

Background: WNK1 binds OSR1 via RFXV motifs and is on intracellular puncta.
Results: C-terminal WNK1 segments bind OSR1 and localize to puncta like endogenous WNK1; changes in tonicity decrease WNK1 mobility.
Conclusion: Complex interplay of protein-protein interactions, changes in tonicity, and localization control the WNK-OSR1 cascade.
Significance: WNK function varies because of family member and splice form expression, protein interactions, and localization.
Keywords: Hypertension, Kidney, Phosphorylation, Protein Kinases, Transport, NKCC, RFXV Motif, WNKs 1–4
Abstract
Two of the four WNK (with no lysine (K)) protein kinases are associated with a heritable form of ion imbalance culminating in hypertension. WNK1 affects ion transport in part through activation of the closely related Ste20 family protein kinases oxidative stress-responsive 1 (OSR1) and STE20/SPS1-related proline-, alanine-rich kinase (SPAK). Once activated by WNK1, OSR1 and SPAK phosphorylate and stimulate the sodium, potassium, two chloride co-transporters, NKCC1 and NKCC2, and also affect other related ion co-transporters. We find that WNK1 and OSR1 co-localize on cytoplasmic puncta in HeLa and other cell types. We show that the C-terminal region of WNK1 including a coiled coil is sufficient to localize the fragment in a manner similar to the full-length protein, but some other fragments lacking this region are mislocalized. Photobleaching experiments indicate that both hypertonic and hypotonic conditions reduce the mobility of GFP-WNK1 in cells. The four WNK family members can phosphorylate the activation loop of OSR1 to increase its activity with similar kinetic constants. C-terminal fragments of WNK1 that contain three RFXV interaction motifs can bind OSR1, block activation of OSR1 by sorbitol, and prevent the OSR1-induced enhancement of ion co-transporter activity in cells, further supporting the conclusion that association with WNK1 is required for OSR1 activation and function at least in some contexts. C-terminal WNK1 fragments can be phosphorylated by OSR1, suggesting that OSR1 catalyzes feedback phosphorylation of WNK1.
Introduction
WNKs (with no lysine (K)) are large protein-serine/threonine kinases found in all multicellular and many unicellular eukaryotes (1–3). WNK1, the first member of the family identified in mammals, was found in searches for novel components of protein kinase cascades (4). WNK1 is expressed ubiquitously, consistent with effects on many cell types (4–7). Alternative splicing of at least eight exons and transcription initiation from at least two start sites generate a myriad of WNK1 variants containing from just under 2000 to more than 2800 residues, including a catalytically inactive form lacking most of the kinase domain (KS-WNK1), which is the predominant form in the kidney distal convoluted tubule (8–10).
WNK family members, numbering four in human and mouse, are distinct from other protein kinases in that their catalytic lysine is shifted from its usual position buried in the N-terminal part of the kinase core to a more exposed position in the glycine-rich loop (4, 11). The strict conservation of the unique catalytic core structure of the WNK family in organisms as diverse as Chlamydomonas, Phycomyces, Arabidopsis, Drosophila, and mammals suggests conserved properties for these kinases that distinguish them from all other members of the eukaryotic protein kinase superfamily.
The discovery that WNK1 and WNK4 are genetically linked to a rare type of hypertension, pseudohypoaldosteronism type 2 (PHA2),5 demonstrated the importance of WNK function in man (6). Our initial characterization of WNK1 revealed that the kinase activity is sensitive to osmotic stress, which gained significance in view of its roles in regulating ion balance (4, 12). The consequences of WNK mutations in PHA2 are hyperkalemia, renal tubular acidosis, and eventually hypertension (7, 13–17). The investigation of the actions of WNKs, in light of the therapeutics of PHA2, led to some of the first and most logical connections to ion transporters and channels, e.g., the sodium chloride co-transporter (NCC), sodium potassium chloride co-transporters (NKCCs), the renal outer medullary potassium channel, and the epithelial sodium channel (18–28). Although a connection between WNKs and ion transport proteins is predictable, the mechanisms driving regulation of transporters by WNKs are more complex than expected, involving both multiple layers of activity regulation and effects on localization.
The biochemical analysis of WNK1 action and the identification of WNKs in genome and kinome screens have suggested mechanisms of WNK action that may be linked to transporter regulation. For example, WNKs have been found in screens of proteins important in endocytosis, which has been shown to be involved in regulation of transporters by WNK1 (29–32). Somewhat more straightforward, WNK1 regulates several protein kinases that modulate ion transport, including the serum- and glucocorticoid-inducible protein kinase, which affects the epithelial sodium channel, and the kinases oxidative stress-responsive 1 (OSR1) and STE20/SPS1-related proline-, alanine-rich kinase (SPAK), that control activities of NKCCs, NCC, and related members of the solute carrier 12 (SLC12) family of co-transporters (20, 24, 25, 33, 34).
Here we further analyze the regulation of OSR1 by WNK1 and the other WNK family members. We show that intracellular association between WNK1 and OSR1 is required for stimulation of OSR1 and NKCC activities by osmotic stress. Changing tonicity alters the mobility of WNK1 in cells. Distinct C-terminal elements of WNK1 are involved in WNK1 localization and in WNK1 binding to OSR1. Expression of some fragments inhibits OSR1 activity and interferes with regulation of ion co-transporters.
EXPERIMENTAL PROCEDURES
Materials
Plasmids encoding WNKs and NKCC2 (SLC12A1) were as described (4, 35–37). The rat WNK1 splice form with 2126 residues (Δ exons 11–12) and fragments therefrom were used throughout. Fragments and mutants were generated by restriction digestion and ligation, PCR, or QuikChange (Stratagene). Ouabain (Na+/K+ inhibitor) and bumetanide (NKCC1/2 inhibitor) were from Sigma. Anti-WNK1 (Q256) was as described (4). Anti-OSR1 (U5566) was as described (38). Anti-Myc epitope was from National Cell Culture Center and anti-FLAG epitope was from Sigma (058K6113). dsRNA oligonucleotides were from Ambion. Recombinant proteins used for kinase assays were purified from Escherichia coli strain BL21 using standard protocols.
Cell Culture and Transfection
HeLa cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% glutamine (20, 34). The cells were transfected with constructs encoding WNK1 fragments using FuGENE 6 (Promega) (1:3 DNA:FuGENE 6) and used for immunoprecipitations and coupled kinase assays. The cells were harvested in 50 mm HEPES, pH 7.7, 150 mm NaCl, 1.5 mm MgCl2, 1 mm EGTA, 10% glycerol, 100 mm NaF, 0.2 mm NaVO4, 50 mm β-glycerophosphate, 0.1% Triton X-100, and phosphatase and protease inhibitors as described (39).
Immunoprecipitation and Immunoblotting
The proteins were immunoprecipitated from 1 mg of soluble lysate protein using 5 μl of indicated antibodies. Following incubation overnight at 4 °C, a 30-μl slurry of protein A-Sepharose was added for an additional 2 h at 4 °C. The beads were washed three times in lysis buffer, and then proteins were eluted using 5× electrophoresis sample buffer. The proteins were resolved by electrophoresis in SDS and transferred to nitrocellulose. Immunoblots were developed using enhanced chemiluminescence or Li-COR infrared imaging.
Immunofluorescence
To quantify the co-localization of WNK1 and OSR1, the cells were transfected with a construct encoding FLAG-OSR1 using Lipofectamine 2000 (Invitrogen). After 24 h, the cells were washed with PBS once and fixed with 4% paraformaldehyde in 60 mm Pipes, 25 mm HEPES, pH 6.9, 10 mm EGTA, and 2 mm MgCl2 at room temperature for 10 min, followed by two washes with PBS at room temperature for 5 min to remove excess paraformaldehyde. The cells were permeabilized with 0.1% Triton X-100 in PBS at 4 °C for 5 min, washed as above, incubated with 10% normal goat serum at room temperature for 30 min, and then co-stained with anti-WNK1 antibody (Cell Signaling, 4947) and anti-FLAG antibody (Sigma clone M2) at 4 °C overnight. The next day, the cells were washed as above and incubated with secondary antibodies, goat anti-rabbit Alexa Fluor-594 and goat anti-mouse Alexa Fluor-488, at room temperature for 1 h. The cells were washed again as above, and coverslips were mounted with Aqua-poly/Mount (Polysciences, Inc., 18606).
