

Background: PIAS proteins are implicated in the regulation of many transcription factors through distinct mechanisms. It remains largely unknown whether PIAS proteins exert metabolic actions.
Results: PIAS1 repressed LXR-dependent up-regulation of lipogenic genes in a SUMOylation-independent manner.
Conclusion: PIAS1 could act as a lipogenic regulator by negatively modulating LXRs.
Significance: These findings reveal a regulatory role for PIAS proteins in lipid metabolism.
Keywords: Fatty Acid Metabolism, Gene Regulation, Lipid Metabolism, Lipogenesis, Sumoylation, Liver X Receptor, Peroxisome Proliferator-activated Receptor γ, Co-activator 1β, Protein Inhibitor of Activated STAT
Abstract
Liver X receptors (LXRs) are nuclear receptors that function to modulate lipid metabolism as well as immune and inflammatory responses. Upon activation by their ligands, LXRs up-regulate a spectrum of gene transcription programs involved in cholesterol and fatty acid homeostasis. However, the mechanisms by which LXR-mediated transcriptional activation is regulated remain incompletely understood. Here, we show that PIAS1, a member of the protein inhibitor of the activated STAT family of proteins with small ubiquitin-like modifier (SUMO) E3 ligase activity, acts to suppress LXR ligand-dependent transcriptional activation of the lipogenic program in hepatocytes. We found that liver mRNA expression levels of Pias1 and Pias3 were inversely associated with those of genes involved in lipogenesis in mouse models with diet-induced or genetic obesity. Overexpression of PIAS1 in primary hepatocytes resulted in a reduction of LXR ligand-induced fatty acid synthesis and suppression of the expression of lipogenic genes, including Srebp1c and Fas. Moreover, PIAS1 was able to interact with LXRβ and repress its transcriptional activity upon ligand stimulation, which did not require PIAS1-promoted SUMO modification of LXRβ. In addition, PIAS1 could also interact with PGC-1β and attenuate its association with LXRβ, blunting the ability of PGC-1β to co-activate LXRβ. Importantly, PIAS1 impaired LXRβ binding to its target DNA sequence. Taken together, our results suggest that PIAS1 may serve as a lipogenic regulator by negatively modulating LXRs in a SUMOylation-independent manner.
Introduction
Liver X receptor (LXR)3-α and LXRβ are nuclear receptors that play key regulatory roles in lipid metabolism (1, 2). Both LXRs are widely expressed in multiple tissues and cell types, with LXRα most highly expressed in liver and intestine (3, 4). LXRs were originally viewed as “orphan” nuclear receptors (5, 6) and have been established to function as nuclear cholesterol sensors because oxysterols were identified as their physiological ligands (6, 7). As ligand-dependent transcription factors, LXRs can form permissive heterodimers with retinoid X receptor and bind to LXR-responsive elements (LXREs) within their target genes (8). Ligand binding of LXR results in the replacement of co-repressors by co-activators, leading to enhanced activation of gene transcription (9). Activated in response to increased intracellular cholesterol levels, LXRs act to induce specific gene expression programs to control various aspects of whole-body cholesterol homeostasis (1, 2, 10–12). LXRs are also currently known as critical regulators of hepatic lipogenesis (1, 2), which can increase hepatic lipogenesis through transcriptional up-regulation of sterol regulatory element-binding protein 1c (SREBP1c), the master lipogenic regulator (13). LXRs were shown to directly bind to two functional LXREs in the promoter region of the Srebp1c gene to increase its expression (13, 14). LXRs have also been shown to regulate key enzymes involved in fatty acid biosynthesis, including fatty-acid synthase (FAS), acetyl-CoA carboxylase (ACC), and stearoyl-CoA desaturase 1 (SCD1) (14–16). In addition, recent studies demonstrated the carbohydrate-response element-binding protein as another LXR target that also mediates LXR-dependent up-regulation of select lipogenic genes (17). In the settings of obesity-associated metabolic disorders, however, it remains to be understood whether LXRs are dysregulated and mechanistically linked to the derangement of lipid metabolism.
An accumulating body of evidence has revealed that like other nuclear receptor family members such as peroxisome proliferator-activated receptor γ (PPARγ), LXRs are able to integrate both metabolic and inflammatory signaling (2, 18). Although up-regulating genes that are involved in reverse cholesterol transport, both LXR isoforms possess anti-inflammatory activity via repressing inflammatory genes in macrophages (19). Because the promoters of these repressed genes do not contain LXREs, an indirect inhibitory mechanism was proposed to mediate the transrepressing actions of LXRs (2). Recently reported studies suggest that LXRs and PPARγ can repress overlapping but distinct subsets of proinflammatory genes by a mechanism that is dependent on small ubiquitin-like modifier (SUMO) conjugation (SUMOylation) of these nuclear receptors (20). Additionally, Lee et al. (21) demonstrated that in IFN-γ-stimulated brain astrocytes, both synthetic and oxysterol derivatives of LXR ligands could suppress STAT1-dependent inflammatory responses; furthermore, this action requires SUMOylation of LXRβ and LXRα, which were catalyzed by two SUMO E3 ligase family members, the protein inhibitor of activated STAT1 (PIAS1) and histone diacetylase 4 (HDAC4), respectively.
