets (white), and platelet aggregates (red) are visualized in the channel. Scale bar, 45 μm; arrows indicate flow direction. Representative of n = 3 experiments. Lipo-S, sulfatide-enriched liposomes.
Dab2 SBM Directly Modulates Adhesive Platelet Aggregate Formation
Next, we asked how effective Dab2 SBM is in altering the size and number of platelet aggregates when compared with Dab2 N-PTB under physiological hemodynamic conditions. Using the microchannel device, we flowed washed platelets onto plasma protein-coated channels, and measurements were carried out as described under “Experimental Procedures.” As expected, the addition of both ADP and sulfatide-enriched liposomes increased the number (Fig. 6A) and cluster size (Fig. 6B) of platelet aggregates when compared with nonstimulated washed platelets (Fig. 6). Accordingly, the addition of Dab2 N-PTB, but not its sulfatide binding-deficient form (Dab2 N-PTB4M), significantly reduced both the number and the size of the aggregates consistent with our previous observations (16). The RGDS peptide also affected platelet aggregation number and size, although to a lesser extent (Fig. 6). Dab2 SBM comparably affected the number and size of clusters. Dab2 SBM inhibitory properties became more apparent when the peptide concentration was increased from 10 to 100 μm (data not shown). Overall, these findings are consistent with our SPR data, in which Dab2 SBM exhibited a weaker affinity for sulfatides (Fig. 3D) when compared with the N-PTB region of the protein (15). Together, our data indicate that Dab2 SBM is an active peptide that can modulate platelet aggregation under physiological conditions and that its regulatory role is associated with sulfatide binding.
FIGURE 6.

Modulation of the number of platelet aggregates by Dab2 SBM. A, bar graphs representing the average count of platelet aggregates found in four different fields on the microchannel at 500 s. Washed platelets were flowed over a microchannel in the absence of ADP and sulfatides or stimulated with ADP and preincubated with sulfatide liposomes. In addition, ADP-stimulated sulfatide-treated platelets were preincubated with the following proteins/peptides (10 μm each): Dab2 N-PTB, Dab2 N-PTB4M, RGDS, and Dab2 SBM. Samples were equilibrated for at least 2 min at 70 s−1 before data collection. B, bar graphs depicting the average area of platelet clusters of four different regions of the microchannel with the experimental conditions described in A. In both panels, * indicates p < 0.05 when compared with the ADP + Lipo-S condition. Error bars indicate S.D. Lipo-S, sulfatide-enriched liposomes.
DISCUSSION
In this study, we obtained the solution structure for a Dab2-derived peptide that encompasses the two sulfatide-binding regions found in the protein. The 15N, 1H HSQC spectrum of Dab2 SBM exhibited poor resonance dispersion in the 1H dimension (data not shown) and this, together with the observed strong CD spectrum minimum at 199 nm, indicated that the peptide was disordered (Fig. 1B). The peptide showed resonances with uniform line width and signal intensity and adopted a helical structure when embedded in DPC micelles. The change in peptide structure suggests that this region contacts sulfatide-enriched membranes in Dab2. The DPC-embedded SBM peptide displayed a long N-terminal disordered region followed by two helical elements. The crystal structure of the mouse Dab2 PTB domain (residues 33–191) reveals the presence of a short helix (residues 37–42) that partially overlaps with the first helix of the DPC-embedded Dab2 SBM (residues 38–43), whereas a long Dab2 PTB β-strand element (residues 48–60) is replaced by a helical element in the micellar Dab2 SBM (residues 48–55). In DPC micelles, Dab2 SBM adopts a helical structure with significant curvature at the center region (Fig. 2C). The surface electrostatic potential of the peptide clearly shows the amphipathic nature of Dab2 SBM (Fig. 2D), a property found in other helical membrane-binding extracellular proteins that bind anionic lipids (45). The amphipathic helical fold in the SBM region of Dab2 could facilitate the platelet sulfatide-mediated membrane localization once the protein is secreted via α-granules.
