

Background: A hierarchy of catalytic steps characterizes multifunctional cytochrome P450 enzymes.
Results: In the post-polyketide oxidative tailoring of mycinamicins by MycG, the two methoxy groups of mycinose are sensors that mediate initial recognition and discriminate between closely related molecules.
Conclusion: Bulky and conformationally restrained macrolide substrates advance to the catalytically productive mode through multiple steps.
Significance: Protein engineering facilitating substrate progression may enhance catalysis.
Keywords: Cytochrome P450, Enzyme Catalysis, Natural Product Biosynthesis, NMR, X-ray Crystallography
Abstract
The majority of characterized cytochrome P450 enzymes in actinomycete secondary metabolic pathways are strictly substrate-, regio-, and stereo-specific. Examples of multifunctional biosynthetic cytochromes P450 with broader substrate and regio-specificity are growing in number and are of particular interest for biosynthetic and chemoenzymatic applications. MycG is among the first P450 monooxygenases characterized that catalyzes both hydroxylation and epoxidation reactions in the final biosynthetic steps, leading to oxidative tailoring of the 16-membered ring macrolide antibiotic mycinamicin II in the actinomycete Micromonospora griseorubida. The ordering of steps to complete the biosynthetic process involves a complex substrate recognition pattern by the enzyme and interplay between three tailoring modifications as follows: glycosylation, methylation, and oxidation. To understand the catalytic properties of MycG, we structurally characterized the ligand-free enzyme and its complexes with three native metabolites. These include substrates mycinamicin IV and V and their biosynthetic precursor mycinamicin III, which carries the monomethoxy sugar javose instead of the dimethoxylated sugar mycinose. The two methoxy groups of mycinose serve as sensors that mediate initial recognition to discriminate between closely related substrates in the post-polyketide oxidative tailoring of mycinamicin metabolites. Because x-ray structures alone did not explain the mechanisms of macrolide hydroxylation and epoxidation, paramagnetic NMR relaxation measurements were conducted. Molecular modeling based on these data indicates that in solution substrate may penetrate the active site sufficiently to place the abstracted hydrogen atom of mycinamicin IV within 6 Å of the heme iron and ∼4 Å of the oxygen of iron-ligated water.
Introduction
Mycinamicins are macrolide antibiotics produced by the actinomycete Micromonospora griseorubida (1). They are composed of a 16-membered macrolactone ring and two sugars, desosamine and mycinose, at the C-5 and C-21 positions respectively (Fig. 1). Wild-type M. griseorubida mainly produces mycinamicins I and II, mycinamicin IV (M-IV),2 and mycinamicin V (M-V) (1), whereas a high producing industrial strain produces mycinamicin I and mycinamicin II as its two major products (2). Mycinamicin II, having strong antimicrobial activities against Gram-positive bacteria and mycoplasma (1), has been developed as a veterinary drug (2). Details of the mycinamicin biosynthetic pathway have been established by the isolation and structural characterization of intermediates (3–5) and by bioconversion studies of genetically modified strains (6–8). More recently, the complete 62-kb nucleotide sequence of the mycinamicin biosynthetic gene cluster comprising 22 open reading frames has been determined (9). Two P450 enzymes, MycCI and MycG, were identified in this cluster. MycCI is the C-21 methyl hydroxylase of mycinamicin VIII, the earliest macrolide in the post-polyketide synthase tailoring pathway, whose optimal activity depends on its native redox partner ferredoxin MycCII (10). In the mid-1990s, dual hydroxylation and epoxidation functions were proposed for a second P450, MycG, based on genetic complementation analysis (8). Its activity was characterized in detail using an in vitro system reconstituted with recombinant MycG, native substrates isolated from fermentation broths, and a surrogate commercial spinach ferredoxin/ferredoxin reductase redox system, which was employed because MycCII does not support the catalytic activity of MycG (10). Collectively, these studies demonstrated that MycG catalyzes sequential hydroxylation and epoxidation reactions at two distinct sites, a tertiary allylic C–H bond (C-14) and an olefin (C12–C13). Premature epoxidation at C12–C13 completely abolishes hydroxylation at C-14, thus terminating the pathway (Fig. 1). Interestingly, P450 Gfs4 in the biosynthesis of the macrolide antibiotic FD-891 in Streptomyces graminofaciens represents yet another example of a single P450 enzyme sequentially introducing both a hydroxyl and an epoxy group on the 16-membered ring macrolide scaffold, but it has reverse order reactivities compared with MycG (Fig. 2) (11).
FIGURE 1.

Final steps of mycinamicin biosynthesis. The solid arrows indicate the major steps in mycinamicin biosynthesis. The dashed arrow demonstrates low conversion from M-III to mycinamicin IX catalyzed by MycG. M-I cannot be hydroxylated by MycG.
FIGURE 2.
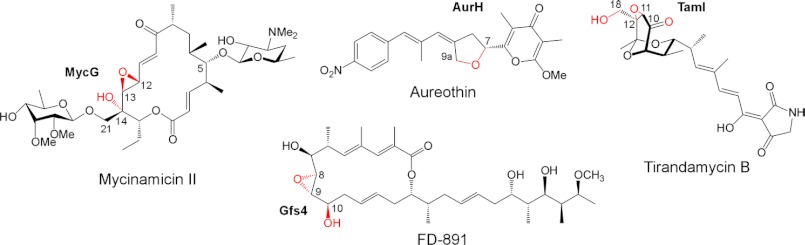
Natural products decorated by bacterial multifunctional P450 enzymes. Oxygen atoms introduced by P450s are highlighted in red.
Other characterized bacterial biosynthetic multifunctional P450s in Streptomyces secondary metabolome include TamI in the tirandamycin biosynthetic pathway of Streptomyces sp. 307-9 (12–14), and AurH in the biosynthetic pathway of aureothin in Streptomyces thioluteus (Fig. 2) (15). TamI operates on a bicyclic ketal moiety of tirandamycin C to catalyze successive epoxidation and hydroxylation reactions in an iterative cascade with the flavin oxidase TamL (14). AurH sequentially installs two C–O bonds into the polyketide backbone of aureothin to yield a tetrahydrofuran ring, a key pharmacophore of this antibiotic (16, 17). A critical difference that sets multifunctional P450s apart from substrate promiscuous enzymes is an apparent hierarchy in the sequence of catalytic steps, suggesting that each step may be a prerequisite for the one that follows (18).
The current data reveal that many fungal P450s are multifunctional enzymes that catalyze up to four consecutive steps on the same substrate molecule (19). For instance, Tri4 (CYP58) in the plant pathogen Fusarium graminearum performs one epoxidation and three hydroxylation steps in the biosynthesis of trichothecenes (20). The trichothecenes are sesquiterpenoid secondary metabolites that are potent mycotoxins of mold-contaminated cereal grains. Another example of a multifunctional fungal P450 is Fum6 (CYP505 family) in the biosynthesis of mycotoxins fumonisins from the maize pathogen Fusarium verticillioides (21–23), which catalyzes two consecutive hydroxylations at adjacent carbon atoms. The biosynthesis of plant hormone gibberellins in the rice pathogen Fusarium fujikuroi involves four multifunctional P450 enzymes that catalyze 10 of the 15 biosynthetic steps (24–26). Fungal P450s are integral membrane proteins, making structural and biophysical characterization challenging. In this regard, understanding the switch of function mechanism in the more accessible bacterial multifunctional P450s should bring considerable new insights to this versatile class of underexplored monooxygenases.