Mammalian PIAS family proteins were initially characterized as inhibitors of STAT-mediated signaling (22, 23), which consist of PIAS1, PIAS3, PIASx, and PIASy (24). PIAS proteins have recently been shown to have SUMO E3 ligase activity (25, 26) and are implicated in regulation of many transcription factors by distinct mechanisms such as promoting protein SUMOylation, blocking the DNA binding ability of transcription factors, or recruiting transcriptional co-repressors or co-activators (24, 27). Although they are well documented to play crucial roles in the regulation of gene activation pathways in inflammation and immunity, it is largely unclear whether PIAS proteins can exert metabolic functions by modulating gene transcription programs involved in lipid homeostasis. Here, we report the association of the expression of two PIAS family members, Pias1 and Pias3, with that of lipogenic genes in livers of diet-induced or genetic mouse models of obesity. We also found a repressive effect of PIAS1 upon LXR ligand-stimulated up-regulation of lipogenic genes and attempted to decipher the molecular basis of PIAS1 actions in the LXR-dependent control of the lipogenic program.
EXPERIMENTAL PROCEDURES
Animals
Male wild-type C57BL/6J mice were purchased from Shanghai Laboratory Animal Co. Ltd. Male C57BL/6J db/db mice were from Model Animal Research Center, Nanjing University, and male C57BL/6J ob/ob mice were from National Center for Drug Screening, Shanghai Institute of Materia Medica, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. Animals were maintained in laboratory cages at a temperature of 23 ± 3 °C and a humidity of 35 ± 5% under a 12-h dark/light cycle with free access to standard chow (Shanghai Laboratory Animals Co.) and water in accredited animal facilities at Shanghai Institutes for Biological Sciences. For diet-induced obesity, male wild-type C57BL/6J mice were fed ad libitum a low fat (LFD, 10 kcal % fat) or a high fat diet (HFD, 60 kcal % fat) (Research Diets Inc.) as described previously (28). Animals were sacrificed under anesthetic conditions, and livers were snap-frozen in liquid nitrogen immediately after resection and stored at −80 °C. All experimental protocols were approved by the Institutional Animal Care and Use Committee at the Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences.
Plasmid Constructs for Protein Expression and Luciferase Assays
For the expression of Myc-tagged LXRs, cDNA fragments encoding mouse LXRα or LXRβ were obtained by RT-PCR and subcloned into pcDNA6-His/Myc (Invitrogen) using the KpnI/XbaI sites. To express the truncated versions of mouse LXRβ protein, LXRβ-DBD or LXRβ-LBD, cDNA segments encoding the desired regions of LXRβ were likewise subcloned into pcDNA6-His/Myc. Expression constructs for HA-tagged LXRα and LXRβ were also generated using the plasmid vector pcDNA3-HA (Invitrogen). The K30R and 3KR (K30R/K395R/K433R) mutant forms of LXRβ were created via PCR-directed mutagenesis (SBS Genetech, China), which were confirmed by DNA sequencing. For the luciferase reporter construct, the promoter region of the mouse Srebp1c gene was obtained by PCR and subsequently subcloned into the pGL3 vector. The expression vector for FLAG-tagged mouse PIAS1 was kindly provided by Dr. Ke Shuai (UCLA). The constructs for the truncated forms of PIAS1 were generated using a PCR-based strategy. The Myc-SUMO-1 construct was provided by Dr. Jihe Zhao (University of Central Florida, College of Medicine). The TK-LXRE3-Luc reporter construct was a generous gift from Dr. David J. Mangelsdorf (University of Texas Southwestern Medical Center), and the expression plasmids for Myc-tagged PGC-1β and PGC-1α were from Dr. Jiandie Lin (University of Michigan). The expression plasmids for SRC-3 and TRβ1 and the TK-Pal-Luc reporter construct containing two TRE-binding sites were as described previously (29). All the oligonucleotide primers used for PCR-based cloning are listed as follows: LXRα-FL, sense 5′-CCAGGGTACCAGGAAGAGATGTCCTTG-3′ and antisense 5′-GTGATCTAGACTCTCGTGGACATCCCAG-3′; LXRβ-FL, sense 5′-TGCAGGTACCTTCGTGACCCACTATGTC-3′ and antisense 5′-TGGCTCTAGACTCTCGTGCACATCCCAG-3′; LXRβ-LBD, sense 5′-CCAGGGTACCATGGCTCAGGAGCTGATG-3′ and antisense 5′-TGGCTCTAGACTCTCGTGCACATCCCAG-3′; LXRβ-DBD, sense 5′-TGCAGGTACCTTCGTGACCCACTATGTC-3′ and antisense 5′-CTGCTCTAGAACTAACTGCTGGATC-3′; LXRα-FL HA, sense AATTAAGCTTAGGAAGAGATGT and antisense GATCTAGACTCTCGTGGACATCCCAG; LXRβ-FL HA, sense 5′-TACCAAGCTTACCCACTATGTCTTC-3′ and antisense 5′-GCTCTCTAGACTCGTGCACATCCCAG-3′; PIAS1-N, sense 5′-AAAATCGATGCGGACAGTGCGGAACTAAAGC-3′ and antisense 5′-AAAGCGGCCGCGCAGCGAAACCCGTAGGCTG-3′; PIAS1-C, sense 5′-AAAATCGATTGTGTCCACTTGGGAAAATG and antisense 5′-AAAGCGGCCGCTCAGTCCAATGAGATAAT-3′; SREBP1c pro, sense 5′-ACATCTCGAGAAAGGGGATCAGGGCAG-3′ and antisense 5′-AAATGTGCAATCCATGGCTCCGTG-3′.