Sulfatides are sphingolipids found at the outer leaflet of the plasma membrane of most eukaryotic cells, and they represent the major lipid component (4–6%) of the myelin sheath in the mammalian nervous system (46). Sulfatides interact with several proteins specifically participating in hemostasis, cell adhesion, differentiation, and signal transduction (18). We have recently shown that the N-terminal Dab2 N-PTB specifically binds sulfatides through basic residues located in two consensus sulfatide-binding motifs (15). We have also reported that sulfatide recognition by Dab2 is required for the protein to regulate the surface expression of P-selectin, which is critical for homo- and heterotypical interactions with leukocytes (16). Our NMR analysis indicates that several hydrophobic residues in the second helix of SBM are perturbed by the presence of sulfatides (Fig. 3, A–C). Particularly, this second helix is rich in lysine residues, which can interact with sulfatides through electrostatic interactions, and together with the surrounding nonpolar residues, facilitate contact of the peptide to the membrane surface. Indeed, the residues in the second helix, which correspond to the major sulfatide-binding region previously described by our group (15), are conserved among Dab proteins (Fig. 1A). Also, our NMR data suggest that the first putative sulfatide-binding motif in Dab2 SBM does not play any major role in interactions with the sphingolipid. This observation is consistent with our previous data, in which one mutation in the first sulfatide-binding site and three mutations in the second sulfatide-binding site are required to abolish sulfatide binding of the Dab2 N-PTB region (15). Also, the major role of the second sulfatide-binding motif in lipid recognition is supported by our paramagnetic quenching experiments, in which the second half of the peptide is not contacting the solvent (Fig. 4). Given that the HSQC spectra do not display the HN side chains of Dab2 SBM lysine residues, we cannot rule out the possibility that the lysine side chains of the first sulfatide-binding motif can contribute to sulfatide recognition.
Our HSQC analysis indicates that Dab2 SBM can also interact with negatively charged lipids such as PtdSer, but poorly with zwitterionic phospholipids (supplemental Fig. S1). In contrast to the permanent residence of sulfatides at the platelet surface, PtdSer is transiently exposed during platelet activation, and its surface exposure depends on certain physiological conditions such as Ca2+ influx from the extracellular medium (47). Therefore, it is possible that Dab2 SBM can interact with both sulfatides and PtdSer, but cell surface sulfatides are likely the major target for the peptide.
A few protein tertiary structures in complex with sulfatides have been determined, including the saposin B/sulfatide (48), CD1a-sulfatide (49), CD1d-sulfatide (50), the Taiwanese cobra cardiotoxin (51), and the glycolipid transfer protein (GLTP)-sulfatide (52) complexes. There are some differences in how these proteins contact sulfatides. Although glycolipid transfer protein contacts sulfatides by a hydrogen-bond interaction network to the negatively charged sulfatide head group and its corresponding backbone, very few hydrogen bonds between the sphingolipid ceramide backbone and saposin B, CD1a, CD1b, and cardiotoxin are observed. In CD1 proteins, the lipid tails contact their hydrophobic pockets via van der Waals interactions, in which the attractive and repulsive forces are the same between the two molecules (53). Interestingly, both saposin B and cardiotoxin dimerize when in contact with sulfatides. In saposin B, dimerization is required for the assembly of a hydrophobic cavity that allows contact to both sulfatide hydrocarbon chains (48). In the case of cardiotoxin, the lipid head group is buried in a pocket formed by two protein monomers with several basic residues contacting both the sulfate and the galactose groups (51). Based on our functional and structural data, we propose that the interaction of Dab2 with sulfatide membranes mainly depends on the integrity of the second sulfatide-binding motif, which is located within the PTB domain. This association is driven by conserved basic and nonpolar residues present in this motif surrounded by other residues outside of the SBM peptide that may contribute to membrane insertion. Basic residues in Dab2 may interact electrostatically with the negatively charged sulfatide head group, whereas neighbor nonpolar residues contribute to membrane interactions. Also, we hypothesize that membrane contact could be accompanied by a conformational change in the second sulfatide-binding motif, which is a β-strand in the PTB domain (11) but adopts a helical structure when it interacts with membrane mimics (Fig. 2). This hypothesis is supported by the observed conformational change of the Dab2 N-PTB region in sulfatide-enriched micelles (17).