Ongoing structural and functional studies are beginning to provide detailed insights into the molecular basis for sequential reactivity and the pattern of oxidation in multifunctional systems. Although no substrate-bound structure has been obtained for AurH, a hypothetical switch of function mechanism has been proposed based on computational docking of two consecutive substrates into x-ray structures of different AurH ligand-free conformers (15). The key role is assigned to glutamine 91, which upon completion of the hydroxylation step changes the conformation to provide an H-bond to the newly installed C7-OH group. In the course of mutual conformational adjustments, the hydroxylated intermediate relocates deeper into the substrate binding pocket to enable the next attack by the activated oxygen species at C-9a, whereas the substrate backbone bends to facilitate tetrahydrofuran ring formation.
Here, we have structurally characterized MycG in complex with each of its two consecutive native substrates, M-IV and M-V, and their much less reactive biosynthetic precursor, M-III, which bears the monomethoxy sugar javose instead of dimethoxylated mycinose (Fig. 1). Despite this single methyl group difference, M-III is only hydroxylated by MycG in trace amounts in vitro (10). Traces of the C14-hydroxylated M-III (mycinamicin IX) and M-III bearing a C12–C13 epoxide from culture broths of the mycF disruption mutant M. griseorubida TPMA0004 are also consistent with marginal activity of MycG against M-III in vivo (27, 28). We provide structural information that suggests a complex recognition strategy by MycG in which the enzyme successfully distinguishes between substrates and the closely related M-III intermediate. However, the precise mechanism of discrimination between the two native substrates M-IV and M-V remained obscure in light of the crystal structures obtained in this work due to the large distances separating the reactive sites from the iron center. Paramagnetically induced 1H spin relaxation NMR measurements indicate that deeper penetration of the active site of MycG by M-IV may take place in solution, and the pattern of observed relaxation effects is consistent with the x-ray binding orientation as predominating in the presence of excess substrate.
EXPERIMENTAL PROCEDURES
MycG Expression and Purification
MycG was cloned, expressed, and purified as an N-terminal His6-tagged protein as described elsewhere (10), with the modification that Tris-HCl buffer was used instead of sodium phosphate in the harvesting and purification protocols. The Phe-286 site-directed mutants were generated following the QuikChange protocol (Stratagene). Purification procedures included affinity chromatography on nickel-nitrilotriacetic resin (Qiagen), S-Sepharose flow through passage, and finally ion-exchange chromatography on Q-Sepharose in a gradient of NaCl (0–0.5 m) concentrations (29). Pure protein fractions were combined and concentrated in 20 mm Tris-HCl, pH 7.5, 0.5 mm EDTA, and ∼200 mm NaCl, as eluted from Q-Sepharose, to ≥1 mm for storage at −80 °C until required. As expected, the sodium dithionite-reduced MycG solution displayed an absorbance peak at 408 nm with a 447-nm peak appearing after CO was bubbled through the solution (30).
Isolation of the MycG Substrates
M-IV and M-V were isolated and purified from the fermentation broth of the wild-type M. griseorubida A11725 strain following the procedures described elsewhere (1). M-III was purified from fermentation broths of the mycF disrupted mutant strain TMPA0004 (27).
Crystallization, Data Collection, and Structure Determination
Prior to crystallization, the protein was diluted to 0.2–0.5 mm by mixing with 10 mm Tris-HCl, pH 7.5, alone or supplemented with 0.5–2.0 mm M-III, M-IV, or M-V. Crystallization conditions in each case were determined using commercial high throughput screening kits available in deep-well format (Hampton Research), a nanoliter drop-setting Mosquito robot (TTP LabTech) operating with 96-well plates, and a hanging drop crystallization protocol. Crystals were generated under a wide variety of screening conditions; however, only a minor subset was amenable to optimization to produce diffraction quality specimens. Crystals of different morphologies from distinct crystallization conditions were further optimized in 24-well plates for diffraction data collection (Table 1). Plate-shaped crystals diffracting in the C2221 space group formed under conditions that included benzamidine hydrochloride. Because of the lateral molecular symmetry, benzamidine dramatically improved the quality of the plate crystals by binding in the symmetry-related positions at the interfaces of protein monomers. Prior to data collection, all crystals were cryo-protected by plunging them into a drop of reservoir solution supplemented with 20–25% glycerol or ethylene glycol and then flash-frozen in liquid nitrogen.
TABLE 1.
Data collection and refinement statistics
PDB, Protein Data Bank; AU, asymmetric unit; c.m.s.d., root mean square deviation.
| MycG |
MycGF286A |
MycGF286V |
||||||
|---|---|---|---|---|---|---|---|---|
| Mycinamicin IV | Mycinamicin IV | Mycinamicin V | Mycinamicin III | Mycinamicin III | No ligand | Mycinamicin IV | Mycinamicin V | |
| PDB code | 2Y98 | 2Y46 | 2Y5N | 2Y5Z | 2YCA | 2YGX | 3ZSN | 4AW3 |
| Data collection | ||||||||
| Space group | P21212 | C2221 | P21 | C2221 | P21212 | P21 | C2221 | P1 |
| Cell dimensions | ||||||||
| a, b, c (Å) | 56.3, 162.0, 50.4 | 58.3, 101.0, 441.5 | 82.8, 57.4, 101.9 | 58.3, 100.9, 439.4 | 56.7, 162.3, 49.8 | 57.2, 162.0, 100.1 | 58.2, 100.9, 440.8 | 55.5, 56.1, 76.4 |
| α, β, γ | 90°, 90°, 90° | 90°, 90°, 90° | 90°, 113.5°, 90° | 90°, 90°, 90° | 90°, 90°, 90 ° | 90°, 89.9°, 90° | 90°, 90°, 90° | 90.4°, 97.2°, 102.1° |
| Molecules in AU | 1 | 3 | 2 | 3 | 1 | 4 | 3 | 2 |
| Wavelength | 1.11587 | 1.11587 | 1.11587 | 1.11587 | 1.11587 | 1.11587 | 1.11587 | 1.11587 |
| Resolution (Å) | 1.65 | 1.83 | 1.62 | 2.06 | 1.80 | 2.39 | 1.90 | 2.05 |
| Rsym or Rmerge (%) | 6.6 (62.7)a | 8.0 (56.0) | 4.0 (30.4) | 8.0 (36.0) | 12.0 (93.0) | 7.0 (32.0) | 8.0 (56.0) | 16.8 (54.0) |
| I/σI | 15.0 (1.6) | 11.1 (1.5) | 16.2 (2.2) | 9.8 (1.5) | 7.6 (1.5) | 10.80 (3.2) | 10.9 (1.7) | 4.1 (1.7) |
| Completeness (%) | 84.9 (40.6) | 83.1 (36.2) | 70.4 (11.1) | 86.3 (42.3) | 99.9 (99.9) | 95.6 (76.6) | 89.2 (64.2) | 96.4 (95.7) |
| Redundancy | 6.4 (3.3) | 5.3 (2.4) | 3.5 (1.9) | 4.4 (1.4) | 4.2 (4.3) | 3.8 (2.6) | 5.4 (3.2) | 2.1 (2.2) |
| Crystallization conditions | 8% PEG 4000, 0.2 m ammonium sulfate, 0.1 m BisTris (pH 5.5) | 12% PEG MME 2000, 5% Tacsimate (pH 7.5), 1% benzamidine HCl | 7% PEG 4000, 0.025 m magnesium acetate | 12.5% PEG MME 2000, 6% Tacsimate (pH 7.0), 0.15 m NaCl, 2% benzamidine HCl | 0.6 m succinic acid (pH 7.0) | 0.6 m succinic acid (pH 7.0), 0.0075 mm CYMAL-7, 2% glycerol | 2.5% PEG 1500, 0.1 m malonate (pH 5.0), 2% benzamidine HCl | 1.6 m ammonium sulfate, 0.1 m MES (pH 6.5), 10% (v/v) 1,4-dioxane |
| Refinement | ||||||||
| No. reflections | 45,438 | 90,775 | 75,373 | 66,285 | 41,348 | 59,628 | 86,665 | 48,500 |
| Rwork/Rfree (%) | 14.8/20.6 | 15.8/23.3 | 11.5/18.8 | 15.0/22.1 | 15.2/21.0 | 18.4/24.7 | 18.5/24.5 | 16.6/23.5 |
| No. atoms | ||||||||
| Protein | 3203 | 9410 | 6439 | 9385 | 3192 | 12227 | 9386 | 6197 |
| Heme | 43 | 129 | 86 | 129 | 43 | 172 | 129 | 86 |
| Substrate | 49 | 147 | 100 | 111 | 48 | n/a | 147 | 200 |
| Solvent | 420 | 979 | 1216 | 878 | 354 | 511 | 1021 | 582 |
| Mean B value | 21.0 | 23.1 | 18.6 | 28.3 | 23.1 | 21.4 | 23.4 | 22.7 |
| B-Factors | ||||||||
| Protein | 19.4 | 21.9 | 15.9 | 27.5 | 21.8 | 21.8 | 23.5 | 22.06 |
| Heme | 13.4 | 11.7 | 9.6 | 16.2 | 14.7 | 10.4 | 12.8 | 13.8 |
| Substrate | 29.9 | 33.4 | 18.9 | 39.0 | 25.9 | n/a | 43.0 | 23.6 |
| Solvent | 33.3 | 34.3 | 33.4 | 38.8 | 25.9 | 21.1 | 31.6 | 32.2 |
| r.m.s.d. | ||||||||
| Bond lengths (Å) | 0.028 | 0.023 | 0.023 | 0.020 | 0.024 | 0.017 | 0.022 | 0.022 |
| Bond angles | 2.1730° | 1.887° | 1.904° | 1.826° | 1.875° | 1.715° | 1.902° | 2.028° |
a Values in parentheses are for the highest resolution shell.