Cell Transfection, Co-immunoprecipitation, and Western Immunoblotting
HEK 293T cells were cultured in DMEM containing 10% fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Transfection of 293T cells was performed with the polyethyleneimine method (Polysciences) (30). Co-immunoprecipitation (co-IP) assays were performed as described previously (30). Briefly, whole-cell lysates were prepared in a lysis buffer (20 mm Tris (pH 7.5), 100 mm NaCl, 0.1% Nonidet P-40, 1 mm EDTA, 1 mm DTT, 10% glycerol, 50 mm NaF, and 10 mm sodium pyrophosphate) containing 1% protease inhibitors mixture (Sigma). After incubation with the desired primary antibody for 18 h at 4 °C via gentle rocking, protein G-Sepharose beads (GE Healthcare) were used to capture the immune complexes via mixing for 2 h at 4 °C on a rotator. Beads were subsequently washed three times with the washing buffer (20 mm Tris-HCl (pH 7.5), 150 mm KCl, 0.5% Nonidet P-40, 1 mm EDTA, and 10% glycerol containing 1% protease inhibitors mixture (Sigma)) before SDS-PAGE and Western immunoblotting analysis. Proteins were transferred to polyvinylidene difluoride (PVDF) membrane filters (Millipore), and after incubation with the desired antibodies, the blots were developed with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) or Immobilon Western chemiluminescent HRP substrate (Millipore).
Chemical Reagents and Antibodies
The LXR agonist T0901317 and the TRβ1 agonist T3 were purchased from Sigma. The monoclonal FLAG antibody was from Sigma, and the monoclonal Myc antibody was a generous gift from Dr. Jinqiu Zhou (Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences). Polyclonal antibodies against Myc, HA, PIAS1, and FAS were all from Cell Signaling. The polyclonal antibody against the mouse PGC-1β was homemade in rabbits using the N-terminal portion of PGC-1β expressed as the recombinant GST fusion protein. The antibody was purified from the collected sera by affinity chromatography using the HiTrap Protein-G HP column (Amersham Biosciences) according to the manufacturer's instructions.
Generation of the Recombinant Adenoviruses
Recombinant adenoviruses that overexpress EGFP or PIAS1 were generated with the AdEasyTM System (Stratagene) according to the manufacturer's instructions, which was described previously in detail (31). Briefly, the DNA fragment for FLAG-PIAS1 was first subcloned into pShuttle-CMV vector, which was then used to produce the recombinant adenoviral plasmid by homologous recombination with pAdEasy-1 in Escherichia coli BJ5183 cells. HEK 293A cells (Invitrogen) were transfected using the linearized recombinant plasmids to generate the recombinant viruses. Before infection of primary hepatocytes, the multiplicity of infection of the viruses was determined according to the manufacturer's instructions.
Isolation of Primary Hepatocytes and Adenovirus Infection
Primary hepatocytes were isolated from male C57BL/6J mice at 8–12 weeks of age according to the procedure that was described previously (32). Collagenase perfusion was conducted after anesthetizing through the portal vein with 50 ml of perfusion buffer (Krebs-Ringer buffer with 3.6 mg/ml glucose, 1 m CaCl2, and 0.66 mg/ml collagenase I (Worthington)) at 37 °C. The liver was aseptically removed and cut, and hepatocytes were filtrated, washed three times with cold Hepatocyte Wash Medium (Invitrogen), and resuspended in 15 ml of cold HepatoZYME-SFM (Invitrogen) medium supplemented with 2 mm l-glutamine, 20 units/ml penicillin, and 20 mg/ml streptomycin. The viability was determined by trypan blue staining, and the hepatocytes were plated at 6 × 105 cells/well in 6-well culture dishes pre-coated with collagen. Cells were cultured for 8 h before infection with the desired adenoviruses at an multiplicity of infection of 40 for 2–3 days. Cells were subsequently treated with DMSO or T0901317 prior to total RNA preparation, protein extraction, or measurement of lipogenesis.