Fluorescence Microscopy and Image Analysis
Fluorescent images were acquired and deconvolved using a Deltavision RT deconvolution microscope (Applied Precision, Issaquah, WA). Co-localization was quantified using the Coloc module of Imaris three-dimensional measurement and analysis software (Andor). For each cell, the boundary of the cell was detected as an intensity isoline in the green channel (WNK1), and the Costes method was used to exclude randomly overlapping voxels and calculate the Pearson coefficient for the whole cell volume and for the cytoplasmic volume only (40). To obtain values for Pearson coefficient specifically in the cytoplasm, a contour surface enclosing the nucleus was created interactively in Imaris, and voxels inside the surface were set to 0. n = 8 for whole cell and for cytoplasm only. For localization of WNK1 fragments in live cells, HeLa cells were transiently transfected with WNK1 fragments fused to the fluorescent protein tdTomato (WNK1 td) as described above and imaged 24 h after transfection.
Fluorescence Recovery after Photobleaching (FRAP)
The cells were transfected with constructs encoding full-length GFP-WNK1 and GFP-WNK1 1800–2126 using Lipofectamine 2000 (Invitrogen) (1:3 DNA: Lipofectamine 2000) for 16 h. FRAP was performed on cells in CO2-independent medium (Invitrogen) in a temperature controlled chamber set to 37 °C. Tonicity was increased or hypertonicity was induced by the addition of 0.2 m sorbitol for 5 min or 0.5 m sorbitol for 30 min; hypotonic conditions were generated by placing cells for 20 min in the medium above diluted 4-fold with water or in medium in which chloride was largely replaced with gluconate: 67.5 mm sodium gluconate, 0.5 mm CaCl2, 0.5 mm MgCl2, 5 mm glucose, 15 mm HEPES, pH 7.4 (modified from Ref. 41). FRAP was carried out using the 488-nm laser line of a Zeiss LSM780 on an AxioExaminer upright stand with a 20× W/1.0 dipping lens. The scan was zoomed to 11×, and a 12 × 12 pixel region of interest was photobleached for two iterations with 100% laser power. Fluorescence recoveries were recorded at 2% laser power for a 30-s duration after photobleaching. Time stacks were analyzed in ImageJ using Image>Stacks>Plot Z-axis Profile to obtain fluorescence intensity changes over time in the bleached region. The time stamps for the images in the stack were obtained with the LSM Toolbox plugin. The data were exported to Excel, and the raw intensity values were normalized to the initial intensity before photobleaching. Single exponentials were fit to the normalized data for each recovery curve using the “one-phase association” algorithm of GraphPad Prism. The mean values for plateau and half-time under different conditions were compared by unpaired t test with Welch's correction.
siRNA
HeLa cells were detached from a dish with trypsin and immediately transfected with 10 nm dsRNA oligonucleotides using Lipofectamine RNAiMax (Invitrogen). After 72 h, protein localization was examined as in the preceding section. Oligonucleotides: hWNK1.1, sense, cagacagugcaguauucacTT; antisense, gugaauacugcacugucugTT (Ambion). For rescue experiments, full-length WNK1 was transfected 24 h after oligonucleotide transfection using Lipofectamine 2000 (Invitrogen).
Kinase Assays
WNK protein kinase activity was assayed with full-length OSR1 K46R. OSR1 activity was assayed with NKCC2 1–175. GST-tagged enzymes and substrates were added to reactions in 10 mm HEPES, pH 8.0, 10 mm MgCl2, 1 mm DTT, and 1 mm benzamidine, and 0.5 mm ATP (5000–13,000 dpm/pmol [γ-32P]ATP; MP Biologicals). WNK1 was used at 20 pmol (0.7 μm) and OSR1 at 40 pmol (1.3 μm) for kinetic analyses. The reactions were incubated at 30 °C for 10 min, terminated with electrophoresis sample buffer, and analyzed on SDS-polyacrylamide gels. The gels were dried and exposed to film for autoradiography. Protein bands were excised and subjected to scintillation counting. Activation of OSR1 by WNK was assayed as above, with the following changes: WNK1 amount was reduced to 5 pmol (0.14 μm), OSR1 to 20 pmol (0.57 μm), and NKCC2 80 pmol (2.28 μm), and radioactive ATP was omitted. The reactions were incubated at 30 °C for 30 min and then transferred to ice. After the addition of NKCC2 and [γ-32P]ATP, second reactions were incubated at 30 °C for 10 min and analyzed as above. Kinetic constants for WNKs were determined with assays as above except with 0.3 mm ATP, 1 pmol of WNK, OSR1 K46R from 1–64 pmol at room temperature for 45 min. The data were analyzed with GraphPad Prism 5 software. For immune complex kinase assays, beads (20 μl) were incubated with 50 μm ATP, 10 mm MgCl2, and 20 mm HEPES, pH 7.4, for 30 min at 30 °C. The proteins were resolved on gels in SDS as above. The gels were dried and exposed to film for autoradiography. Incorporation of radioactivity was quantified by scintillation counting of the bands excised from the gel.
Reconstitution and Assay of NKCC Activity
Endogenous WNK1 was knocked down as above. HeLa cells were detached from plates, and 10 nm WNK1.1 or scrambled oligonucleotides were introduced with Lipofectamine RNAiMAX in Opti-MEM (Invitrogen). After 24 h, a construct encoding full-length WNK1 was transfected into the cells using Lipofectamine 2000. After 24 h, the cells were trypsinized and plated on 24-well plates. The next day, the cells were washed twice in 140 mm NaCl, 5 mm KCl, 1 mm MgCl2, 1 mm CaCl2, 10 mm HEPES, pH 7.4, 10 mm glucose, 10 mm sodium pyruvate, and 0.1% bovine serum albumin. After 30 min, 86Rb (107 cpm/ml; PerkinElmer Life Sciences) and 0.5 mm ouabain were added with or without 10 μm bumetanide. The cells were incubated for 5 min at 37 °C and then washed twice in cold 100 mm MgCl2, 10 mm HEPES buffered to pH 7.4 with solid Tris. The cells were lysed in 2% SDS. Liquid scintillation counting was performed on 0.1 ml of each lysate, and protein concentration was determined using the Pierce MicroBCA protein assay kit. 86Rb uptake with and without bumetanide was measured in triplicate. Bumetanide-sensitive 86Rb uptake was taken as NKCC activity. To assay effects of WNK1 fragments on NKCC activity, the fragments were transfected in HeLa cells using FuGENE 6. After 24 h, the cells were detached and replated on 24-well plates, and the next day, 86Rb uptake was measured as above.
Statistical Analysis
Statistical comparison between two groups was made using the two-tailed unpaired t test. Multiple comparisons were determined using one-way analysis of variance followed by Tukey's multiple comparison tests. p values less than 0.05 and 0.01 were considered significant for single and multiple comparisons, respectively.
RESULTS
Co-localization of OSR1 with WNK1
WNK1 co-immunoprecipitates and co-purifies with OSR1, and we have previously reported their localization to cytoplasmic puncta by immunofluorescence with antibodies to the two endogenous proteins (33, 42–45). Here, we examined the extent of co-localization of the proteins in the absence of osmotic stress (Fig. 1). To avoid using two rabbit antibodies, we expressed a tagged form of OSR1 for which a mouse antibody was available so that we could eliminate possible secondary antibody cross reactivity in the quantitation. As we showed previously, both WNK1 and OSR1 are distributed in a particulate pattern in the cytoplasm (Fig. 1A); OSR1, but not WNK1, is also found in the nucleus (45). For comparison, one panel shows staining of endogenous OSR1 (Fig. 1B).
FIGURE 1.

WNK1 localization. A, cells were transiently transfected with a construct encoding OSR1-FLAG. After 24 h, the cells were co-stained with antibodies to endogenous WNK1 (red) and to FLAG (green) for immunofluorescent co-localization (Co-loc) analysis by Imaris software Co-Loc module. Co-localized objects are shown in yellow. Scale bar, 5 μm. B, endogenous OSR1 in HeLa cells was immunostained (green), and some of the OSR1 signals were distributed in the nucleus (DAPI staining in blue). Scale bar, 5 μm. C, HeLa cells were transiently transfected for 24 h with constructs encoding different WNK1 fragments fused to tdTomato. Fluorescent signals were examined in live cells (red). Scale bar, 5 μm. D, cells transfected as in C except with GFP-1800–2126. The cells were immunostained with antibodies to endogenous WNK1 (red). The co-localization channel (yellow) was generated as described in Fig. 1A. White arrowheads point to examples of co-localized objects. Scale bar, 5 um.
Co-localization data were analyzed quantitatively using a three-dimensional statistical algorithm rather than the typical two-dimensional color merge. The Imaris software CoLoc function uses the Costes method to distinguish true co-localization from random overlap (40). The quantitative assessment of co-localization comes from the Pearson correlation coefficient, r. The range of r is from −1 to 1. If two images co-localize from random overlap, r will be near 0. If two images co-localize in a nonrandom fashion, r will be a positive number, approaching 1 for perfect co-localization. The mean correlation coefficient for WNK1 and OSR1 in the cytoplasm was 0.54 for eight images analyzed, indicating a substantial degree of co-localization. As shown by the map of co-localized voxels (Fig. 1A, Co-loc, yellow puncta), WNK1 co-localized with much of the OSR1 that was present in the cytoplasm. If the nucleus is included, the Pearson coefficient is little changed (0.53).