Diffraction data were collected at 100–110 K at beamline 8.3.1, Advanced Light Source, Lawrence Berkeley National Laboratory. Data indexing, integration, and scaling were conducted using MOSFLM (31) and the programs implemented in the ELVES software suite (32). The first crystal structure determined was for the MycG·M-IV complex at a resolution of 1.65 Å by molecular replacement using diffraction data processed in the P21212 space group, with Rmerge of 6.6% and atomic coordinates of MoxA (PDB code 2Z36) (33) as a search model. The MycG model was built using the BUCCANEER (34, 35) and COOT (36) programs. Refinement was performed by using REFMAC5 software (35, 37) until R and Rfree converged to 14.8 and 20.6%, respectively. The final coordinates were used as the molecular replacement model for the entire series of MycG structures determined in this work. Data collection and refinement statistics are shown in Table 1.
1H M-IV R1 Relaxation Experiments
To determine the orientation of M-IV bound in the active site in solution, 1H R1 relaxation rates were measured by nuclear magnetic resonance (NMR) for the resonances of M-IV in the presence of varying amounts of MycG. A 2 mm sample of M-IV was prepared in 50 mm KPi, pH 7.3, 50 mm NaCl, 1 mm EDTA, and 0.2 mm dithiothreitol in 90% H2O, 10% D2O. 1H and 13C resonance assignments for M-IV were made using two-dimensional DQF-COSY, 1H,13C HSQC, and NOESY datasets. A series of 1H inversion recovery experiments with recovery times of 0.05, 0.1, 0.2, 0.3, 0.5, 0.6, 0.8, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, and 4.0 s were performed in the presence of increasing concentrations of MycG, with molar ratios of MycG to M-IV of 0, 1:50, 1:25, and 1:10. All NMR experiments were performed at a magnetic field strength of 18.78 tesla (800.13 MHz 1H) on a Bruker Avance spectrometer equipped with a cryoprobe and pulsed-field gradients. NMR data were processed and relaxation times calculated using TopSpin 3.0 (Bruker Biospin). All experiments were performed in triplicate, with reported relaxation rates (R1) the averaged results of the three sets of data.
The paramagnetic contribution R1(para) to the observed 1H R1(obs) relaxation rates were calculated using Mathematica 7 (Wolfram Research) by fitting to Equation 1 (38),
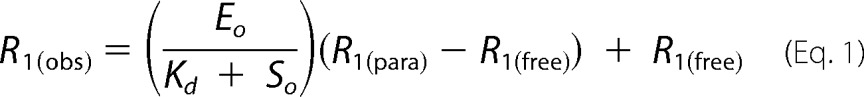 |
where R1(free) is the observed relaxation rate of a given M-IV resonance in the absence of MycG; Eo is concentration of MycG, and So is the concentration of M-IV. A KD value for the M-IV·MycG complex of 700 nm was used for the fit. Goodness of fit was confirmed graphically, with no greater than a 5% deviation from linearity of the plotted data in points used for distance calculations.
Distances r from the heme iron to individual M-IV protons were calculated using the Solomon-Bloembergen Equation 2 (39, 40),
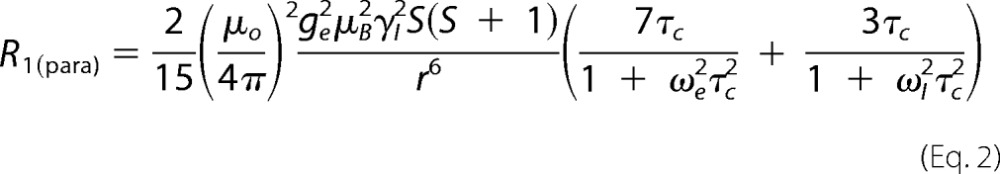 |
where μo is the permeability of free space; μB is the Bohr magneton; ge is the electron g value; γI is the gyromagnetic ratio of the nuclear spin (1H); r6 is the sixth power of the electron nuclear distance; S is the net electronic spin (taken as S = ½); ωe is the electron transition frequency in radial units, and ωI is the 1H frequency. The correlation time τc is assumed to be dominated by the electronic relaxation time, and the value for low spin cytochrome P450cam (τc = 5.4 × 10−11 s) was used (41).
Modeling of the Solution Complex
The crystallographic structure PDB code 2Y98 was used as a starting point for modeling the solution complex of MycG with M-IV. The coordinates of M-IV were divided into three subunits, mycinose, macrolactone, and desosamine, and the antechamber module of AmberTools 1.5 was used to obtain appropriate bonding and atomic charges for each subunit. The XLeap graphical editor interface of AmberTools 1.5 was used to combine the three subunits into a single molecule and to generate the parameter and topology files of the MycG·M-IV complex for Amber 11 (42). Charges were neutralized with potassium ions to obtain overall charge of 0 for the complex. The desosamine dimethylamino group was left unprotonated. The sander module of Amber 11 was used for minimization and molecular dynamics modeling of the complex restrained by the Fe-M-IV distances calculated from relaxation measurements as described above. Simulations were performed in vacuum using a continuum dielectric. SHAKE restraints were applied to all bonds involving hydrogen. Only active site waters were retained as discrete molecules, with a single water molecule constrained as a distal pocket ligand for the heme iron.
A set of 22 distance restraints between the heme iron and M-IV protons was used for modeling the solution complex (see Table 2). A soft potential gradient over a 0.3 Å range was used for distance restraints in the modeling, with the calculated distance defining the midpoint of the potential. No penalty was applied for closer approach to the metal center than the restrained distance.
TABLE 2.