Determination of Fatty Acid Synthesis in Hepatocytes
Fatty acid synthesis in the primary hepatocytes was measured according to the described procedure (33) with some modifications. Briefly, adenovirally infected hepatocytes were washed twice with PBS after culturing overnight and then incubating with 2 ml of high glucose DMEM plus [14C]acetate at 0.5 μCi per well. The wells were gassed with 95% O2 and 5% CO2, and the plates were sealed with Parafilm before incubating overnight. Cells were washed twice with PBS and then mixed with 2.5 ml of 10% KOH in methanol and 1.0 ml of distilled water per well. The mixture was heated at 90 °C for 3 h and then extracted with 4 ml of petroleum ether. Three milliliters of the lower aqueous layer was mixed first with 1.0 ml of 10 m H2SO4 and then with 4 ml of petroleum ether. The mixture was subsequently subjected to centrifugation, and 3 ml of the upper layer was obtained and dried under low heat. Levels of 14C-labeled fatty acids were measured by a scintillation counter.
Luciferase Reporter Assays
For luciferase reporter assays, 293T cells were co-transfected for 48 h with the desired plasmids, including 300 ng of TK-LXRE3-Luc, SREBP1c-Pro-Luc, or TK-Pal-Luc reporter together with 10 ng of Renilla plasmid DNA per well. Cells were treated with DMSO, 5 μm T0901317, or 100 nm T3 for 12 h before measurement of luciferase activities. Luciferase activity was determined according to the manufacturer's instructions, which was normalized to Renilla.
RT-PCR Analysis
Total RNA was extracted from cells or livers with TRIzol (Invitrogen). After reverse transcription by Moloney murine leukemia virus reverse transcriptase (Invitrogen), regular PCR was performed with the TaKaRa Taq kit (Takara). Quantitative real time PCR was conducted with an ABI Prism 7500 sequence detection system, using Power SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacturer's recommendations (Applied Biosystems). Cyclophilin was used as an internal control for normalization. The oligonucleotide primers for each target gene examined are as follows: Pias1, sense 5′-GCTCGTGGGCTCCAATGA-3′ and antisense 5′-CTGGTTGTGGGACGCTACCT-3′; Pias3, sense 5′-GATCCGGAATCCAGACCATTC-3′ and antisense 5′-ACATGAGTGACACCCGGAGACT-3′; Lxrα, sense 5′-GCAGGACCAGCTCCAAGTAG-3′ and antisense 5′-GGCTCACCAGCTTCATTAGC-3′; Lxrβ, sense 5′-ATTAAGGAAGAGGGGCAGGA-3′ and antisense 5′-TGACCACGATGTAGGCAGAG-3′; Srebp1c, sense 5′-GGAGCCATGGATTGCACATT-3′ and antisense 5′-GGAAGTCACTGTCTTGGTTGTTGA-3′; Acc1, sense 5′-TGAATCTCACGCGCCTACTATG-3′ and antisense 5′-ATGACCCTGTTGCCTCCAAAC-3′; Fas, sense 5′AAGTTGCCCGAGTCAGAGAA-3′ and antisense 5′-CGTCGAACTTGGAGAGATCC-3′; Scd1, sense 5′-GCGATACACTCTGGTGCTCA-3′ and antisense 5′-CCCAGGGAAACCAGGATATT-3′; and Cyclophilin A, sense 5′-ATGGCAAATGCTGGACCAAA-3′ and antisense 5′-CATGCCTTCTTTCACCTTCCC-3′.
Immunofluorescent Staining Analysis
293T cells were grown on glass coverslips and transfected with the desired plasmids for 48 h. Cells were then fixed for 10 min with 4% paraformaldehyde in PBS, permeabilized with 0.1% Triton X-100 in PBS for 10 min, and incubated sequentially with the desired primary and secondary antibodies. Confocal images were obtained with an LSM510 confocal microscope (Zeiss, Jena, Germany). An argon laser (488 nm) was used for excitation of Alexa 488-conjugated antibodies and a helium/neon laser (543 nm) for Alexa 546-conjugated antibodies. Red/green/blue (RGB) images were processed using the LSM510 software.
Chromatin Immunoprecipitation (ChIP) Analysis
ChIP assays were performed using the agarose ChIP kit (Pierce) following the manufacturer's instructions. Briefly, transfected 293T cells were subjected to cross-linking with 1% formaldehyde. Glycine solution was added, and the nuclear extract was prepared. Chromatin·LXRβ complexes were immunoprecipitated with the polyclonal anti-HA antibody by incubating at 4 °C overnight on a rocking platform, followed by incubating with protein G-Sepharose beads (GE Healthcare) at 4 °C for 1 h with gentle rocking. After washing five times with the washing buffer, the chromatin·LXRβ·antibody complexes were eluted with the elution buffer from the beads and were subjected to PCR analysis. For the secondary ChIP analysis, the eluted complexes were diluted in IP dilution buffer and incubated with anti-FLAG or anti-Myc antibody before PCR analysis. The oligonucleotide primers used for ChIP assays are as follows: SREBP1c(−239 to ∼−477), sense 5′-TCAGGGTGCCAGCGAACC-3′ and antisense 5′-GCTCGAGTTTCACCCCGC-3′; TK-Pal-Luc, sense 5′-ATAAGCTTCGAGCTCGCCCGG-3′ and antisense 5′-GTGTTCGAGGCCACACGCGT-3′.