Imaris also calculates an estimate of the percentage of each protein co-localized by multiplying the number of voxels by the intensity of each voxel, making the assumption that intensity is proportional to protein concentration. Using data for the whole cell, the co-localized fraction is predicted to be 62% of WNK1 and 66% of OSR1; for cytoplasm only, the value for WNK1 is 67% and for OSR1 70%. In addition to the fact that WNK1 activates OSR1, their co-localization suggests that the actions of each may be influenced by the presence of the other.
Localization of WNK1 Fragments
We examined the localizations of a series of WNK1 fragments in an effort to identify a region of WNK1 that is responsible for its punctate distribution (Fig. 1C). Placing a fluorescent tag at the N terminus of any WNK1 construct that we have tested other than residues 1800–2126, including the full-length protein, results in a diffuse signal in resting cells. WNK1 fragments that included the C-terminal coiled coil and that were expressed with C-terminal fluorescent tags mimicked the punctate distribution of the endogenous protein. As indicated above, WNK1 fragments containing the C-terminal coiled-coil (residues 1300–2126, 1200–2126, and 1800–2126) showed a perinuclear punctate localization. The fragments lacking this C-terminal coiled-coil (residues 1–660, 193–1600, and 1300–1600) appeared diffuse. Endogenous WNK1 was similarly distributed in a perinuclear punctate pattern. We compared the localization of GFP-1800–2126 directly with endogenous WNK1. Using an antibody to an N-terminal sequence of WNK1, we were able to distinguish endogenous WNK1 signals (Fig. 1D, red) and exogenously expressed GFP-1800–2126 (Fig. 1D, green). The co-localization (yellow puncta) was determined by the method mentioned above. Thus, we tested GFP-WNK1 1800–2126 as a mimic of endogenous WNK1 for the following live cell imaging experiments.
Effect of Hyperosmotic Stress on GFP-WNK1 and GFP-WNK1 1800–2126
WNK1 is activated by both hypertonic and hypotonic conditions (4, 12, 25, 33). Thus, we examined effects of both conditions on WNK1 localization. In contrast to endogenous WNK1, which appears in a punctate pattern under isotonic conditions, full-length GFP-WNK1 was mostly diffuse in the cytoplasm with a few indistinct puncta as reported previously (43, 45). After photobleaching, the diffuse material recovered to 87% of the initial value with a mean half-time of less than 0.5 s, indicating that the protein was highly mobile (Table 1 and Fig. 2D). In cultures equilibrated with 0.2 m sorbitol for 5 min, the localization of the full-length protein was primarily punctate. The recovery of fluorescence after photobleaching of individual puncta was less complete (69%) and more than 10-fold slower than in isotonic conditions (half-time = 5.3 s). Visual inspection of the time sequences showed that recovery of fluorescence involved exchange of WNK1 onto persistent structures rather than formation of new puncta (Fig. 2, A–C).
TABLE 1.
Summary of FRAP data
The data records showing change in fluorescence intensity over time (in seconds, s) after photobleaching were fit to a single exponential to obtain the projected plateau value and the half-time of recovery. Mean plateau and half-time (± S.E.) for multiple data records are reported for each condition. The values of plateau and half-time for individual data records were used for unpaired t tests with Welch's correction to determine whether the mean values for the various conditions were significantly different. ND, not determined.
| Construct | Condition | Plateau | Plateau different from untreated? (p < 0.05) | Plateau different from 0.2 m sorbitol? (p < 0.05) | Half-time | n | Haltime different from untreated? (p < 0.05) | Haltime different from 0.2 m sorbitol? (p < 0.05) |
|---|---|---|---|---|---|---|---|---|
| s | s | |||||||
| GFP-1800–2126 | Untreated | 0.76 ± 0.024 | 2.0 ± 0.31 | 15 | ||||
| 0.2 m sorbitol | 0.65 ± 0.027 | Yes (p = 0.0056) | 4.2 ± 0.57 | 16 | Yes (p < 0.0001) | |||
| 0.5 m sorbitol | Immobile | Yes | Yes | 5 | Yes | Yes | ||
| Gluconate | 0.75 ± 0.024 | No | No | 2.0 ± 0.33 | 10 | No | No | |
| Dilution | 0.64 ± 0.02 | Yes (p = 0.002) | No | 4.6 ± 1.0 | 5 | No | No | |
| GFP-WNK1-FL | Untreated diffuse | 0.87 ± 0.022 | 0.4 ± 0.09 | 3 | ||||
| 0.2 m sorbitol | 0.69 ± 0.047 | Yes (p = 0.0051) | 5.3 ± 0.63 | 11 | Yes (p < 0.0001) | |||
| Gluconate | 0.65 ± 0.035 | Yes (p = 0.0002) | No | 4.4 ± 1.1 | 11 | Yes (p = 0.0048) | No | |
| Dilution | 0.51 ± 0.048 | Yes (p = 0.001) | Yes (p = 0.022) | 4.4 ± 0.58 | 5 | Yes (p = 0.0024) | No | |
| GFP | Untreated | 100% | 3 | |||||
| 0.5 m sorbitol | 0.82 ± 0.046 | ND | 4.1 ± 0.89 | 7 | Yes | ND |
FIGURE 2.

Analysis of WNK1 localization and mobility by FRAP. WNK1 reversibly associates with the surface of persistent punctate structures. A–C, three frames from a FRAP experiment on a cell expressing GFP-WNK1 and equilibrated with 0.2 m sorbitol. A box indicates bleached region. A, immediately before bleaching. B, immediately after bleaching for 70 ms. C, 38 s after photobleaching. D, representative FRAP curves for GFP-WNK1 and GFP-WNK1 1800–2126. Full-length GFP-WNK1 (FL) expressed in cells under isotonic conditions recovers rapidly to more than 80% of the prebleach value (black). Both 0.2 m sorbitol (sorb, red) and gluconate (purple) substantially reduce the percentage of recovery and also reduce the half-time of the recovery by ∼10-fold. In contrast, the recovery kinetics of GFP-WNK1 1800–2126 (Cterm) are similar to WNK1 in 0.2 m sorbitol, even in isotonic conditions (green and blue). The lines represent nonlinear least squares fit of a single exponential to the data. E, immune complex kinase assay of WNK1 using OSR1 K46R from cells exposed to the two different hypo-osmotic conditions. Fold activity is relative to that in untreated cells. Untr, untreated; hypo, hypo-osmotic; Cterm, C-terminal.
Unlike the full-length GFP-tagged protein, GFP-WNK1 1800–2126, containing the conserved coiled coil and the balance of the C terminus, was primarily punctate, even under isotonic conditions, consistent with the appearance of the endogenous protein (Fig. 1, A, C, and D). Thus, we also analyzed its behavior. After photobleaching, these puncta recover to 76% of the initial value with a mean half-time of 2 s, significantly slower than full-length GFP-WNK1 (Table 1 and Fig. 2D). After equilibration in 0.2 m sorbitol, the mobility of GFP-WNK1 1800–2126 was further reduced and was not significantly different from the full-length protein under the same conditions (65% recovery, mean half-time = 4.2 s). When equilibrated in 0.5 m sorbitol, GFP-WNK1 1800–2126 was essentially immobile on the timescale of the experiment. By comparison, GFP alone remained mobile in 0.5 m sorbitol, recovering to 81% of its initial value, although the mean half-time for recovery (4.1 s) was substantially longer than in isotonic conditions where the recovery was too fast to measure (Table 1). The immobility of GFP-WNK1 1800–2126 relative to GFP alone under these highly hypertonic conditions is apparently not due solely to the reduced water content of the cell.
Effect of Hypo-osmotic Stress on GFP-WNK1 and GFP-WNK1 1800–2126
Equilibration with either hypotonic medium induced full-length GFP-WNK1 to become punctate. The fluorescence recovery of the protein on these puncta (65% recovery with a mean half-time = 4.4 s) was very similar to that in the presence of 0.2 m sorbitol (Table 1 and Fig. 2D). GFP-WNK1 1800–2126 was variably affected by hypotonic medium. In gluconate-containing medium, the fluorescence recovery of puncta (75% recovery with mean half-time = 2 s) was nearly identical with that under isotonic conditions (Table 1). On the other hand, medium made hypotonic by dilution reduced the mobility of GFP-WNK1 1800–2126 to values (64% recovery and mean half-time = 4.6 s) very similar to those in 0.2 m sorbitol. Because two different hypotonic media were tested, we compared the activity of endogenous WNK1 under the two conditions (Fig. 2E). In both cases WNK1 activity was increased a little more than 2-fold.