1H resonance assignments for M-IV
Measurements were performed at 298 K in D2O, pH 7.4, with R1(para) relaxation rates extracted using Equation 1 and distance restraints calculated with Equation 2 for modeling of the M-IV complex with MycG. Relaxation rates and distance restraints were calculated using a KD for the complex of 700 nm.
| Proton | δ | R1(para) | Fe-H restraint |
|---|---|---|---|
| ppm | s−1 | Å | |
| 2 | 5.92 | 10.15 | 13.9 |
| 3 | 6.57 | 9.71 | 14.0 |
| 4 | 2.74 | 6.75 | 14.9 |
| 8 | 2.48 | 8.98 | 14.2 |
| 10 | 6.50 | 8.21 | 14.4 |
| 11 | 7.05 | 10.21 | 13.9 |
| 12 | 6.23 | 9.84 | 13.9 |
| 13 | 5.99 | 7.88 | 14.5 |
| 14 | 2.61 | 10.52 | 13.8 |
| 17 | 0.82 | 8.94 | 14.2 |
| 21 | 3.64 | 11.69 | 13.6 |
| 22 | 4.37 | 7.09 | 14.7 |
| 24 | 3.35 | 4.76 | 15.7 |
| 25 | 1.45 | 1.51 | 19.1 |
| 26 | 3.69 | 6.63 | 14.9 |
| 27 | 1.07 | 4.87 | 15.7 |
| 30 | 4.53 | 10.50 | 13.8 |
| 31 | 3.10 | 9.30 | 14.1 |
| 33 | 3.26 | 9.35 | 14.1 |
| 34 | 3.65 | 9.07 | 14.1 |
| 35 | 1.15 | 5.20 | 15.5 |
| 36 | 3.47 | 10.90 | 13.7 |
| 37 | 3.39 | 8.33 | 14.3 |
After 2000 steps of minimization of the starting complex, the temperature of the simulation was increased in the course of 6000 steps (δt = 2 fs) from 0 to 300 K. A production run at 300 K of 10,000 steps with coordinates saved every 100 steps yielded 100 structures, of which 10 showed no restraint violation penalties.
RESULTS
Overall Structure and Substrate Orientations
MycG structures were determined alone (to a resolution of 2.4 Å) and in complex with the natural substrates M-IV and M-V and their biosynthetic precursor M-III to a resolution of 2.0 Å or higher (Table 1). MycG has the typical P450 scaffold that is known to accommodate substrates of broad structural variety. To date, six P450 monooxygenases operating on the macrolactone core of different antibiotics have been structurally characterized bound to their native substrates as follows: EryF (CYP107A1) (43, 44) and EryK (CYP113A1) (45) (both in erythromycin biosynthesis); PikC (CYP107L1; methymycin/neomethymycin and pikromycin biosynthesis) (46–48); PimD (pimaricin biosynthesis) (49); PteC (CYP105P1; filipin biosynthesis) (50, 51), and P450 EpoK (CYP167A; epothilone biosynthesis) (52). Similarly to EryK, MycG operates on a doubly glycosylated macrolactone, with the difference that the glycosylation pattern of erythromycin D, the EryK substrate, features both sugar units attached along the same side of the macrolactone ring opposite the hydroxylation site, whereas in mycinamicins the two sugar units oppose each other on the macrolactone ring, creating a 2-fold rotation ambiguity along the short axis of the elongated mycinamicin molecule (Fig. 3). Mycinose linked to the macrolactone ring adjacently to the sites of oxidation at C-14 and C12–C13 sterically hinders access to these sites, presenting an additional challenge to the catalytically productive positioning of the bulky mycinamicins.
FIGURE 3.

Doubly glycosylated macrolactone substrates of EryK and MycG.
In the crystals, both M-IV and M-V are bound orthogonally to the heme plane in extended “mycinose-in desosamine-out” binding orientation between the α-helical and β-sheet sub-domains. The two methoxy groups of the mycinose moiety contacted the heme macrocycle serving as sensors that probe the macrocycle surface, whereas the second sugar moiety, desosamine, extends toward the protein surface inclined against the short FG-loop with its tertiary amino group (Fig. 4). These protein structures superimpose well with the substrate-free form (represented by the four MycG monomers in the asymmetric unit of 2YGX), suggesting that the substrate-free binding site tends to stay surface-exposed, and that the initial substrate recognition in this site likely occurs rapidly, perhaps as fast as solution diffusion. However, in this orientation substrate reactive sites are further away from the Fe catalytic center than the distances required to interact with iron-oxo or iron-peroxy species. The C-14 hydroxylation site of M-IV is within 8.9 Å and the olefin epoxidation site C12–C13 in M-V is within 10.0 Å of the heme iron (Fig. 4, B and C).
FIGURE 4.
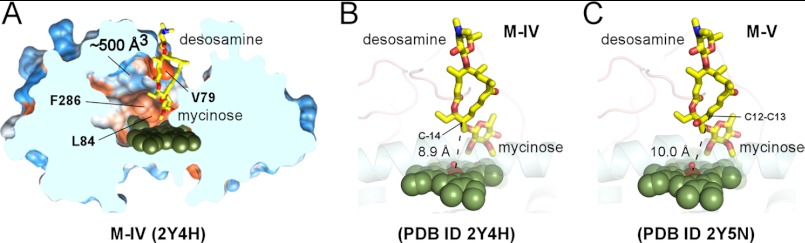
Orthogonal “mycinose-in desosamine-out” substrate-binding mode. A, slice through the bulk solvent-exposed substrate recognition site shows M-IV (yellow sticks) and the protein surface featuring Phe-286, Leu-84, Val-79, and the large void space, including ∼500 Å3 cavity fully enclosed in the protein interior. Hydrophobic areas of the surface are in orange and hydrophilic areas in blue. The M-IV (B) and M-V (C) substrates (yellow sticks) are shown bound orthogonal to the heme plane. Mycinose methoxy group tentacles approach the heme macrocycle at van der Waals distances, preventing access of C-14 and C12–C13 to the iron center. The heme iron axial water ligand is shown as a small red sphere. Both substrates are virtually superimposable. No apparent discrimination between M-IV and M-V occurs in this orientation, suggesting initial recognition rather than catalytic function. In all figures, heme is shown as dark green spheres with the iron atom in brown. Oxygen atoms are shown in red and nitrogen in blue. Images were generated using PyMOL (59) or CHIMERA (60). PDB code codes of the structures are shown in parentheses.
By contrast, two distinct orientations were observed for the biosynthetic precursor M-III. In the first orientation, “javose-in desosamine-out” was similar to the substrate orientation described above, with the qualification that the javose sugar was not defined in the scattered electron density. In the second orientation, “desosamine-in javose-out,” M-III was aligned virtually parallel to the heme with the desosamine sugar accommodated in the buried pocket and the javose extending toward the protein exterior (Fig. 5). Consistent with the poor functional activity of M-III, the edge of the macrolactone ring opposite the modification sites faced the heme macrocycle.
FIGURE 5.

Parallel “desosamine-in javose-out” M-III-binding mode. A, slice through the parallel to the heme-binding site accommodating M-III. B, M-III is well defined by the 2Fo − Fc electron density map contoured at 1.0 σ in the 2YCA structure (blue mesh). Selected side chains within 5 Å of the M-III are shown in gray. The glycerol molecule (cyan) is bound in the vicinity of desosamine, partially filling the void space of the buried cavity.
No major protein conformational differences have been observed between the complexes resolved in this study. Other than the striking behavior of the short Thr-284–Phe-286 fragment, which samples multiple conformations across the range of MycG structures (Fig. 6A), and a few single residues, Val-79, Phe-168, and Tyr-187, all are directly involved in the substrate binding. The iron axial water ligand was present in all the structures, although spectral transition of the heme iron from the low spin to the high spin spin state occurs in solution upon mixing with the substrates (10).
FIGURE 6.