Statistical Analysis
All data are presented as the mean ± S.E. Statistical analysis was conducted using unpaired two-tailed t test or two-way analysis of variance (ANOVA) followed by Bonferroni's post test with GraphPad Prism 5.0. p < 0.05 was considered to be statistically significant.
RESULTS
Expression of Hepatic Pias1 and Pias3 Is Inversely Correlated with That of Lipogenic Genes in Obese Mice
To gain insights into the possible metabolic actions of PIAS proteins, we first tested by quantitative RT-PCR analysis whether hepatic Pias1 and Pias3 genes exhibit altered expression patterns in the states of obesity. Interestingly, the mRNA expression of both Pias1 and Pias3 was significantly up-regulated (Fig. 1A) in livers of mice fed an HFD as compared with the LFD (28). This HFD-induced up-regulation of Pias1 and Pias3 was paralleled with marked reductions in the mRNA abundance of Srebp1c and the lipogenic enzymes Acc1, Fas, and Scd1 (Fig. 1A), consistent with our previous finding that HFD feeding resulted in suppressed expression of lipogenic genes (34). Next, we examined the expression of these genes in two genetic mouse models of obesity, the db/db mouse with deficient leptin receptor and the ob/ob mouse lacking leptin. As compared with their wild-type littermates, Pias1 and Pias3 were significantly down-regulated in livers of db/db or ob/ob mice (Fig. 1, B and C). On the one hand, elevated mRNA levels were observed for Srebp1c, Acc1, Fas and Scd1, which was in accordance with dysregulation of the lipogenic pathway as a result of deficient leptin signaling (34). On the other hand, neither Lxrα nor Lxrβ showed significant alterations in their mRNA expression levels in the states of obesity resulting from HFD feeding or abrogation of leptin signaling (Fig. 1, A–C). Consistently, hepatic PIAS1 protein expression was also dramatically reduced (by ∼75%) in ob/ob mice (Fig. 1D), which was accompanied by a marked increase in the protein expression level of FAS (by ∼5-fold). These results thus revealed an inverse association of Pias1 or Pias3 expression with the lipogenic gene expression program, raising the possibility that PIAS proteins may exert a regulatory action in the control of lipogenesis.
FIGURE 1.

Expression of Pias1 and Pias3 is negatively associated with lipogenic gene expression in livers of diet-induced or genetic mouse models of obesity. A–C, mRNA abundance of Pias1 and Pias3 and genes involved in fatty acid synthesis, Srebp1c, Acc1, Fas, and Scd1, as well as Lxrα and Lxrβ, was assessed by quantitative real time RT-PCR. Total RNA was prepared from livers of mice at 16 weeks of age that were fed a low-fat diet (LFD) or high-fat diet (HFD) for 8 weeks (n = 6/group) (A), db/db mice and their wild-type (WT) littermates at 8 weeks of age (n = 6/group) (B), or ob/ob mice and their WT littermates at 8 weeks of age (n = 5/group) (C). Cyclophilin A was used as an internal control. The expression level of each gene was normalized to that of LFD-fed mice or WT littermates (set as 1.0). D, Western immunoblot analysis of the expression of PIAS1 and FAS proteins in livers of ob/ob mice and their WT littermates at 8 weeks of age. Immunoblotting was performed using antibodies against PIAS1 and FAS, and α-tubulin was used as a loading control. Each lane represents the liver extract from one individual animal. Relative protein levels were determined by densitometric quantification of the immunoblots and normalized to α-tubulin. Data in all panels are shown as means ± S.E. *, p < 0.05 versus LFD-fed control mice or WT littermates by unpaired two-tailed Student's t test.
PIAS1 Overexpression Blunts LXR Agonist-induced Up-regulation of Lipogenesis
Given the crucial role for LXRs in lipogenic regulation and the implication of PIAS1 protein in mediating LXR-dependent suppression of inflammatory genes (21), we asked if PIAS1 could affect LXR-dependent control of lipogenesis. To test this idea, we examined the effect of adenovirally overexpressed FLAG-tagged PIAS1 protein (Fig. 2A) in mouse primary hepatocytes in the absence or presence of T0901317 (T0), a synthetic agonist of LXRs (16, 35). In hepatocytes infected with the control adenovirus Ad-EGFP, T0901317 treatment increased the biosynthesis of fatty acids, whereas overexpression of PIAS1 significantly reduced it in cells infected with Ad-FLAG-PIAS1 (Fig. 2B). While exhibiting no significant effects upon the endogenous LXRα or LXRβ protein expression level (Fig. 2A) as well as their mRNA abundance with or without agonist stimulation (Fig. 2C), PIAS1 overexpression significantly attenuated T0901317-stimulated up-regulation of Srebp1c as well as Acc1, Fas, and Scd1 (Fig. 2D). Notably, PIAS1 overexpression could also decrease the basal expression levels of Srebp1c and Fas in the absence of T0901317 induction. These data suggest that PIAS1 may play a negative role in the control of lipogenesis by attenuating LXR-dependent up-regulation of the lipogenic gene expression program.