Effects of WNK1 Fragments Containing RFXV Motifs on OSR1 Binding and Activity
In addition to its core kinase domain, OSR1 contains two regions conserved in SPAK and orthologues called PF1 and PF2 domains (named for a common domain in PASK (alternate name for SPAK) and Fray (Drosophila protein)). PF1 is essential for the kinase domain fold. The PF2 domain binds specifically to RFXV motifs in WNKs, transporters, and other proteins (42, 44, 46–48). Depending on splice form, WNK1 contains as many as six RFXV motifs (Fig. 3A). One of these motifs was previously shown to interact strongly with cellular OSR1 and to be useful for affinity purification (44, 49). We tested the potential impact of four of the other motifs on binding and activation of OSR1.
FIGURE 3.
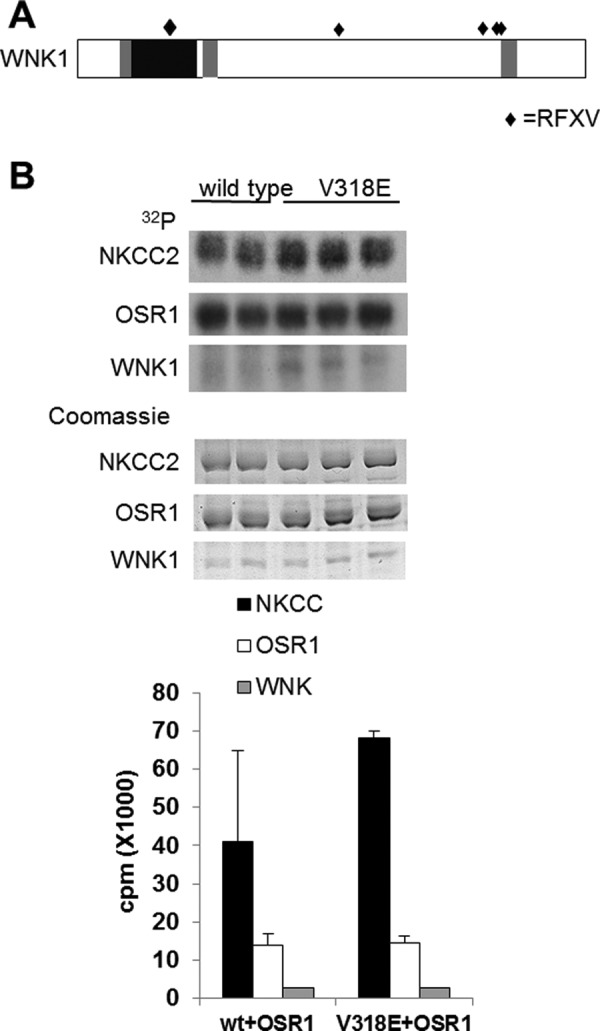
RFXV motif in the kinase domain of WNK1 is dispensable for its action. A, schematic showing the domain arrangement of the rat isoform of WNK1 showing the position of the RFXV motifs (filled diamonds). B, recombinant wild type (in duplicate) or V318E mutant (in triplicate) kinase domain of WNK1 was used in a coupled kinase assay toward OSR1 and its substrate NKCC2 1–175. Autoradiograms (upper panel) and Coomassie-stained gels (lower panel) are shown. Incorporated radioactivity was quantified using liquid scintillation counting and is plotted on the graph.
One of the RFXV motifs is in the WNK1 kinase domain and was previously identified as a region of charge difference on the substrate binding surfaces of WNK1 and WNK4, in that valine in WNK1 is replaced by glutamate in WNK4 (50). Because this region affected phosphorylation of the substrate synaptotagmin 2 by WNK1, we considered the possibility that recognition of OSR1 might be affected by this motif. We assayed the ability of the kinase domain of WNK1 V318E to phosphorylate and activate OSR1 in vitro. A slight increase in autophosphorylation was observed with V318E WNK1 compared with control. Altering this motif does not alter OSR1 phosphorylation or activation under these conditions (Fig. 3B).
Three RFXV motifs are clustered within a stretch of just over 100 residues near the WNK1 C terminus. We originally identified a fragment of WNK1 containing these clustered motifs as an OSR1 binding partner (42). To determine whether we could use fragments of WNK1 to block interaction of OSR1 with endogenous WNK1, we expressed fragments of different lengths with and without the three RFXV motifs in cells. We first tested the relative ability of these fragments to co-immunoprecipitate with OSR1 compared with endogenous WNK1. WNK1 1570–2126 co-precipitated with OSR1 and displaced the majority of the endogenous WNK1 from the OSR1 immunoprecipitates, as assessed by immunoblotting (Fig. 4, A and B). This fragment contains the three C-terminal RFXV motifs, a coiled coil, and the rest of the C terminus (Fig. 3A). Longer fragments also bound OSR1, but not as well as residues 1570–2126 (Fig. 4A). The fragments retaining the RFXV motifs but lacking either the C-terminal region, WNK1 1570–1820, or both the coiled coil and the most C-terminal region, WNK1 1570–1758, bound OSR1 much less well (10–30%). No binding of OSR1 to the WNK1 C-terminal residues 1804–2126 was detected (Fig. 4B). The larger WNK1 fragments were phosphorylated in the OSR1 kinase reactions (Fig. 4A), and WNK1 fragments containing residues 1301–1600 and 1601–1850 were both phosphorylated by recombinant OSR1 in vitro, supporting the idea that, once activated, OSR1 phosphorylates WNK1. Because some endogenous WNK1 was present in the precipitates, it is also possible that some of the phosphorylation of the WNK1 fragments is catalyzed by WNK1 itself. A band corresponding to endogenous WNK1 was also phosphorylated roughly in proportion to its amount present in the OSR1 immunoprecipitates.
FIGURE 4.
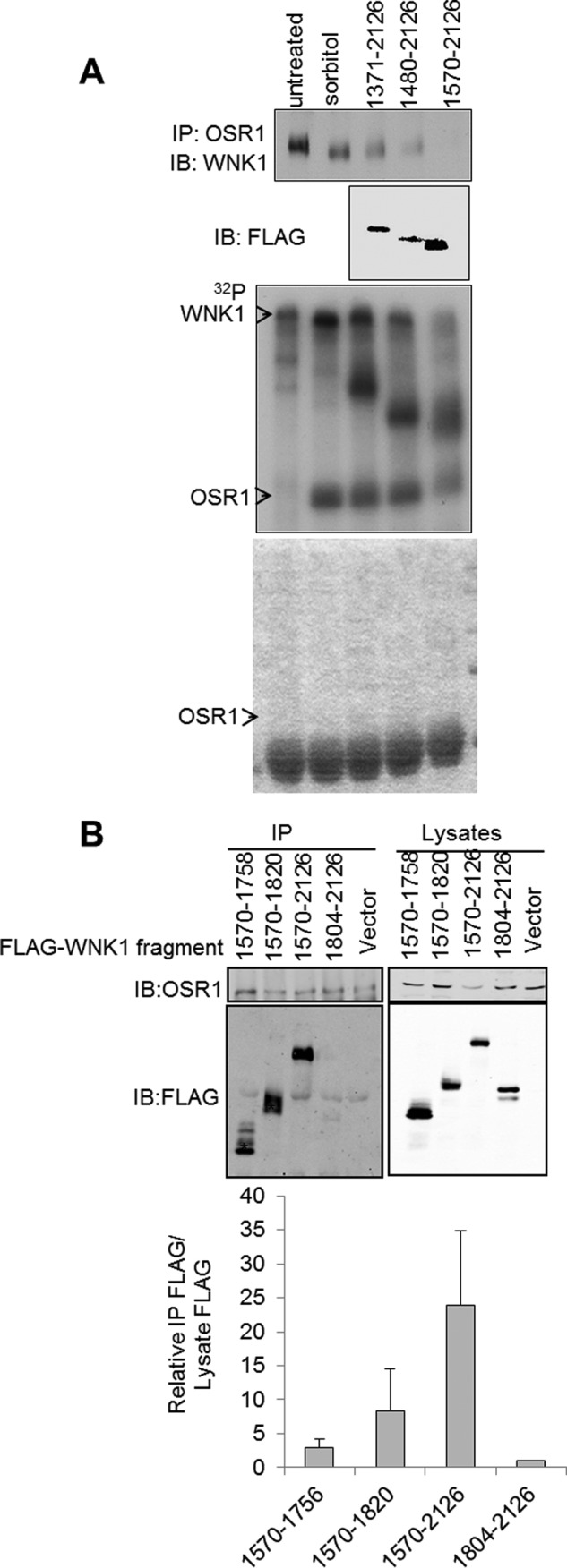
WNK1 binds to OSR1 via its C terminus. A, HeLa cells were either untransfected or transfected with FLAG-tagged WNK1 C-terminal fragments. Endogenous OSR1 was immunoprecipitated (IP) from cell lysates, and co-immunoprecipitating WNK1 and FLAG-tagged fragments were monitored using immunoblotting (IB). An in vitro kinase assay was performed using the immunoprecipitates, and the autoradiogram is shown. The bottom panel shows the Coomassie Blue stain of the kinase assay gel in gray scale. B, HeLa cells were transfected with the indicated FLAG-tagged WNK1 fragments. OSR1 was immunoprecipitated from these cells, and the fragments co-immunoprecipitating with OSR1 were detected using immunoblotting for FLAG. The expression of the fragments was detected by immunoblotting the lysates. The data shown are averaged from five experiments analyzed using LiCOR imaging. Because the expression of the fragments was not equal, the intensity of the FLAG band in the immunoprecipitates was normalized to the intensity of the FLAG band in the lysates (corresponding to level of expression) with the value for 1804–2126 arbitrarily set to 1.