Interactions in the initial recognition complex. M-IV (A), M-V (B and D), and M-III (C) (yellow sticks) are shown fitted to the 2Fo − Fc electron density map (blue mesh) contoured at 1.0 σ. A, superimposition of two alternative conformations for the Thr-284–Phe-286 fragment in the MycG·M-IV complex is shown in dark blue lines. Conformational fluctuations of the Thr-284–Phe-286 fragment correlate with the residual electron density in the Fo − Fc map (pink mesh) contoured at 2.5 σ. B, M-V is shown surrounded by the selected residues (gray sticks) within 5 Å, including the N terminus of the symmetry related molecule (light pink). To enhance clarity, not all amino acid residues are shown. The heme iron axial water ligand is shown as a small red sphere. C, macrolactone and desosamine sugars in M-III are clearly defined in the 2Fo − Fc electron density map (blue mesh) contoured at 0.6 σ, whereas the javose moiety explores the space above the heme macrocycle and thus could not be positioned in the residual Fo − Fc map (pink mesh) contoured at 2.5 σ. D, F286V mutant with M-V bound in two alternative conformations, A and B. Conformation B points toward the hydrophobic cavity gated by residue 286.
M-IV and M-V Orthogonal Mycinose-in Desosamine-out Binding Mode
A prominent feature of MycG is its spacious active site, which is reminiscent of certain xenobiotic metabolizing human P450 enzymes with complex substrate-binding patterns (reviewed in Ref. 53). A large void volume of ∼1500 Å3 partially filled with structured water molecules is adjacent to the space occupied by the substrate (Fig. 4A). In the orthogonal mode, both M-IV and M-V, in addition to mycinose-mediated contacts with the heme macrocycle, make well defined multiple interactions within <5 Å of the amino acid residues grouped in the four protein regions including the following: BC-loop: Arg-75, Glu-77, and Val-79–Ser-85; the FG-loop: Phe-168–Ser-170, Ala-172, Val-174–Ala-176, Glu-178, Met-179, and Ala-183; the I-helix: Gly-230, Val-233, Ala-234, Glu-237, and Ser-238; the turn of the C-terminal β-sheet: Leu-386 and Leu-387 (Fig. 6B); and finally, the stand-alone Phe-286, which deeply protrudes into the void volume in the substrate binding site. The adjacent residues, Thr-284 and Ala-285, are conformationally variable, whereas no alternative conformations were apparent for Phe-286 (Fig. 6A), perhaps due to its extensive contacts with Leu-84 and the stabilizing influence of the salt bridge formed between the downstream Arg-288 and the heme propionate side chain. Conformational variability of the Thr-284–Ala-285 fragment correlates with the presence of scattered electron density in the vicinity of heme, the latter suggesting ambiguity in substrate binding. Only in the chain A of the MycG·M-V complex (PDB code 2Y5N) was no ambiguity observed. However, the N terminus of the symmetry-related chain B interacts with the substrate in this structure and thus may contribute to stabilization of the complex (Fig. 6B).
Analyses of crystals of different space groups, including those having one, two, three, or four molecules in the asymmetric unit (Table 1), largely excluded the possibility of crystal packing influence on the substrate binding orientation. Despite the different crystal packing contacts, all the MycG molecules bound substrates in the same orientation indicating that the observed orthogonal binding mode in the surface-exposed site is not an artifact of crystallization but rather represents an essential step of initial recognition in the substrate binding process.
M-III Orthogonal Binding Mode
The mycinose sugar has been shown to be essential for MycG function, as only minor conversion to hydroxylated product occurred in reconstituted assays in vitro when M-IV was substituted by its biosynthetic precursor M-III (10, 28) or in vivo by the mycF-disrupted deletion mutant of M. griseorubida (Fig. 1) (10, 28). Unlike physiological substrates, M-III was found in two distinct, virtually orthogonal orientations in the 2Y5Z and 2YCA structures. In 2Y5Z, the macrolactone and desosamine sugar of M-III were unambiguously localized in exactly the same place as in the MycG·substrate complexes described above, with the qualification that their occupancies were set to lower values to satisfy weaker electron density. However, the position of the monomethoxylated javose moiety in this complex could not be defined in any of the three molecules in the asymmetric unit due to noninterpretable electron density (Fig. 6C). The flexible dihedral angles that include the covalent linkage of javose to the macrolactone and the insufficient spatial constraints due to the lack of interactions between the missing methoxy group and the heme macrocycle apparently result in unrestricted rotation of javose as judged by the shapeless patches of electron density in the vicinity of heme. The atomic coordinates for javose were not used in the refinement and are not present in the PDB entry 2Y5Z. This finding indicates that the interactions of both methoxy groups of mycinose with the heme macrocycle are key to formation of the initial recognition complex in the surface-exposed site.
M-III Parallel Desosamine-in Javose-out Binding Mode
Unlike that of 2Y5Z, the electron density of the 2YCA structure clearly delineates the whole M-III molecule. In a single MycG monomer constituting an asymmetric unit, M-III is aligned parallel to the heme macrocycle making different points of contact with the active site (Fig. 5B). The desosamine sugar is accommodated in the buried pocket adjacent to the heme macrocycle and is held in position largely by both the main chain atoms of the Leu-280–Phe-286 fragment (which in this complex adopts a single well defined conformation) and the amino acid side chains of Leu-280, Gly-281, Thr-284, Ala-285, Phe-286, Thr-311, Gly-338, Leu-386, and Leu-387. As a result of M-III binding, the Phe-286 aromatic ring rotates away from Leu-84 and is equidistant between Leu-84 and Val-79, whereas Val-79 shifts ∼2.5 Å toward the aromatic ring. Being deeper than required to bind desosamine alone, the cavity also accommodates a glycerol molecule (cyan in Fig. 5B). By contrast, the javose moiety extends toward the protein exterior and interacts with the side chains of Gly-81, Gly-82, Tyr-187, Asp-226, Ile-229, Gly-230, and Val-233. Consistent with the poor functional activity of M-III, the reactive centers C-14 or C12–C13 are not exposed to the heme iron in 2YCA structure.
Role of Phe-286 in Substrate Binding
To explore a role of Phe-286 in substrate stabilization in the orthogonal mycinose-in desosamine-out binding mode, we generated a series of the Phe-286 mutants by substituting phenylalanine with leucine, valine, alanine, or glycine. None of the mutations abolished MycG catalytic function, but the catalytic activity of mutants declined gradually with a decrease of the substituent size, from phenylalanine to glycine (data not shown). Co-crystal structures were determined for the Phe-286 to glycine or alanine mutants with M-IV, and F286V mutant co-crystallized with M-V. In the alanine or glycine mutants, M-IV was again found in the orthogonal to the heme binding orientation as represented by the PDB code 3ZSN (Table 1).
By contrast, M-V, also orthogonally oriented in the F286V mutant (PDB code 4AW3), revealed a second alternative conformation for the mycinose sugar (conformation B in Fig. 6D). In this new conformation, mycinose has swung toward Val-286, thus pointing to the buried cavity previously shown to accommodate desosamine of M-III in the parallel binding mode. Assuming that M-V would advance further in this direction to enter the cavity, its macrolactone portion could turn to adopt an orientation parallel to the heme, exposing the reactive C-H and the double bond to the iron center.
Modeling of the Solution Complex and Comparison with Crystallographic Results
Longitudinal (R1) relaxation rates of the assigned 1H resonances of M-IV in the presence of sub-stoichiometric amounts of oxidized MycG can be interpreted in terms of relative distances between the paramagnetic center (the heme iron) and the individual protons of M-IV. There are a number of approximations involved in such calculations, perhaps the most important being the assumption that the unpaired electron spin is localized on the heme iron. Second, the calculated paramagnetic contribution R1(para) to the observed relaxation rate depends upon the KD value of the enzyme·substrate complex, so the accuracy of the calculated distances depend in turn upon the accuracy of the KD value used. Nevertheless, even the availability of relative distances is valuable, as these restraints can provide evidence of the preferred orientation of the substrate when bound in the active site of the enzyme.