FIGURE 2.

Hepatic overexpression of PIAS1 attenuates LXR agonist-induced fatty acid synthesis and up-regulation of lipogenic genes. Primary hepatocytes were infected for 48 h with recombinant adenoviruses encoding EGFP (Ad-EGFP) or FLAG-tagged PIAS1 (Ad-FLAG-PIAS1). Cells were subsequently incubated with DMSO or LXR agonist T0901317 (T0) at 5 μm for 12 h. A, Western immunoblot analysis of PIAS1, LXRα, and LXRβ proteins using the indicated antibodies. α-Tubulin was used as a loading control. B, hepatocytes were cultured in the presence of [14C]acetate for 17 h. Fatty acid synthesis was measured by the level of conversion of [14C]acetate into 14C-labeled intracellular fatty acids (n = 3 independent experiments). C and D, mRNA abundance of Lxrα and Lxrβ (C) or Srebp1c, Acc1, Fas, and Scd1 (D) was analyzed by quantitative RT-PCR using cyclophilin as an internal control (n >3 independent experiments). Data in B–D are shown as means ± S.E. *, p < 0.05 versus T0901317-treated Ad-EGFP-infected cells; #, p < 0.05 versus DMSO-treated Ad-EGFP-infected cells by two-way ANOVA.
PIAS1 Interacts with LXRs
To explore the molecular mechanism by which PIAS1 may down-regulate LXR-mediated lipogenic actions, we investigated whether PIAS1 can interact with LXRs. Co-immunoprecipitation experiments showed that in co-transfected 293T cells, FLAG-tagged PIAS1 protein could associate with both LXRα and LXRβ but not with thyroid hormone receptor TRβ1, indicating that PIAS1 could selectively interact with LXRs (Fig. 3A). Similarly, PIAS1 protein could be also immunoprecipitated with LXRα or LXRβ when using the anti-Myc antibody to pull down Myc-tagged LXRs (Fig. 3B). Importantly, co-IP experiments in mouse primary hepatocytes using anti-PIAS1 antibody showed that PIAS1 protein could endogenously associate with LXRα and LXRβ, which were appreciably enhanced upon T0901317 treatment (Fig. 3C). Further co-IP assays indicated that the C-terminal portion containing the LBD of LXRβ, but not its N-terminal segment with the DBD, was able to interact with PIAS1 protein (Fig. 3D). In addition, co-IP analysis using truncated forms of PIAS1 revealed that the N-terminal portion of PIAS1 containing the SAP and PINIT domains (PIAS1-N), but not the C-terminal segment with a partial RLD domain (PIAS1-C), could associate with LXRβ (Fig. 3E). Thus, PIAS1 could associate with LXRs and may thereby exert its effect upon LXR-activated gene transcription.
FIGURE 3.

PIAS1 interacts with LXRs. A and B, HEK 293T cells were co-transfected for 48 h with empty vector (−) or plasmids encoding FLAG-PIAS1, Myc-LXRα, Myc-LXRβ, or FLAG-TRβ1 as indicated. Cell lysates were immunoprecipitated (IP) with anti-FLAG or anti-PIAS1 antibody (A) or with anti-Myc antibody (B). Immunoblotting was conducted using antibodies against FLAG or Myc. C, primary hepatocytes were treated with DMSO or T0901317 (T0) at 5 μm for 12 h. Cell lysates were immunoprecipitated with control IgG or anti-PIAS1 antibody. Immunoblotting was performed using antibodies against LXRα, LXRβ, or PIAS1. D, schematic diagram of the full-length (FL) and truncated versions of LXRβ analyzed for its association with PIAS1. DNA-binding domain (DBD), black box; ligand-binding domain (LBD), gray box. 293T cells were co-transfected with the indicated plasmids. Immunoprecipitation and immunoblotting were likewise performed as in A. E, schematic diagram of the full-length (FL) and truncated versions of PIAS1 analyzed for its interaction with LXRβ. The domain structure of PIAS1 is as follows: SAP domain (SAP), PINIT amino acid motif (PINIT), RING finger-like zinc-binding domain (RLD), highly acidic domain (AD), and serine- and threonine-rich region (S/T). 293T cells were co-transfected with the indicated plasmids. Immunoprecipitation and immunoblotting were likewise performed as in B. All results are representative of at least two independent experiments.