Because DPC micelles present a thickness comparable with that of a plasma membrane (54), the topology of SBM relative to the sulfatide-enriched micellar surface and peptide insertion was investigated using paramagnetic probes including Mn2+, 5-DSA, and 16-DSA. The disordered N terminus of Dab2 SBM, comprising residues 24–39, is exposed to the solvent, and the two helices lie approximately parallel to the sulfatide-enriched micelle surface. Indeed, both 5-DSA and 16-DSA affected most of the hydrophobic residues (Ala-52, Leu-54, and Ile-55) of Dab2 SBM, located at the C-terminal half of the peptide. Other NMR resonances that became broadened by the paramagnetic probes are Lys-44, Lys-49, Lys-51, and Lys-53, which likely interacted with the negatively charged sulfatide head group. In fact, Lys-49, Lys-51, and Lys-53 belong to the second sulfatide-binding motif, whose resonances were perturbed in our HSQC titration studies (Fig. 2A). The observed amphipathic nature of Dab2 SBM and the paramagnetic quenching data suggest that the basic and nonpolar residues in the second helix are partially immersed in the sulfatide-enriched micelle and that the residues in the first helix play a minor role in membrane interactions. Taken together, we propose that Dab2 SBM employs electrostatic and hydrophobic interactions with the sulfatide head group and with the acyl chain region of the lipid, respectively, which are essential for driving the peptide to the membrane surface to anchor it in a parallel orientation. Thus, although Dab2 SBM lies at the surface of these membrane mimics (Fig. 4), the Dab2 N-PTB region penetrates sulfatide-enriched micelles (17). This difference could be attributed to the lack of additional membrane-binding residues in SBM that map within the N-PTB region. The absence of deep membrane penetration of Dab2 SBM is reflected by the reduced binding affinity of the peptide for sulfatides when compared with that of the N-PTB region (15). The difference in affinity is also evidenced in the strength of the inhibition of platelet aggregation by these regions of Dab2 (Fig. 5). Nonetheless, SBM is an active peptide and inhibits platelet aggregation in a sulfatide-dependent manner.
The formation of thrombus initiates in platelet attachment, cohesion, and aggregation on the surface of extracellular matrices. Surface platelet proteins including fibronectin, Von Willebrand factor, and fibrinogen regulate thrombus formation. Fibrinogen interacts with the αIIbβ3 integrin receptor and induces platelet aggregation (55). One of the fibrinogen regions critical for both integrin receptor recognition and cell-cell interaction is the tetrapeptide sequence RGD(S/F) (amino acids 572–575) (56). RGD peptides inhibit fibrinogen binding to activated platelets and, consequently, platelet aggregation (57). However, these peptides have been reported to promote in vitro endothelial cell detachment (58), causing some concern about its toxicity in vivo. Moreover, RGD peptides have low activity and poor stability in plasma (59). Milk-derived peptides, such as those from casein and lactoferrin (60, 61), have been reported to show anti-thrombotic activity, although it is not known whether they can be effective in vivo. Here, we report that Dab2 SBM peptide exhibits anti-aggregatory activity comparable with that observed with the fibrinogen-derived RGDS peptide (Fig. 6). Further studies will be devoted to address the biological activity, toxicity, and stability of Dab2 SBM in in vivo models.
In summary, we have solved the three-dimensional structure of a Dab2-derived peptide embedded in DPC micelles, which exhibited sulfatide binding properties. We have identified the sulfatide-interacting residues and defined the orientation of the peptide in the membrane. The Dab2 SBM antiplatelet activity is weaker than the corresponding N-PTB region but comparable with the antiplatelet integrin receptor blocker, the RGDS peptide. Thus, these studies will provide the basis for the design of promising SBM-derived low-molecular mass antiplatelet agents.
Supplementary Material
Acknowledgment
We thank Dr. Janet Webster for assistance during preparation of the manuscript.
This work was supported by American Heart Association Grant 11BGIA6160000 (to D. G. S. C.).

This article contains supplemental Fig. S1.
The atomic coordinates and structure factors (code 2lsw) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The coordinates reported in this paper have been deposited in the BioMagResBank under accession code 18849.