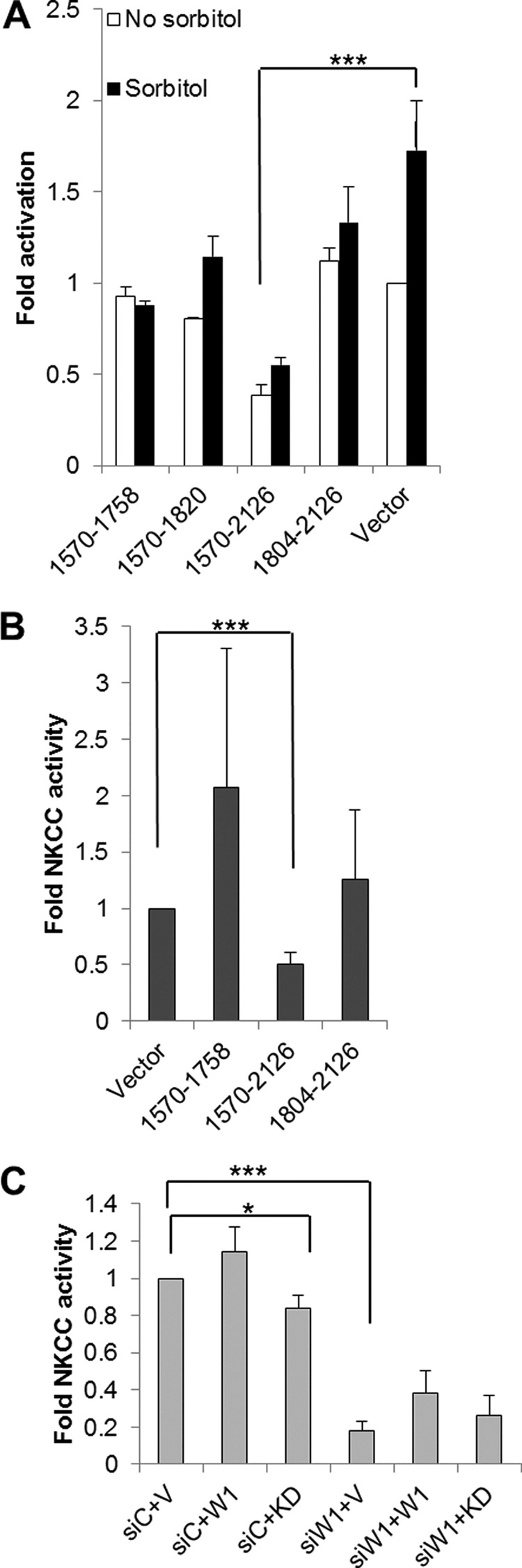
We examined functional consequences of inhibiting WNK1-OSR1 binding using the fragment WNK1 1570–2126, which was most effective in blocking their association. Of the fragments tested, this one also inhibited OSR1 activation most effectively (Fig. 5A). The other C-terminal WNK1 fragments had less significant effects on OSR1 phosphorylation and activation.
FIGURE 5.
WNK1 C-terminal fragments affect OSR1 and NKCC activity and the kinase activity of WNK1 is required for NKCC activity. A, FLAG-tagged WNK1 C-terminal fragments were expressed in HeLa cells. The cells were unstimulated or treated with sorbitol, and endogenous OSR1 was immunoprecipitated. Activation of OSR1 under basal (white bars) and sorbitol-stimulated (black bars) cells was measured using in vitro kinase assays. Activity averaged from two separate experiments was plotted relative to that in vector transfected control cells. Comparing activity in the presence of WNK1 1570–2126 to vector control. ***, p < 0.05. B, activity of endogenous NKCC1 was measured using uptake of radiolabeled 86Rb in HeLa cells expressing FLAG-tagged WNK1 fragments. Uptake was averaged from three separate experiments each carried out in triplicate relative to activity in cells transfected with vector control. Uptake in the presence of WNK1 1570–2126 was compared with vector control. ***, p < 0.05. C, endogenous WNK1 was depleted in HeLa cells using siRNA (siW1, three bars on the right). A scrambled siRNA (siC, three bars on the left) was used as control. The cells were transfected with empty vector (V) or vectors encoding full-length wild type WNK1 (W1) or kinase dead WNK1 (KD). The activity of endogenous NKCC1 was assayed using uptake of radiolabeled 86Rb. An average of three separate experiments with kinase-dead (KD) and five separate experiments with wild type WNK1 (W1) plotted after normalizing to the scrambled siRNA with empty vector (siC+V). We tested whether kinase-dead WNK1 interferes with endogenous activity and compared siC+KD to siC+V; *, p = 0.08. We tested whether WNK1 is required for NKCC activity and compared siWNK1+V to siC+V; ***, p = 7.4473E-10s.
To determine whether the inhibitory effect of WNK1 fragments containing the RFXV motifs extended to the physiological response of ion uptake by NKCCs, we assayed the activity of the ubiquitous NKCC, NKCC1, in HeLa cells using uptake of radiolabeled 86Rb. Expression of WNK1 1570–2126 caused a significant reduction in rubidium uptake compared with vector alone under iso-osmotic conditions or in the presence of sorbitol. Results with shorter fragments, WNK1 1570–1758 or 1804–2126, were more variable (Fig. 5B). These results suggest that WNK1 1570–2126 can be used as an inhibitor to sequester OSR1 from endogenous WNK1, thereby preventing OSR1 phosphorylation and activation.
Depletion of endogenous WNK1 with siRNA caused a significant reduction in NKCC activity, confirming our earlier result that WNK1 is required to regulate NKCC in HeLa cells (42). WNK1 knockdown has provided important information about function, but rescue by re-expression for structure/function analysis has proven difficult except in single cell assays. This may be due in part to the fact that WNK1 overexpression causes membrane phenotypes. Thus, it is necessary to achieve uniform expression near the endogenous expression level and that is difficult. We attempted to demonstrate rescue of NKCC activity by expression of wild type rat WNK1 after knockdown of endogenous WNK1 in HeLa cells. In some individual experiments rat WNK1 did appear to rescue NKCC activity; however, the results of a dozen experiments did not demonstrate a statistically significant increase in transport compared with the knockdown (Fig. 5C). In lieu of a rescue assay, we expressed kinase-dead WNK1 to determine whether a long form of the protein lacking kinase activity could compete with the endogenous protein. Modest, but statistically significant, inhibition was observed (p = 0.08), consistent with our earlier findings showing that WNK1 catalytic activity is required to stimulate OSR1 (42).
OSR1 Can Be Activated by All Four WNKs
A number of factors have contributed to controversies surrounding the assessment of physiological functions of WNKs and the relationship to their biochemical properties. There are conflicting findings on control of NCC activity by WNKs 1 and 4 in the kidney (51–53). Their actions are confounded by the expression of more than one isoform of both WNK1 and SPAK; in each case, in addition to one or more catalytically active forms, there is an unusual splice form lacking catalytic activity but retaining most other conserved elements (54). In addition, there remains a poor understanding of the defects conferred by the WNK1 and WNK4 mutations associated with PHA2 hypertension. To accumulate biochemical information to assist in resolving these questions, we wished to determine the ability of the WNK kinase domains to activate OSR1 in vitro to define the relative activities of the WNKs toward this substrate. The results should be of use in determining mechanistic differences in the actions of the full-length proteins in tissues.
We analyzed kinetics of OSR1 activation by the catalytic domains all four WNKs. We showed previously that mutation of the essential phosphorylation site, Thr-185 to aspartate alone or together with other acidic substitutions in activation loop residues induces up to ∼20-fold increase in OSR1 activity relative to wild type protein. Phosphorylation of OSR1 by WNK1 induces more than a 100-fold increase in OSR1 activity (Fig. 6, A and B), consistent with earlier reports (25, 33, 42). All four WNK family members phosphorylated OSR1 leading to similarly increased activity toward NKCC. We assessed kinetic constants for the four WNK family members and found Km values in the range of 1–4 micromolar for WNKs 1, 3, and 4 (Fig. 6C). The values of kcat ranged from 0.63 to 1.2 min−1. The data with WNK2 were not of sufficient quality to obtain reliable constants but seem to fall in the same range.