Table 2 provides 1H resonance assignments, R1(para) extracted from titration data and calculated distances from the heme iron that were used as modeling restraints. Fig. 7 highlights the positions of M-IV 1H resonances that show the greatest sensitivity to paramagnetic effects on R1. Qualitatively, 1H relaxation patterns indicate that in the presence of excess substrate, the preferred binding orientation of M-IV places the mycinose and macrolide ring closer than the desosamine to the heme iron (mycinose-in desosamine-out orientation). Also, the orientation of the macrolactone places the sites of hydroxylation (C-14) closest to the heme. Protons on the desosamine ring were either unaffected or only modestly affected by the metal center (see Table 2).
FIGURE 7.

Structural distribution of maximum contributions of paramagnetism to R1 relaxation in M-IV. A, proton numbering scheme. Proton numbers refer to the carbon atoms to which the proton is bound. B, protons (dark red) that are affected so as to yield calculated restraint distances <14 Å from the heme iron are highlighted with dotted dark red spheres.
A comparison between the crystallographic structure PDB code 2Y98 (green) and a structure resulting from NMR modeling using paramagnetic restraints (blue) is shown in Fig. 8. Fig. 8A shows superimposition of the backbones of both structures, whereas Fig. 8B highlights differences in the position of M-IV in the two structures. The paramagnetic restraint model was found to have no violations of distance restraints and provides a plausible model for the solution complex that rationalizes the observed pattern of paramagnetic effects on M-IV. The deeper penetration of M-IV into the active site required a number of secondary structural features to be displaced (Fig. 8A). The largest displacements are to residues in the B helix and B-C loop (residues 63–88) and the F-G loop (residues 169–176), the most inherently flexible P450 regions, with smaller displacements of the F and G helices capping the active site and the β3 sheet. These displacements enlarge the active site sufficiently so that mycinose moves laterally in the active site toward the B-C loop, away from the buried cavity gated by Phe-286. This serves to draw the macrolactone ring (including the C-14 proton) considerably closer to the heme iron in the solution model compared with the crystallographic structure.
FIGURE 8.

X-ray structure compared with the relaxation-derived NMR model. A, superposition of crystallographic structure 2Y98 (in green) with the NMR model (in blue) that shows no violations of relaxation-based distance restraints. The largest differences are seen in the B helix, the β3-sheet (including residues 284–286), and the B-C and F-G loops. Heme is in light pink sticks; M-IV is in magenta light lines. B, expansion of superposition displayed in A showing repositioning of the M-IV NMR model (green, heavy lines) relative to the x-ray structure (magenta light lines). Also shown are the positions of Phe-286 side chains, the B-C loop (residues 72–86), and the heme (magenta light lines). Distances shown (8.9 Å in 2Y98 and 6.8 Å in NMR model) are from the heme iron atom to C-14, the site of hydroxylation on M-IV. This corresponds to a distance of 6.25 Å in 2Y89 and 4.15 Å in the NMR model between the distal iron ligand water oxygen (not shown) and the abstracted hydrogen at C-14. See text for further details.
DISCUSSION
Specificity in P450 catalysis is dictated by the local chemical environment of the enzyme active site, which results in precise positioning of the substrate with respect to the iron catalytic center. Within bonding distances of the activated oxygen species, steric and stereoelectronic factors, rather than chemical reactivity of the reaction sites, determine regio- and stereo-selectivity of the oxidative reactions (49, 54, 55). To determine whether the MycG substrates consecutively bind in two discrete positions while progressing through the hydroxylation/epoxidation cascade, the binding modes of M-IV and M-V and their biosynthetic precursor M-III were extensively explored in this study using the x-ray and NMR techniques. A complex recognition pattern has emerged from this analysis.
The orthogonal mycinose-in desosamine-out substrate-binding mode consistently observed in multiple co-structures reported in this work could represent a preliminary step en route to a catalytically productive orientation. In favor of this hypothesis is the large void volume in the active site, the conformational ambiguity of the Thr-284–Ala-285 fragment and the patches of scattered electron density that suggest the possibility of alternative substrate-binding modes. Toward this end, the binding behavior of the biosynthetic precursor M-III demonstrates a very different desosamine-in javose-out orientation that is parallel to the heme co-factor. Finally, the NMR relaxation data indicate that in solution M-IV may penetrate the active site sufficiently to place the abstracted hydrogen atom at C-14 within 6 Å of the heme iron. However, the paramagnetic relaxation model did not reveal the basis for epoxidation across the C12–C13 double bond.
As evidenced by the MycG-M-III structures, javose is less preferable than mycinose as an initial recognition marker. The unproductive desosamine-in javose-out orientation of M-III likely results from the failure of javose to establish effective interactions with the heme. For substrate to reach a catalytically productive mode, mycinose instead of desosamine should lead the way, which is consistent both with the x-ray structures and the relaxation behavior of M-IV substrate in solution. This is in turn consistent with desosamine being predominantly distal to the heme iron in the M-IV complex.
Rapid binding of substrate in the surface-exposed recognition site apparently is followed by its translocation to the catalytically competent orientation. Attainment of the latter state is likely coupled with conformational fluctuations of the Thr-284–Phe-286 region, which demonstrates conformational ambiguity across the structures and may depend on Phe-286 due to its central location and slower rate of fluctuation compared with adjacent Thr-284–Ala-285. Behavior of the Phe-286 mutants indicates that neither additional space in the active site introduced by the Phe-286 substitutions to leucine, valine, or alanine nor protein flexibility gained in F286G mutant facilitated M-IV relocation deeper in the catalytic pocket.
By contrast, mycinose in M-V has swung toward Val-286 in the F286V mutant, in the direction opposite to the one predicted by the proton relaxation NMR model (Fig. 6D). This suggests the possibility of translocation to the void space of the buried cavity. Thus, opening access to the buried space might be a necessary but not sufficient step en route to the epoxidation-compatible binding mode that would place the double bond parallel with respect to the heme iron. The possibility of substrate relocation in two different directions assumed by the NMR model and by the co-structure of the F286V mutant might provide a plausible explanation for both catalytic functions of MycG.
Interestingly, work in our laboratories has recently shown that removal of substrate from CYP101A1 results in a significant inward displacement of the β3 sheet, which is structurally homologous to the Phe-286 region in MycG (56). The current modeling results suggest that the same region is displaced out of the active site to accommodate the substrate in an orientation conducive to the observed chemistry. However, it is likely that further reorganization, perhaps driven by the binding of a redox partner as is the case with CYP101A1 (57), is required to reach the catalytically competent conformation of the enzyme. Change in the heme redox state may be another plausible factor gating access to a catalytically productive orientation.
Based on the combined efforts of crystallography and the paramagnetic NMR relaxation data described in this report, the hypothesis of sequential translocation of the substrate molecule from the recognition site to the catalytic site has emerged. We interpret this translocation as a synergism between conformational changes of MycG and limited flexibility of the substrate molecule. Mycinose rotation around the bond connecting it to the macrolactone appears significant for sampling conformations that eventually would enable the substrate molecule to translocate to the catalytic site. Transition of the sugar moiety between the transient and catalytic binding pockets resembles in principle the mechanism previously observed for PikC substrates (47). In that system a single D50N mutation in PikC releases electrostatic interactions in the transient site and facilitates relocation of the desosamine sugar moiety to the buried catalytic pocket. This observation has found practical application in a PikCD50N mutant fused to a surrogate electron transporter (58), which has been developed as a highly effective C–H bond activation catalyst for hydroxylation of diverse macrolide and carbolide substrates with antibiotic properties (48). Based on our knowledge of PikC, AurH, and now MycG, it would not be surprising if a multistep progression to the catalytically productive mode turns out to be a common feature of the bulky and conformationally restrained substrates such as glycosylated macrolactones. Evidence for multistep substrate binding has also been suggested for other P450 enzymes, including drug-metabolizing P450s from human liver, that feature large substrate-binding sites (reviewed in Ref. 53). In cases where substrate translocation is the limiting step, protein engineering strategies could be applied to facilitate substrate progression to the active site and thus enhance P450 catalysis.