- Dab2
- Disabled-2
- DPC
- dodecylphosphocholine
- DSA
- doxylstearic acid
- HSQC
- heteronuclear single-quantum coherence
- N-PTB
- N-terminal region of Dab2 containing the PTB domain
- PTB
- phosphotyrosine-binding domain
- PtdSer
- phosphatidylserine
- SBM
- sulfatide-binding motif peptide
- PtdIns(4,5)P2
- phosphatidylinositol (4,5)-bisphosphate.
REFERENCES
- 1. Hocevar B. A., Smine A., Xu X. X., Howe P. H. (2001) The adaptor molecule Disabled-2 links the transforming growth factor β receptors to the Smad pathway. EMBO J. 20, 2789–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morris S. M., Cooper J. A. (2001) Disabled-2 colocalizes with the LDLR in clathrin-coated pits and interacts with AP-2. Traffic 2, 111–123 [DOI] [PubMed] [Google Scholar]
- 3. Mishra S. K., Keyel P. A., Hawryluk M. J., Agostinelli N. R., Watkins S. C., Traub L. M. (2002) Disabled-2 exhibits the properties of a cargo-selective endocytic clathrin adaptor. EMBO J. 21, 4915–4926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morris S. M., Arden S. D., Roberts R. C., Kendrick-Jones J., Cooper J. A., Luzio J. P., Buss F. (2002) Myosin VI binds to and localizes with Dab2, potentially linking receptor-mediated endocytosis and the actin cytoskeleton. Traffic 3, 331–341 [DOI] [PubMed] [Google Scholar]
- 5. Mok S. C., Wong K. K., Chan R. K., Lau C. C., Tsao S. W., Knapp R. C., Berkowitz R. S. (1994) Molecular cloning of differentially expressed genes in human epithelial ovarian cancer. Gynecol. Oncol. 52, 247–252 [DOI] [PubMed] [Google Scholar]
- 6. Sheng Z., Sun W., Smith E., Cohen C., Sheng Z., Xu X. X. (2000) Restoration of positioning control following Disabled-2 expression in ovarian and breast tumor cells. Oncogene 19, 4847–4854 [DOI] [PubMed] [Google Scholar]
- 7. Cho S. Y., Jeon J. W., Lee S. H., Park S. S. (2000) p67 isoform of mouse Disabled-2 protein acts as a transcriptional activator during the differentiation of F9 cells. Biochem. J. 352, 645–650 [PMC free article] [PubMed] [Google Scholar]
- 8. Hasson T. (2003) Myosin VI: two distinct roles in endocytosis. J. Cell Sci. 116, 3453–3461 [DOI] [PubMed] [Google Scholar]
- 9. DiNitto J. P., Lambright D. G. (2006) Membrane and juxtamembrane targeting by PH and PTB domains. Biochim. Biophys. Acta 1761, 850–867 [DOI] [PubMed] [Google Scholar]
- 10. Uhlik M. T., Temple B., Bencharit S., Kimple A. J., Siderovski D. P., Johnson G. L. (2005) Structural and evolutionary division of phosphotyrosine binding (PTB) domains. J. Mol. Biol. 345, 1–20 [DOI] [PubMed] [Google Scholar]
- 11. Yun M., Keshvara L., Park C. G., Zhang Y. M., Dickerson J. B., Zheng J., Rock C. O., Curran T., Park H. W. (2003) Crystal structures of the Dab homology domains of mouse Disabled-1 and -2. J. Biol. Chem. 278, 36572–36581 [DOI] [PubMed] [Google Scholar]
- 12. Stolt P. C., Jeon H., Song H. K., Herz J., Eck M. J., Blacklow S. C. (2003) Origins of peptide selectivity and phosphoinositide binding revealed by structures of Disabled-1 PTB domain complexes. Structure 11, 569–579 [DOI] [PubMed] [Google Scholar]
- 13. Stolt P. C., Vardar D., Blacklow S. C. (2004) The dual-function Disabled-1 PTB domain exhibits site independence in binding phosphoinositide and peptide ligands. Biochemistry 43, 10979–10987 [DOI] [PubMed] [Google Scholar]
- 14. Huang C. L., Cheng J. C., Stern A., Hsieh J. T., Liao C. H., Tseng C. P. (2006) Disabled-2 is a novel αIIb-integrin-binding protein that negatively regulates platelet-fibrinogen interactions and platelet aggregation. J. Cell Sci. 119, 4420–4430 [DOI] [PubMed] [Google Scholar]