FIGURE 6.

All four WNKs have similar action on OSR1. A, autoradiograms showing phosphorylation of OSR1 by recombinant kinase domain proteins of all four WNKs in the presence of γ-32P-labeled ATP (top panel) and autophosphorylation of recombinant kinase domain proteins of all four WNKs (bottom panel). Coomassie staining showing equal loading of proteins is shown (top panel). Quantification of phosphorylation of OSR1 by all four WNKs is shown as a bar graph (bottom panel). B, autoradiograms showing activation of OSR1 as seen by phosphorylation of its substrate NKCC2 in the presence of recombinant kinase domains of all four WNKs (top panel) and phosphorylation of OSR1 by all four WNKs (bottom panel). Coomassie staining showing equal loading is shown (top panel). Quantification of NKCC2 phosphorylation (open bars) and OSR1 phosphorylation (closed bars) is shown (bottom panel). C, Michaelis-Menten curves showing the kinetics of phosphorylation of OSR1 by WNKs 1, 3, and 4. The substrate concentration at half-maximal velocity (Km) is calculated for each kinase and is shown on the graph.
DISCUSSION
Several different experimental strategies initially revealed that WNK1 and its substrate kinase OSR1 were tightly interacting partners (24, 33, 42–44). Here we find that in interphase cells, WNK1 and OSR1 display a significant co-localization validated by immunofluorescence with a Pearson coefficient above 0.5. Both proteins appear in a punctate pattern in multiple cell types, HeLa, HT29, MCF-7, even in the absence of osmotic stress. A recent study in kidney in which SPAK was knocked out showed that the typical apical localization of these proteins in distal tubule was replaced by a punctate distribution very similar to what is observed here (55). From our immunofluorescence studies, we concluded that overexpression of GFP-tagged WNK1 does not consistently reflect the endogenous localization, and by comparison with the study noted above, it seems likely that in highly specialized tissues WNK1, OSR1 and other tight binding partners will display localizations unique to their tissue-specific functions.
We are using information on their localizations as a starting point for defining which WNK1 functions might be mediated through OSR1 and which ones are independent of OSR1. For example, we recently showed that WNK1 is required for proper function of the mitotic spindle (50). This property of the protein does not require its binding partner, OSR1, and OSR1 is not co-localized with WNK1 during mitosis.
Association of OSR1/SPAK with WNK1 has been assumed to be due to the multiple RFXV motifs present in its C terminus. Because WNK family members and splice variants contain different numbers of these motifs, OSR1/SPAK interactions will likely be strongly influenced by number and organization of RFXV sequences. One of the RFXV motifs that appears unimportant for OSR1 interaction is located on the protein substrate binding surface in the WNK catalytic domain. Val-318, within this RFXV motif, is important for its interaction with the substrate synaptotagmin 2 (50) and perhaps other substrates as well. Also of note, the similar motif RVXF has been shown to confer binding of a variety of proteins to phosphoprotein phosphatase 1 (56).
In contrast, the clustered RFXV motifs nearer the WNK1 C terminus can displace endogenous WNK1 from immunoprecipitated OSR1, confirming their importance in forming a stable WNK1-OSR1 complex. The inclusion of the C-terminal coiled-coil increased the effectiveness of inhibition by the RFXV-containing segment, suggesting that the encoded localization information enhanced inhibition by increasing its concentration in the vicinity of the WNK1-OSR1 complex. The idea that RFXV fragments may be useful complements to siRNA strategies to dissociate WNK1 and OSR1 to explore their regulation and functions is supported by the capacity of a portion of WNK1 containing three clustered RFXV motifs to inhibit NKCC activity. Our results suggest that additional factors may be involved, because all fragments containing the clustered motifs are not equally effective in blocking interaction of OSR1 with endogenous WNK1. Further use of this approach should help to reveal roles of WNK-OSR1/SPAK signaling complexes in physiologic processes in addition to ion homeostasis. The diversity in number and sequence context of RFXV motifs among the different WNK family members also suggests a wealth of potential mechanisms for their regulation, including competition among motifs on different WNKs and sequestering or revealing motifs by post-translational modification as a feedback mechanism or through inputs from distinct signaling events. One such mechanism, whereby phosphorylation promotes dissociation, has already been identified (44). Several years ago, Delpire and Gagnon (47) found evidence that OSR1 and SPAK PF2 domains could interact with many proteins that contain RFXV motifs. It is well established that RFXV motifs in NKCCs bind OSR1 and SPAK, raising the question of how substrate and regulator interactions are juggled by these kinases. A detailed accounting of events that control OSR1/SPAK binding to relevant RFXV motifs will almost certainly be required to understand their physiological regulation fully.
Localization of endogenous WNK1 is mimicked by WNK1 GFP-1800–2126. Because endogenous WNK1 is distributed in a perinuclear punctate pattern (45) and can form dimers or oligomers (12, 49, 57), it seems possible that the C-terminal fragment might interact with endogenous WNK1 through the coiled-coil domain. The behavior of WNK1 1800–2126 upon exposure to changes in tonicity also most closely parallels behavior of the endogenous protein. This C-terminal region contains a coiled coil and an additional short segment conserved among WNK family members. Although the WNK1-associated structures have not yet been identified, apparently this region contains the information necessary to direct the fragment to its punctate location.
WNK1 associates with the outside of punctate structures in cells. Recovery of fluorescence following photobleaching appears to involve exchange of WNK1 molecules on existing puncta rather than the formation of new puncta. Co-localization experiments have not provided a definitive identification of these structures; nevertheless, they appear to be vesicles, because in hypotonic medium, conditions that might cause vesicles to swell, lumens were visible in some of the structures. As suggested by Alessi and co-workers (43), hypertonicity induces tighter binding of WNK1 to these punctate structures, reducing its mobility. Perhaps a surprise, we find that reduced mobility is also observed with hypotonicity. In both cases, the reduction in mobility appears to be larger than would be expected because of tonicity-induced changes in diffusion, based in part on the comparison with GFP alone. Both increased and decreased tonicity enhance WNK1 activity. However, new methods will be required to determine whether there is a functional relationship between the apparent mobility of WNK1 in cells and its catalytic activity.
What data are available to draw conclusions about the biological activity of WNK1 in the kidney and elsewhere? The conclusion that NCC is regulated positively by OSR1 and SPAK is accepted based in part on the utility of NCC inhibitors in disease therapy (7, 13–16). Increased WNK1 expression is thought to be responsible for WNK1-dependent PHA2; however, insufficient information has yet been collected about which, how many, and where WNK1 splice forms are overexpressed in this disease. A strong contribution in this direction comes from a recent study on splice form expression, which revealed very large differences, in the range of 100-fold, in KS-WNK1 expression across different segments of the mouse nephron (10). The magnitude of changes in KS-WNK1 expression underscores the difficulty in determining which form or forms of WNK1 dominate, even in different regions of the kidney. From more defined, but less physiologically based cell culture studies, it is evident that manipulation of the amounts of WNK proteins expressed will alter the biological outcomes. This problem confounds animal studies in which the relative expression of WNK family members is disturbed. The presence of isoforms not only of WNK1 but also of SPAK that lack catalytic activity yet apparently retain the capacity to bind their target proteins (58, 59) also challenges data interpretation. We showed previously that autoinhibitory domains of one WNK have the potential to inhibit other family members and that WNKs are likely to exist in complexes probably as tetramers (12). Those results suggested that KS-WNK1 might not only compete for binding to OSR1/SPAK but also directly inhibit the catalytic activity of any wild type WNK splice forms. The discovery of a WNK1-KS-WNK1 complex supports this idea (58). Diverse observations have been reported on actions of WNKs on NCC and NKCC activities in tissue and reconstituted systems (51, 60, 61). Bringing clarity to this problem will require further analysis on multiple levels.
To define the potential contribution of catalytic differences among the four WNKs for OSR1, we analyzed the capacities of the isolated kinase domains of the WNKs to activate OSR1. We conclude that functional differences that have been observed do not seem to arise from differential OSR1 recognition by WNK kinase domains. This result is consistent with a study showing that all known WNK3 splice forms have similar effects on SLC12 family members in a reconstituted system (62). If in tissue settings WNK variants are shown to have different effects on NCC and related co-transporters, the explanation will lie not in inherent catalytic capacity but in the ratios of active and inactive forms, localization and mobility differences, relative expression of the independent noncatalytic activator Mo25, perhaps, or other competing processes (63, 64). Findings here, together with earlier literature, allow us to conclude that all WNKs containing complete kinase domains can activate OSR1 and will do so given appropriate spatial setting and absence of inhibitory constraint.