Acknowledgments
We thank Potter Wickware for critical reading of the manuscript and the staff members James Holton, George Meigs, and Jane Tanamachi of beamline 8.3.1 at the Advanced Light Source, Lawrence Berkeley National Laboratory, for assistance with data collection. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the United States Department of Energy under Contract DE-AC02-05CH11231. Molecular graphics images were produced in part using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by National Institutes of Health Grant P41 RR001081).
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 GM078553 (to D. H. S. and L. M. P.) and GM44191 (to T. C. P.). This work was also supported by the Hans W. Vahlteich Professorship (to D. H. S.).
- M-IV
- mycinamicin IV
- M-III
- mycinamicin III
- M-V
- mycinamicin V
- PDB
- Protein Data Bank.
REFERENCES
- 1. Satoi S., Muto N., Hayashi M., Fujii T., Otani M. (1980) Mycinamicins, new macrolide antibiotics. I. Taxonomy, production, isolation, characterization, and properties. J. Antibiot. 33, 364–376 [DOI] [PubMed] [Google Scholar]
- 2. Takenaka S., Yoshida K., Yamaguchi O., Shimizu K., Morohoshi T., Kinoshita K. (1998) Enhancement of mycinamicin production by dotriacolide in Micromonospora griseorubida. FEMS Microbiol. Lett. 167, 95–100 [DOI] [PubMed] [Google Scholar]
- 3. Kinoshita K., Satoi S., Hayashi M., Harada K., Suzuki M., Nakatsu K. (1985) Mycinamicins, new macrolide antibiotics. VIII. Chemical degradation and absolute configuration of mycinamicins. J. Antibiot. 38, 522–526 [DOI] [PubMed] [Google Scholar]
- 4. Kinoshita K., Imura Y., Takenaka S., Hayashi M. (1989) Mycinamicins, new macrolide antibiotics. XI. Isolation and structure elucidation of a key intermediate in the biosynthesis of the mycinamicins, mycinamicin VIII. J. Antibiot. 42, 1869–1872 [DOI] [PubMed] [Google Scholar]
- 5. Kinoshita K., Satoi S., Hayashi M., Nakatsu K. (1989) Mycinamicins, new macrolide antibiotics. X. X-ray crystallography and the absolute configuration of mycinamicin IV. J. Antibiot. 42, 1003–1005 [DOI] [PubMed] [Google Scholar]
- 6. Suzuki H., Takenaka S., Kinoshita K., Morohoshi T. (1990) Biosynthesis of mycinamicins by a blocked mutant of Micromonospora griseorubida. J. Antibiot. 43, 1508–1511 [DOI] [PubMed] [Google Scholar]
- 7. Kinoshita K., Takenaka S., Suzuki H., Morohoshi T., Hayashi M. (1992) Mycinamicins, new macrolide antibiotics. XIII. Isolation and structures of novel fermentation products from Micromonospora griseorubida (FERM BP-705). J. Antibiot. 45, 1–9 [DOI] [PubMed] [Google Scholar]
- 8. Inouye M., Takada Y., Muto N., Beppu T., Horinouchi S. (1994) Characterization and expression of a P-450-like mycinamicin biosynthesis gene using a novel Micromonospora-Escherichia coli shuttle cosmid vector. Mol. Gen. Genet. 245, 456–464 [DOI] [PubMed] [Google Scholar]
- 9. Anzai Y., Saito N., Tanaka M., Kinoshita K., Koyama Y., Kato F. (2003) Organization of the biosynthetic gene cluster for the polyketide macrolide mycinamicin in Micromonospora griseorubida. FEMS Microbiol. Lett. 218, 135–141 [DOI] [PubMed] [Google Scholar]
- 10. Anzai Y., Li S., Chaulagain M. R., Kinoshita K., Kato F., Montgomery J., Sherman D. H. (2008) Functional analysis of MycCI and MycG, cytochrome P450 enzymes involved in biosynthesis of mycinamicin macrolide antibiotics. Chem. Biol. 15, 950–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kudo F., Motegi A., Mizoue K., Eguchi T. (2010) Cloning and characterization of the biosynthetic gene cluster of 16-membered macrolide antibiotic FD-891. Involvement of a dual functional cytochrome P450 monooxygenase catalyzing epoxidation and hydroxylation. ChemBioChem 11, 1574–1582 [DOI] [PubMed] [Google Scholar]
- 12. Carlson J. C., Li S., Burr D. A., Sherman D. H. (2009) Isolation and characterization of tirandamycins from a marine-derived Streptomyces sp. J. Nat. Prod. 72, 2076–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carlson J. C., Fortman J. L., Anzai Y., Li S., Burr D. A., Sherman D. H. (2010) Identification of the tirandamycin biosynthetic gene cluster from Streptomyces sp. 307-9. ChemBioChem. 11, 564–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carlson J. C., Li S., Gunatilleke S. S., Anzai Y., Burr D. A., Podust L. M., Sherman D. H. (2011) Tirandamycin biosynthesis is mediated by co-dependent oxidative enzymes. Nat. Chem. 3, 628–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zocher G., Richter M. E., Mueller U., Hertweck C. (2011) Structural fine-tuning of a multifunctional cytochrome P450 monooxygenase. J. Am. Chem. Soc. 133, 2292–2302 [DOI] [PubMed] [Google Scholar]
- 16. He J., Hertweck C. (2003) Iteration as programmed event during polyketide assembly; molecular analysis of the aureothin biosynthesis gene cluster. Chem. Biol. 10, 1225–1232 [DOI] [PubMed] [Google Scholar]
- 17. Müller M., He J., Hertweck C. (2006) Dissection of the late steps in aureothin biosynthesis. ChemBioChem 7, 37–39 [DOI] [PubMed] [Google Scholar]
- 18. Podust L. M., Sherman D. H. (2012) Diversity of P450 enzymes in the biosynthesis of natural products. Nat. Prod. Rep. 29, 1251–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crešnar B., Petrič S. (2011) Cytochrome P450 enzymes in the fungal kingdom. Biochim. Biophys. Acta 1814, 29–35 [DOI] [PubMed] [Google Scholar]
- 20. Tokai T., Koshino H., Takahashi-Ando N., Sato M., Fujimura M., Kimura M. (2007) Fusarium Tri4 encodes a key multifunctional cytochrome P450 monooxygenase for four consecutive oxygenation steps in trichothecene biosynthesis. Biochem. Biophys. Res. Commun. 353, 412–417 [DOI] [PubMed] [Google Scholar]
- 21. Seo J. A., Proctor R. H., Plattner R. D. (2001) Characterization of four clustered and coregulated genes associated with fumonisin biosynthesis in Fusarium verticillioides. Fungal Genet. Biol. 34, 155–165 [DOI] [PubMed] [Google Scholar]
- 22. Bojja R. S., Cerny R. L., Proctor R. H., Du L. (2004) Determining the biosynthetic sequence in the early steps of the fumonisin pathway by use of three gene-disruption mutants of Fusarium verticillioides. J. Agric. Food Chem. 52, 2855–2860 [DOI] [PubMed] [Google Scholar]
- 23. Proctor R. H., Plattner R. D., Desjardins A. E., Busman M., Butchko R. A. (2006) Fumonisin production in the maize pathogen Fusarium verticillioides. Genetic basis of naturally occurring chemical variation. J. Agric. Food Chem. 54, 2424–2430 [DOI] [PubMed] [Google Scholar]
- 24. Tudzynski B., Rojas M. C., Gaskin P., Hedden P. (2002) The gibberellin 20-oxidase of Gibberella fujikuroi is a multifunctional monooxygenase. J. Biol. Chem. 277, 21246–21253 [DOI] [PubMed] [Google Scholar]
- 25. Tudzynski B., Hedden P., Carrera E., Gaskin P. (2001) The P450-4 gene of Gibberella fujikuroi encodes ent-kaurene oxidase in the gibberellin biosynthesis pathway. Appl. Environ. Microbiol. 67, 3514–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rojas M. C., Hedden P., Gaskin P., Tudzynski B. (2001) The P450-1 gene of Gibberella fujikuroi encodes a multifunctional enzyme in gibberellin biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 98, 5838–5843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsukada S., Anzai Y., Li S., Kinoshita K., Sherman D. H., Kato F. (2010) Gene targeting for O-methyltransferase genes, mycE and mycF, on the chromosome of Micromonospora griseorubida producing mycinamicin with a disruption cassette containing the bacteriophage phi C31 attB attachment site. FEMS Microbiol. Lett. 304, 148–156 [DOI] [PubMed] [Google Scholar]