PIAS1 Suppresses the Transactivation Activity of LXRs
To determine whether PIAS1 protein can affect the transcriptional activity of LXRs, we employed luciferase reporter assays. Co-transfection experiments in 293T cells showed that T0901317 treatment prominently stimulated the transcriptional activity of both LXRα and LXRβ toward the TK-LXRE3-luciferase reporter (Fig. 4A), which contains three copies of LXRE within the thymidine kinase promoter (6). While significantly reducing their transcriptional activities in the absence of ligand stimulation, co-expression of PIAS1 resulted in more marked decreases of T0901317-dependent transactivation by LXRα or LXRβ (Fig. 4A). Similar repressive effects of co-expressed PIAS1 on the ligand-activated transcriptional activity of endogenous LXRs were also observed in HepG2 cells (Fig. 4B). Furthermore, when assessed in HepG2 cells using the Srebp1c promoter-luciferase reporter, overexpression of the full-length PIAS1 as well as the truncated PIAS1-N form that is presumably SUMO E3 ligase-deficient because of deletion of the acidic domain and part of the RING finger-like zinc-binding domain (36, 37), but not the PIAS1-C form, resulted in significant reductions in T0901317-induced transcriptional activation of the Srebp1c promoter (Fig. 4C). These results demonstrated that PIAS1 could exert a negative action upon LXR-activated gene expression.
FIGURE 4.

Overexpression of PIAS1 results in suppression of LXR-dependent transcriptional activity. A and B, schematic is shown for TK-LXRE3-Luc, the luciferase (Luc) reporter under the control of the thymidine kinase promoter containing three repeated LXR-responsive or binding elements (LXRE3). A, 293T cells were co-transfected for 36 h with the TK-LXRE3-Luc construct and empty vector (−) or the FLAG-PIAS1 plasmid together with plasmids encoding Myc-LXRα or Myc-LXRβ as indicated. B, HepG2 cells were co-transfected for 36 h with the TK-LXRE3-Luc and empty vector (−) or the FLAG-PIAS1 plasmid. C, schematic is shown for the luciferase reporter under the control of the Srebp1c promoter (SREBP1c-Pro-Luc) that harbors two LXREs as indicated. HepG2 cells were co-transfected for 36 h with the SREBP1c-Pro-Luc together with empty vector (−) or the plasmid encoding FLAG-PIAS1-FL, -N, or -C. Transfected cells were treated with DMSO or 5 μm T0901317 for 12 h, and cell extracts were analyzed for luciferase activities. All data are presented as means ± S.E. from three independent experiments after normalization to the value of DMSO-treated empty vector control (set as 1). *, p < 0.05 versus T0901317 (T0)-treated empty vector control; #, p < 0.05 versus DMSO-treated empty vector control by two-way ANOVA.
PIAS1 Regulates LXRβ Activity in a SUMOylation-independent Fashion
As PIAS1 is known to be a SUMO E3 ligase that promotes the LXRβ transrepression of inflammatory genes by a mechanism that requires SUMOylation of LXRβ (21), we asked if PIAS1 could exert its suppressive effect on LXRβ activity by a SUMOylation-dependent mechanism. Consistent with the reported findings (21), T0901317 treatment could stimulate SUMO-1 modification of LXRβ in co-transfected 293T cells (Fig. 5A), which was markedly enhanced by the co-expression of PIAS1 (Fig. 5B). On the basis of the ΨKXE (where Ψ denotes Leu, Ile, Val, or Phe, and X denotes any amino acid) consensus sequence for SUMO modification (38), there are three putative Lys residues, i.e. Lys-30, Lys-395, and Lys-433, of LXRβ that could potentially undergo SUMOylation. Mutational analysis by Lys to Arg substitution revealed that Lys-30 was one of the major sites for PIAS1-promoted SUMO-1 modification, as indicated by the reduced SUMO-1 conjugation of the K30R mutant (Fig. 5C). Substitution mutation of all three Lys residues (3KR) nearly completely abolished PIAS1-dependent SUMO-1 modification (Fig. 5C). To determine whether SUMOylation of LXRβ affects its ability to interact with PIAS1, we conducted co-IP assays in 293T cells. SUMOylated LXRβ retained its ability to associate with PIAS1, whereas the SUMOylation-defective LXRβ-3KR mutant could fully interact with PIAS1 (Fig. 5D). Then we examined the importance of SUMO modifications of LXRβ to its transactivation activity using the TK-LXRE3-luciferase reporter assays. SUMOylation-defective LXRβ mutants, LXRβ-K30R and LXRβ-3KR, had comparable transcriptional activity in the presence of SUMO-1 expression to that of wild-type LXRβ when stimulated by T0901317, which could be repressed to a similar extent by PIAS1 overexpression (Fig. 5E). Consistently, overexpression of PIAS1 could inhibit the transcriptional activity of both LXRβ-WT and LXRβ-3KR without co-expression of SUMO-1 (Fig. 5F). These results indicate that PIAS1-mediated SUMO modification is not required for PIAS1 suppression of LXRβ transactivation.
FIGURE 5.