Acknowledgments
We thank members of the Cobb lab for suggestions about this work and Dionne Ware for administrative assistance. Image and image analysis were carried out with the assistance of the University of Texas Southwestern Live Cell Imaging Facility, a Shared Resource of the Harold C. Simmons Cancer Center.
This work was supported, in whole or in part, by National Institutes of Health Grant GM53032 (to M. H. C.). This work was also supported by National Cancer Institute Cancer Center Support Grant 1P30 CA142543-01 and Robert A. Welch Foundation Grant I1243 (to M. H. C.).
- PHA2
- pseudohypoaldosteronism type 2
- OSR1
- oxidative stress-responsive 1
- NCC
- sodium chloride co-transporter
- NKCC
- sodium potassium chloride co-transporter
- SPAK
- STE20/SPS1-related proline-, alanine-rich kinase
- SLC12
- solute carrier 12
- FRAP
- fluorescence recovery after photobleaching.
REFERENCES
- 1. Ruiz-Pérez V. L., Murillo F. J., Torres-Martínez S. (1995) PkpA, a novel Phycomyces blakesleeanus serine/threonine protein kinase. Curr. Genet. 28, 309–316 [DOI] [PubMed] [Google Scholar]
- 2. Nakamichi N., Murakami-Kojima M., Sato E., Kishi Y., Yamashino T., Mizuno T. (2002) Compilation and characterization of a novel WNK family of protein kinases in Arabiodpsis thaliana with reference to circadian rhythms. Biosci. Biotechnol. Biochem. 66, 2429–2436 [DOI] [PubMed] [Google Scholar]
- 3. Choe K. P., Strange K. (2007) Evolutionarily conserved WNK and Ste20 kinases are essential for acute volume recovery and survival after hypertonic shrinkage in Caenorhabditis elegans. Am. J. Physiol. Cell Physiol. 293, C915–C927 [DOI] [PubMed] [Google Scholar]
- 4. Xu B., English J. M., Wilsbacher J. L., Stippec S., Goldsmith E. J., Cobb M. H. (2000) WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J. Biol. Chem. 275, 16795–16801 [DOI] [PubMed] [Google Scholar]
- 5. Veríssimo F., Jordan P. (2001) WNK kinases, a novel protein kinase subfamily in multi-cellular organisms. Oncogene 20, 5562–5569 [DOI] [PubMed] [Google Scholar]
- 6. Wilson F. H., Disse-Nicodème S., Choate K. A., Ishikawa K., Nelson-Williams C., Desitter I., Gunel M., Milford D. V., Lipkin G. W., Achard J. M., Feely M. P., Dussol B., Berland Y., Unwin R. J., Mayan H., Simon D. B., Farfel Z., Jeunemaitre X., Lifton R. P. (2001) Human hypertension caused by mutations in WNK kinases. Science 293, 1107–1112 [DOI] [PubMed] [Google Scholar]
- 7. Xie J., Craig L., Cobb M. H., Huang C. L. (2006) Role of WNKs in the pathogenesis of Gordon's syndrome. Pediat. Nephrol. 21, 1231–1236 [DOI] [PubMed] [Google Scholar]
- 8. Xu Q., Modrek B., Lee C. (2002) Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res. 30, 3754–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Delaloy C., Lu J., Houot A. M., Disse-Nicodeme S., Gasc J. M., Corvol P., Jeunemaitre X. (2003) Multiple promoters in the WNK1 gene. One controls expression of a kidney-specific kinase-defective isoform. Mol. Cell. Biol. 23, 9208–9221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vidal-Petiot E., Cheval L., Faugeroux J., Malard T., Doucet A., Jeunemaitre X., Hadchouel J. (2012) A new methodology for quantification of alternatively spliced exons reveals a highly tissue-specific expression pattern of WNK1 isoforms. PLoS One 7, e37751- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Min X., Lee B. H., Cobb M. H., Goldsmith E. J. (2004) Crystal structure of the kinase domain of WNK1, a kinase that causes a hereditary form of hypertension. Structure 12, 1303–1311 [DOI] [PubMed] [Google Scholar]
- 12. Lenertz L. Y., Lee B. H., Min X., Xu B. E., Wedin K., Earnest S., Goldsmith E. J., Cobb M. H. (2005) Properties of WNK1 and implications for other family members. J. Biol. Chem. 280, 26653–26658 [DOI] [PubMed] [Google Scholar]
- 13. Rossier B. C., Pradervand S., Schild L., Hummler E. (2002) Epithelial sodium channel and the control of sodium balance. Interaction between genetic and environmental factors. Annu. Rev. Physiol. 64, 877–897 [DOI] [PubMed] [Google Scholar]
- 14. Subramanya A. R., Yang C. L., McCormick J. A., Ellison D. H. (2006) WNK kinases regulate sodium chloride and potassium transport by the aldosterone-sensitive distal nephron. Kidney Int. 70, 630–634 [DOI] [PubMed] [Google Scholar]
- 15. Kahle K. T., Wilson F. H., Lalioti M., Toka H., Qin H., Lifton R. P. (2004) WNK kinases. Molecular regulators of integrated epithelial ion transport. Curr. Opin. Nephrol. Hypertens. 13, 557–562 [DOI] [PubMed] [Google Scholar]
- 16. Bergaya S., Vidal-Petiot E., Jeunemaitre X., Hadchouel J. (2012) Pathogenesis of pseudohypoaldosteronism type 2 by WNK1 mutations. Curr. Opin. Nephrol. Hypertens. 21, 39–45 [DOI] [PubMed] [Google Scholar]
- 17. McCormick J. A., Ellison D. H. (2011) The WNKs. Atypical protein kinases with pleiotropic actions. Physiol. Rev. 91, 177–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilson F. H., Kahle K. T., Sabath E., Lalioti M. D., Rapson A. K., Hoover R. S., Hebert S. C., Gamba G., Lifton R. P. (2003) Molecular pathogenesis of inherited hypertension with hyperkalemia. The Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc. Natl. Acad. Sci. U.S.A. 100, 680–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang C. L., Angell J., Mitchell R., Ellison D. H. (2003) WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J. Clin. Invest. 111, 1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu B. E., Stippec S., Chu P. Y., Lazrak A., Li X. J., Lee B. H., English J. M., Ortega B., Huang C. L., Cobb M. H. (2005) WNK1 activates SGK1 to regulate the epithelial sodium channel. Proc. Natl. Acad. Sci. U.S.A. 102, 10315–10320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kahle K. T., Wilson F. H., Leng Q., Lalioti M. D., O'Connell A. D., Dong K., Rapson A. K., MacGregor G. G., Giebisch G., Hebert S. C., Lifton R. P. (2003) WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat. Genet. 35, 372–376 [DOI] [PubMed] [Google Scholar]
- 22. Richardson C., Rafiqi F. H., Karlsson H. K., Moleleki N., Vandewalle A., Campbell D. G., Morrice N. A., Alessi D. R. (2008) Activation of the thiazide-sensitive Na+-Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J. Cell Sci. 121, 675–684 [DOI] [PubMed] [Google Scholar]
- 23. Lazrak A., Liu Z., Huang C. L. (2006) Antagonistic regulation of ROMK by long and kidney-specific WNK1 isoforms. Proc. Natl. Acad. Sci. U.S.A. 103, 1615–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gagnon K. B., England R., Delpire E. (2006) Volume sensitivity of cation-chloride cotransporters is modulated by the interaction of two kinases. SPAK and WNK4. Am. J. Physiol. Cell Physiol. 290, C134–C142 [DOI] [PubMed] [Google Scholar]
- 25. Moriguchi T., Urushiyama S., Hisamoto N., Iemura S., Uchida S., Natsume T., Matsumoto K., Shibuya H. (2005) WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J. Biol. Chem. 280, 42685–42693 [DOI] [PubMed] [Google Scholar]
- 26. Yang C. L., Liu X., Paliege A., Zhu X., Bachmann S., Dawson D. C., Ellison D. H. (2007) WNK1 and WNK4 modulate CFTR activity. Biochem. Biophys. Res. Commun. 353, 535–540 [DOI] [PubMed] [Google Scholar]
- 27. Dorwart M. R., Shcheynikov N., Wang Y., Stippec S., Muallem S. (2007) SLC26A9 is a Cl− channel regulated by the WNK kinases. J. Physiol. 584, 333–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rafiqi F. H., Zuber A. M., Glover M., Richardson C., Fleming S., Jovanović S., Jovanović A., O'Shaughnessy K. M., Alessi D. R. (2010) Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol. Med. 2, 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pelkmans L., Fava E., Grabner H., Hannus M., Habermann B., Krausz E., Zerial M. (2005) Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature 436, 78–86 [DOI] [PubMed] [Google Scholar]