- 15. Drahos K. E., Welsh J. D., Finkielstein C. V., Capelluto D. G. (2009) Sulfatides partition Disabled-2 in response to platelet activation. PLoS One 4, e8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Welsh J. D., Charonko J. J., Salmanzadeh A., Drahos K. E., Shafiee H., Stremler M. A., Davalos R. V., Capelluto D. G., Vlachos P. P., Finkielstein C. V. (2011) Disabled-2 modulates homotypic and heterotypic platelet interactions by binding to sulfatides. Br. J. Haematol. 154, 122–133 [DOI] [PubMed] [Google Scholar]
- 17. Alajlouni R., Drahos K. E., Finkielstein C. V., Capelluto D. G. (2011) Lipid-mediated membrane binding properties of Disabled-2. Biochim. Biophys. Acta 1808, 2734–2744 [DOI] [PubMed] [Google Scholar]
- 18. Snook C. F., Jones J. A., Hannun Y. A. (2006) Sphingolipid-binding proteins. Biochim. Biophys. Acta 1761, 927–946 [DOI] [PubMed] [Google Scholar]
- 19. Merten M., Thiagarajan P. (2001) Role for sulfatides in platelet aggregation. Circulation 104, 2955–2960 [DOI] [PubMed] [Google Scholar]
- 20. Roberts D. D., Ginsburg V. (1988) Sulfated glycolipids and cell adhesion. Arch. Biochem. Biophys. 267, 405–415 [DOI] [PubMed] [Google Scholar]
- 21. Aruffo A., Kolanus W., Walz G., Fredman P., Seed B. (1991) CD62/P-selectin recognition of myeloid and tumor cell sulfatides. Cell 67, 35–44 [DOI] [PubMed] [Google Scholar]
- 22. Gautier A., Mott H. R., Bostock M. J., Kirkpatrick J. P., Nietlispach D. (2010) Structure determination of the seven-helix transmembrane receptor sensory rhodopsin II by solution NMR spectroscopy. Nat. Struct. Mol. Biol. 17, 768–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wishart D. S., Bigam C. G., Yao J., Abildgaard F., Dyson H. J., Oldfield E., Markley J. L., Sykes B. D. (1995) 1H, 13C, and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6, 135–140 [DOI] [PubMed] [Google Scholar]
- 24. Tengel T., Sethson I., Francis M. S. (2002) Conformational analysis by CD and NMR spectroscopy of a peptide encompassing the amphipathic domain of YopD from Yersinia. Eur. J. Biochem. 269, 3659–3668 [DOI] [PubMed] [Google Scholar]
- 25. Piotto M., Saudek V., Sklenár V. (1992) Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2, 661–665 [DOI] [PubMed] [Google Scholar]
- 26. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 27. Goddar T. D., Kneller D. G. (2008) SPARKY 3, University of California, San Francisco CA [Google Scholar]
- 28. Schwieters C. D., Kuszewski J. J., Tjandra N., Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 29. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marrink S. J., Tieleman D. P., Mark A. E. (2000) Molecular dynamics simulations of the kinetics of spontaneous micelle formation. J. Phys. Chem. B 104, 12165–12173 [Google Scholar]
- 31. Song J. W., Cavnar S. P., Walker A. C., Luker K. E., Gupta M., Tung Y. C., Luker G. D., Takayama S. (2009) Microfluidic endothelium for studying the intravascular adhesion of metastatic breast cancer cells. PLoS One 4, e5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Beswick V., Guerois R., Cordier-Ochsenbein F., Coïc Y. M., Tam H. D., Tostain J., Noël J. P., Sanson A., Neumann J. M. (1999) Dodecylphosphocholine micelles as a membrane-like environment: new results from NMR relaxation and paramagnetic relaxation enhancement analysis. Eur. Biophys. J. 28, 48–58 [DOI] [PubMed] [Google Scholar]