- 28. Anzai Y., Tsukada S., Sakai A., Masuda R., Harada C., Domeki A., Li S., Kinoshita K., Sherman D. H., Kato F. (2012) Function of cytochrome P450 enzymes MycCI and MycG in Micromonospora griseorubida, a producer of the macrolide antibiotic mycinamicin. Antimicrob. Agents Chemother. 56, 3648–3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. von Kries J. P., Warrier T., Podust L. M. (2010) Identification of small molecule scaffolds for P450 inhibitors. Curr. Protoc. Microbiol. 16, 17.4.1–17.4.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Omura T., Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J. Biol. Chem. 239, 2379–2385 [PubMed] [Google Scholar]
- 31. Leslie A. G. (1992) Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 ESF-EAMCB Newslett. Protein Crystallogr. No. 26 [Google Scholar]
- 32. Holton J., Alber T. (2004) Automated protein crystal structure determination using ELVES. Proc. Natl. Acad. Sci. U.S.A. 101, 1537–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yasutake Y., Imoto N., Fujii Y., Fujii T., Arisawa A., Tamura T. (2007) Crystal structure of cytochrome P450 MoxA from Nonomuraea recticatena (CYP105). Biochem. Biophys. Res. Commun. 361, 876–882 [DOI] [PubMed] [Google Scholar]
- 34. Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 35. Collaborative Computational Project No. 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 36. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 37. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 38. Modi S., Primrose W. U., Boyle J. M., Gibson C. F., Lian L. Y., Roberts G. C. (1995) NMR studies of substrate binding to cytochrome P450 BM3. Comparisons to cytochrome P450cam. Biochemistry 34, 8982–8988 [DOI] [PubMed] [Google Scholar]
- 39. Bloembergen N. (1957) Proton relaxation times in paramagnetic solutions. J. Chem. Phys. 27, 572–573 [Google Scholar]
- 40. Solomon I. (1955) Relaxation processes in a system of 2 spins. Phys. Rev. 99, 559–565 [Google Scholar]
- 41. Philson S. B., Debrunner P. G., Schmidt P. G., Gunsalus I. C. (1979) The effect of cytochrome P-450cam on the NMR relaxation rate of water protons. J. Biol. Chem. 254, 10173–10179 [PubMed] [Google Scholar]
- 42. Case D. A., Darden T. A., Cheatham T. E., III, Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B. P., Wang B., Hayik S., Roitberg A., Seabra G., Kolossváry I., Wong K. F., Paesani F., Vanicek J., Liu J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M. J., Cui G., Roe D. R., Mathews D. H., Seetin M. G., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A., Kollman P. A. (2010) AMBER 11. University of California, San Francisco [Google Scholar]
- 43. Cupp-Vickery J. R., Poulos T. L. (1995) Structure of cytochrome P450eryF involved in erythromycin biosynthesis. Nat. Struct. Biol. 2, 144–153 [DOI] [PubMed] [Google Scholar]
- 44. Cupp-Vickery J. R., Han O., Hutchinson C. R., Poulos T. L. (1996) Substrate-assisted catalysis in cytochrome P450 eryF. Nat. Struct. Biol. 3, 632–637 [DOI] [PubMed] [Google Scholar]
- 45. Savino C., Montemiglio L. C., Sciara G., Miele A. E., Kendrew S. G., Jemth P., Gianni S., Vallone B. (2009) Investigating the structural plasticity of a cytochrome P450. Three-dimensional structures of P450 EryK and binding to its physiological substrate. J. Biol. Chem. 284, 29170–29179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sherman D. H., Li S., Yermalitskaya L. V., Kim Y., Smith J. A., Waterman M. R., Podust L. M. (2006) The structural basis for substrate anchoring, active site selectivity, and product formation by P450 PikC from Streptomyces venezuelae. J. Biol. Chem. 281, 26289–26297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li S., Ouellet H., Sherman D. H., Podust L. M. (2009) Analysis of transient and catalytic desosamine-binding pockets in cytochrome P-450 PikC from Streptomyces venezuelae. J. Biol. Chem. 284, 5723–5730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li S., Chaulagain M. R., Knauff A. R., Podust L. M., Montgomery J., Sherman D. H. (2009) Selective oxidation of carbolide C–H bonds by an engineered macrolide P450 mono-oxygenase. Proc. Natl. Acad. Sci. U.S.A. 106, 18463–18468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kells P. M., Ouellet H., Santos-Aberturas J., Aparicio J. F., Podust L. M. (2010) Structure of cytochrome P450 PimD suggests epoxidation of the polyene macrolide pimaricin occurs via a hydroperoxoferric intermediate. Chem. Biol. 17, 841–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu L. H., Fushinobu S., Ikeda H., Wakagi T., Shoun H. (2009) Crystal structures of cytochrome P450 105P1 from Streptomyces avermitilis. Conformational flexibility and histidine ligation state. J. Bacteriol. 191, 1211–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu L. H., Fushinobu S., Takamatsu S., Wakagi T., Ikeda H., Shoun H. (2010) Regio- and stereospecificity of filipin hydroxylation sites revealed by crystal structures of cytochrome P450 105P1 and 105D6 from Streptomyces avermitilis. J. Biol. Chem. 285, 16844–16853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nagano S., Li H., Shimizu H., Nishida C., Ogura H., Ortiz de Montellano P. R., Poulos T. L. (2003) Crystal structures of epothilone D-bound, epothilone B-bound, and substrate-free forms of cytochrome P450epoK. J. Biol. Chem. 278, 44886–44893 [DOI] [PubMed] [Google Scholar]
- 53. Isin E. M., Guengerich F. P. (2008) Substrate binding to cytochromes P450. Anal. Bioanal. Chem. 392, 1019–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Johnston J. B., Ouellet H., Podust L. M., Ortiz de Montellano P. R. (2011) Structural control of cytochrome P450-catalyzed ω-hydroxylation. Arch. Biochem. Biophys. 507, 86–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen M. S., White M. C. (2010) Combined effects on selectivity in Fe-catalyzed methylene oxidation. Science 327, 566–571 [DOI] [PubMed] [Google Scholar]
- 56. Asciutto E. K., Young M. J., Madura J., Pochapsky S. S., Pochapsky T. C. (2012) Solution structural ensembles of substrate-free cytochrome P450(cam). Biochemistry 51, 3383–3393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wei J. Y., Pochapsky T. C., Pochapsky S. S. (2005) Detection of a high barrier conformational change in the active site of cytochrome P450cam upon binding of putidaredoxin. J. Am. Chem. Soc. 127, 6974–6976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li S., Podust L. M., Sherman D. H. (2007) In vitro characterization of a self-sufficient biosynthetic cytochrome P450 PikC fused to a heterologous reductase domain RhFRED. J. Am. Chem. Soc. 129, 12940–12941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
- 60. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]