- 30. Cha S. K., Huang C. L. (2010) WNK4 kinase stimulates caveola-mediated endocytosis of TRPV5 amplifying the dynamic range of regulation of the channel by protein kinase C. J. Biol. Chem. 285, 6604–6611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng C. J., Huang C. L. (2011) Activation of PI3-kinase stimulates endocytosis of ROMK via Akt1/SGK1-dependent phosphorylation of WNK1. J Am. Soc. Nephrol. 22, 460–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He G., Wang H. R., Huang S. K., Huang C. L. (2007) Intersectin links WNK kinases to endocytosis of ROMK1. J. Clin. Invest. 117, 1078–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vitari A. C., Deak M., Morrice N. A., Alessi D. R. (2005) The WNK1 and WNK4 protein kinases that are mutated in Gordon's hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem. J. 391, 17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu B. E., Stippec S., Lazrak A., Huang C. L., Cobb M. H. (2005) WNK1 activates SGK1 by a phosphatidylinositol 3-kinase-dependent and non-catalytic mechanism. J. Biol. Chem. 280, 34218–34223 [DOI] [PubMed] [Google Scholar]
- 35. Xu B. E., Min X., Stippec S., Lee B. H., Goldsmith E. J., Cobb M. H. (2002) Regulation of WNK1 by an autoinhibitory domain and autophosphorylation. J. Biol. Chem. 277, 48456–48462 [DOI] [PubMed] [Google Scholar]
- 36. Xu B. E., Stippec S., Lenertz L., Lee B. H., Zhang W., Lee Y. K., Cobb M. H. (2004) WNK1 activates ERK5 by an MEKK2/3-dependent mechanism. J. Biol. Chem. 279, 7826–7831 [DOI] [PubMed] [Google Scholar]
- 37. Coppola D., Khalil F., Eschrich S. A., Boulware D., Yeatman T., Wang H. G. (2008) Down-regulation of Bax-interacting factor-1 in colorectal adenocarcinoma. Cancer 113, 2665–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen W., Yazicioglu M., Cobb M. H. (2004) Characterization of OSR1, a member of the mammalian Ste20p/germinal center kinase subfamily. J. Biol. Chem. 279, 11129–11136 [DOI] [PubMed] [Google Scholar]
- 39. Xu B., Wilsbacher J. L., Collisson T., Cobb M. H. (1999) The N-terminal ERK binding site of MEK1 is required for efficient feedback phosphorylation by ERK2 in vitro and ERK activation in vivo. J. Biol. Chem. 274, 34029–34035 [DOI] [PubMed] [Google Scholar]
- 40. Costes S. V., Daelemans D., Cho E. H., Dobbin Z., Pavlakis G., Lockett S. (2004) Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 86, 3993–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hannemann A., Christie J. K., Flatman P. W. (2009) Functional expression of the Na-K-2Cl cotransporter NKCC2 in mammalian cells fails to confirm the dominant-negative effect of the AF splice variant. J. Biol. Chem. 284, 35348–35358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Anselmo A. N., Earnest S., Chen W., Juang Y. C., Kim S. C., Zhao Y., Cobb M. H. (2006) WNK1 and OSR1 regulate the Na+, K+, 2Cl− cotransporter in HeLa cells. Proc. Natl. Acad. Sci. U.S.A. 103, 10883–10888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zagórska A., Pozo-Guisado E., Boudeau J., Vitari A. C., Rafiqi F. H., Thastrup J., Deak M., Campbell D. G., Morrice N. A., Prescott A. R., Alessi D. R. (2007) Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J. Cell Biol. 176, 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vitari A. C., Thastrup J., Rafiqi F. H., Deak M., Morrice N. A., Karlsson H. K., Alessi D. R. (2006) Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem. J. 397, 223–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tu S. W., Bugde A., Luby-Phelps K., Cobb M. H. (2011) WNK1 is required for mitosis and abscission. Proc. Natl. Acad. Sci. U.S.A. 108, 1385–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Piechotta K., Lu J., Delpire E. (2002) Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J. Biol. Chem. 277, 50812–50819 [DOI] [PubMed] [Google Scholar]
- 47. Delpire E., Gagnon K. B. (2007) Genome-wide analysis of SPAK/OSR1 binding motifs. Physiol. Genomics 28, 223–231 [DOI] [PubMed] [Google Scholar]
- 48. Villa F., Goebel J., Rafiqi F. H., Deak M., Thastrup J., Alessi D. R., van Aalten D. M. (2007) Structural insights into the recognition of substrates and activators by the OSR1 kinase. EMBO Rep. 8, 839–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thastrup J. O., Rafiqi F. H., Vitari A. C., Pozo-Guisado E., Deak M., Mehellou Y., Alessi D. R. (2012) SPAK/OSR1 regulate NKCC1 and WNK activity. Analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem. J. 441, 325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee B. H., Min X., Heise C. J., Xu B. E., Chen S., Shu H., Luby-Phelps K., Goldsmith E. J., Cobb M. H. (2004) WNK1 phosphorylates synaptotagmin 2 and modulates its membrane binding. Mol. Cell 15, 741–751 [DOI] [PubMed] [Google Scholar]
- 51. Yang C. L., Zhu X., Wang Z., Subramanya A. R., Ellison D. H. (2005) Mechanisms of WNK1 and WNK4 interaction in the regulation of thiazide-sensitive NaCl cotransport. J. Clin. Invest. 115, 1379–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. San-Cristobal P., Ponce-Coria J., Vázquez N., Bobadilla N. A., Gamba G. (2008) WNK3 and WNK4 amino-terminal domain defines their effect on the renal Na+-Cl− cotransporter. Am. J. Physiol. Renal Physiol. 295, F1199–F1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Subramanya A. R., Yang C. L., Zhu X., Ellison D. H. (2006) Dominant-negative regulation of WNK1 by its kidney-specific kinase-defective isoform. Am. J. Physiol. Renal Physiol. 290, F619–F624 [DOI] [PubMed] [Google Scholar]
- 54. Ahlstrom R., Yu A. S. (2009) Characterization of the kinase activity of a WNK4 protein complex. Am. J. Physiol. Renal Physiol. 297, F685–F692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Grimm P. R., Taneja T., Liu J., Coleman R., Chen Y. Y., Delpire E., Wade J. B., Welling P. A. (2012) SPAK, OSR1 and Cab39/MO25 form an interdependent signaling system which regulates thiazide-sensitive salt transport, distal tubule mass and blood pressure. FASEB J. 26, 867.37 [Google Scholar]
- 56. Heroes E., Lesage B., Gornemann J., Beullens M., Van M. L., Bollen M. (2012) The PP1 binding code. A molecular-lego strategy that governs specificity. FEBS J., in press [DOI] [PubMed] [Google Scholar]
- 57. Wang Z., Yang C. L., Ellison D. H. (2004) Comparison of WNK4 and WNK1 kinase and inhibiting activities. Biochem. Biophys. Res. Commun. 317, 939–944 [DOI] [PubMed] [Google Scholar]
- 58. Wade J. B., Fang L., Liu J., Li D., Yang C. L., Subramanya A. R., Maouyo D., Mason A., Ellison D. H., Welling P. A. (2006) WNK1 kinase isoform switch regulates renal potassium excretion. Proc. Natl. Acad. Sci. U.S.A. 103, 8558–8563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McCormick J. A., Mutig K., Nelson J. H., Saritas T., Hoorn E. J., Yang C. L., Rogers S., Curry J., Delpire E., Bachmann S., Ellison D. H. (2011) A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab. 14, 352–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang C. L., Zhu X., Ellison D. H. (2007) The thiazide-sensitive Na-Cl cotransporter is regulated by a WNK kinase signaling complex. J. Clin. Invest. 117, 3403–3411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hannemann A., Flatman P. W. (2011) Phosphorylation and transport in the Na-K-2Cl cotransporters, NKCC1 and NKCC2A, compared in HEK-293 cells. PLoS One 6, e17992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cruz-Rangel S., Melo Z., Vázquez N., Meade P., Bobadilla N. A., Pasantes-Morales H., Gamba G., Mercado A. (2011) Similar effects of all WNK3 variants on SLC12 cotransporters. Am. J. Physiol. Cell Physiol. 301, C601–C608 [DOI] [PubMed] [Google Scholar]
- 63. Richardson C., Sakamoto K., de los Heros P., Deak M., Campbell D. G., Prescott A. R., Alessi D. R. (2011) Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J. Cell Sci. 124, 789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Filippi B. M., de los Heros P., Mehellou Y., Navratilova I., Gourlay R., Deak M., Plater L., Toth R., Zeqiraj E., Alessi D. R. (2011) MO25 is a master regulator of SPAK/OSR1 and MST3/MST4/YSK1 protein kinases. EMBO J. 30, 1730–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]