- 33. Muranyi A., Evenäs J., Stenberg Y., Stenflo J., Drakenberg T. (2000) Characterization of the EGF-like module pair 3–4 from vitamin K-dependent protein S using NMR spectroscopy reveals dynamics on three separate time scales and extensive effects from calcium binding. Biochemistry 39, 15742–15756 [DOI] [PubMed] [Google Scholar]
- 34. Huisman J. G., Carotenuto A., Labrijn A. F., Papavoine C. H., Laman J. D., Schellekens M. M., Koppelman M. H., Hilbers C. W. (2000) Recognition properties of V3-specific antibodies to V3 loop peptides derived from HIV-1 gp120 presented in multiple conformations. Biochemistry 39, 10866–10876 [DOI] [PubMed] [Google Scholar]
- 35. Roberts D. D., Haverstick D. M., Dixit V. M., Frazier W. A., Santoro S. A., Ginsburg V. (1985) The platelet glycoprotein thrombospondin binds specifically to sulfated glycolipids. J. Biol. Chem. 260, 9405–9411 [PubMed] [Google Scholar]
- 36. Fauvel J., Chap H., Roques V., Levy-Toledano S., Douste-Blazy L. (1986) Biochemical characterization of plasma membranes and intracellular membranes isolated from human platelets using Percoll gradients. Biochim. Biophys. Acta 856, 155–164 [DOI] [PubMed] [Google Scholar]
- 37. Shattil S. J., Anaya-Galindo R., Bennett J., Colman R. W., Cooper R. A. (1975) Platelet hypersensitivity induced by cholesterol incorporation. J. Clin. Invest. 55, 636–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deleted in proof
- 39. Boesze-Battaglia K., Schimmel R. (1997) Cell membrane lipid composition and distribution: implications for cell function and lessons learned from photoreceptors and platelets. J. Exp. Biol. 200, 2927–2936 [DOI] [PubMed] [Google Scholar]
- 40. Lerch M., Kamimori H., Folkers G., Aguilar M. I., Beck-Sickinger A. G., Zerbe O. (2005) Strongly altered receptor binding properties in PP and NPY chimeras are accompanied by changes in structure and membrane binding. Biochemistry 44, 9255–9264 [DOI] [PubMed] [Google Scholar]
- 41. Mozsolits H., Wirth H. J., Werkmeister J., Aguilar M. I. (2001) Analysis of antimicrobial peptide interactions with hybrid bilayer membrane systems using surface plasmon resonance. Biochim. Biophys. Acta 1512, 64–76 [DOI] [PubMed] [Google Scholar]
- 42. Kim H. J., Howell S. C., Van Horn W. D., Jeon Y. H., Sanders C. R. (2009) Recent advances in the application of solution NMR spectroscopy to multi-span integral membrane proteins. Prog. Nucl. Magn. Reson. Spectrosc. 55, 335–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liang B., Bushweller J. H., Tamm L. K. (2006) Site-directed parallel spin-labeling and paramagnetic relaxation enhancement in structure determination of membrane proteins by solution NMR spectroscopy. J. Am. Chem. Soc. 128, 4389–4397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Basani R. B., D'Andrea G., Mitra N., Vilaire G., Richberg M., Kowalska M. A., Bennett J. S., Poncz M. (2001) RGD-containing peptides inhibit fibrinogen binding to platelet αIIbβ3 by inducing an allosteric change in the amino-terminal portion of αIIb. J. Biol. Chem. 276, 13975–13981 [DOI] [PubMed] [Google Scholar]
- 45. Segrest J. P., Garber D. W., Brouillette C. G., Harvey S. C., Anantharamaiah G. M. (1994) The amphipathic α helix: a multifunctional structural motif in plasma apolipoproteins. Adv. Protein Chem. 45, 303–369 [DOI] [PubMed] [Google Scholar]
- 46. Eckhardt M. (2008) The role and metabolism of sulfatide in the nervous system. Mol. Neurobiol. 37, 93–103 [DOI] [PubMed] [Google Scholar]
- 47. Williamson P., Bevers E. M., Smeets E. F., Comfurius P., Schlegel R. A., Zwaal R. F. (1995) Continuous analysis of the mechanism of activated transbilayer lipid movement in platelets. Biochemistry 34, 10448–10455 [DOI] [PubMed] [Google Scholar]
- 48. Ahn V. E., Faull K. F., Whitelegge J. P., Fluharty A. L., Privé G. G. (2003) Crystal structure of saposin B reveals a dimeric shell for lipid binding. Proc. Natl. Acad. Sci. U.S.A. 100, 38–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zajonc D. M., Elsliger M. A., Teyton L., Wilson I. A. (2003) Crystal structure of CD1a in complex with a sulfatide self antigen at a resolution of 2.15 Ä. Nat. Immunol. 4, 808–815 [DOI] [PubMed] [Google Scholar]
- 50. Zajonc D. M., Maricic I., Wu D., Halder R., Roy K., Wong C. H., Kumar V., Wilson I. A. (2005) Structural basis for CD1d presentation of a sulfatide derived from myelin and its implications for autoimmunity. J. Exp. Med. 202, 1517–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang C. H., Liu J. H., Lee S. C., Hsiao C. D., Wu W. G. (2006) Glycosphingolipid-facilitated membrane insertion and internalization of cobra cardiotoxin: the sulfatide·cardiotoxin complex structure in a membrane-like environment suggests a lipid-dependent cell-penetrating mechanism for membrane binding polypeptides. J. Biol. Chem. 281, 656–667 [DOI] [PubMed] [Google Scholar]
- 52. Malinina L., Malakhova M. L., Teplov A., Brown R. E., Patel D. J. (2004) Structural basis for glycosphingolipid transfer specificity. Nature 430, 1048–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moody D. B., Zajonc D. M., Wilson I. A. (2005) Anatomy of CD1-lipid antigen complexes. Nat. Rev. Immunol. 5, 387–399 [DOI] [PubMed] [Google Scholar]
- 54. Lauterwein J., Bösch C., Brown L. R., Wüthrich K. (1979) Physicochemical studies of the protein-lipid interactions in melittin-containing micelles. Biochim. Biophys. Acta 556, 244–264 [DOI] [PubMed] [Google Scholar]
- 55. Mackman N. (2008) Triggers, targets and treatments for thrombosis. Nature 451, 914–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Plow E. F., Ginsberg M. H. (1981) Specific and saturable binding of plasma fibronectin to thrombin-stimulated human platelets. J. Biol. Chem. 256, 9477–9482 [PubMed] [Google Scholar]
- 57. Andrieux A., Hudry-Clergeon G., Ryckewaert J. J., Chapel A., Ginsberg M. H., Plow E. F., Marguerie G. (1989) Amino acid sequences in fibrinogen mediating its interaction with its platelet receptor, GPIIbIIIa. J. Biol. Chem. 264, 9258–9265 [PubMed] [Google Scholar]
- 58. Dejana E., Lampugnani M. G., Giorgi M., Gaboli M., Marchisio P. C. (1990) Fibrinogen induces endothelial cell adhesion and spreading via the release of endogenous matrix proteins and the recruitment of more than one integrin receptor. Blood 75, 1509–1517 [PubMed] [Google Scholar]
- 59. Cox D., Aoki T., Seki J., Motoyama Y., Yoshida K. (1994) The pharmacology of the integrins. Med. Res. Rev. 14, 195–228 [DOI] [PubMed] [Google Scholar]
- 60. Jollès P., Lévy-Toledano S., Fiat A. M., Soria C., Gillessen D., Thomaidis A., Dunn F. W., Caen J. P. (1986) Analogy between fibrinogen and casein: effect of an undecapeptide isolated from κ-casein on platelet function. Eur. J. Biochem. 158, 379–382 [DOI] [PubMed] [Google Scholar]
- 61. Léonil J., Mollé D. (1990) Liberation of tryptic fragments from caseinomacropeptide of bovine κ-casein involved in platelet function. Kinetic study. Biochem. J. 271, 247–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.