PIAS1 affects the transcriptional activity of LXRβ in a SUMO modification-independent fashion. A, LXR agonist promoted SUMO1 conjugation to LXRβ. 293T cells were transiently co-transfected for 30 h with Myc-SUMO-1 along with HA-LXRβ-WT as indicated. Cells were then treated with DMSO or 5 μm T0901317 for 12 h. Cell lysates were prepared in the presence of 20 mm N-ethylmaleimide, and Western immunoblotting was performed using anti-HA or anti-Myc antibody. SUMO1-conjugated HA-LXRβ is indicated. B, PIAS1 enhanced SUMO1 modification of LXRβ. 293T cells were co-transfected for 30 h with HA-LXRβ-WT with or without Myc-SUMO-1 or FLAG-PIAS1 as indicated. Cells were then treated with 5 μm T0901317 for 12 h, and cell lysates were analyzed by immunoblotting using the tag antibodies. C, lysine residue 30 of LXRβ was the major site for PIAS1-promoted SUMO1 modification. 293T cells were co-transfected with the indicated plasmids, including those encoding HA-tagged WT or K30R and 3KR mutant forms of LXRβ. Cells were likewise treated and analyzed by immunoblotting. D, 293T cells were co-transfected for 48 h with the indicated plasmids and then treated with 5 μm T0901317 for 12 h. Immunoprecipitation (IP) was performed with anti-FLAG, and immunoblotting was conducted using the tag antibodies. Results in A–D are representative of at least two independent experiments. E and F, PIAS1-mediated SUMO1 modification was not required to suppress the transcriptional activity of LXRβ. 293T cells were co-transfected with the TK-LXRE3-Luc construct and the indicated plasmids, with (E) or without (F) Myc-SUMO1. Cells were subsequently treated with either DMSO or 5 μm T0901317 for 12 h before luciferase assays. Data are presented as means ± S.E., with the value for each LXRβ construct normalized to DMSO-treated control without FLAG-PIAS1 (set as 1). *, p < 0.05; #, p < 0.05 versus T0901317 (T0)- or DMSO-treated cells without FLAG-PIAS1 by two-way ANOVA. N.S., no significance.
PIAS1 Does Not Alter the Cellular Localization of LXRβ
Next, we investigated whether PIAS1 association or PIAS1-promoted SUMOylation could change the subcellular localization of LXRβ. Interestingly, immunofluorescent staining analysis of transfected 293T cells revealed similar nuclear localization patterns for PIAS1 and the wild-type and 3KR forms of LXRβ (Fig. 6A), all with an appearance of enriched distribution around the nuclear membrane. In contrast to LXRβ and PIAS1, whose localization was restricted to the nucleus, SUMO-1 was distributed both in the nucleus and cytoplasm (Fig. 6A). When examined in co-transfected 293T cells, PIAS1 and LXRβ (both the wild-type and 3KR mutant) exhibited similar intra-nuclear co-localization, which were unaltered by the co-expression of SUMO-1 (Fig. 6B). These data suggest that PIAS1 may not influence the nuclear localization of LXRβ.
FIGURE 6.

PIAS1 has no apparent effect on the cellular localization of LXRβ. 293T cells were transfected (A) or co-transfected (B) for 48 h with the indicated plasmids. Cells were subsequently treated with 5 μm T0901317 for 12 h and subjected to fluorescent immunostaining analysis using antibodies against HA, FLAG, or Myc. The nuclei of cells were visualized by Hoechst staining. Representative confocal microscopy images are shown from two independent experiments.
PIAS1 Associates with PGC-1β to Promote Its SUMOylation and Attenuate Its Ability to Co-activate LXRβ
PGC-1α and PGC-1β are two members of the PPARγ co-activator-1 (PGC-1) family of transcriptional co-activators of nuclear receptors (39, 40). PGC-1β has been documented to function as a critical regulator of lipogenesis in the liver, largely by co-activation of SREBP1c as well as LXRβ (41). To determine whether PIAS1 affects LXRβ-regulated gene expression in the context of its co-activation by PGC-1β, we first conducted co-IP experiments in co-transfected 293T cells. PIAS1 was able to interact with both PGC-1β and PGC-1α but not with steroid receptor co-activator-3 (SRC-3), suggesting the specificity of PIAS1 interaction with PGC-1s (Fig. 7A). Of interesting note, the PIAS1-N form but not the PIAS1-C form of PIAS1 could associate with PGC-1β (Fig. 7B). Similar to the observations with LXRβ, PIAS1 could also increase the SUMO1 modification level of PGC-1β in co-transfected 293T cells (Fig. 7C). Moreover, co-IP experiments showed that SUMOylated PGC-1β retained its ability to interact with PIAS1 (Fig. 7D) as well as LXRβ (Fig. 7E). However, Srebp1c promoter-luciferase reporter assays showed that the full-length PIAS1 and truncated PIAS1-N proteins, but not the PIAS1-C protein, could markedly blunt the co-activation activity of PGC-1β on LXRβ in co-transfected HepG2 cells when stimulated with T0901317 (Fig. 7F). Given that the truncated PIAS1-N is most likely to lack the E3 ligase activity, these results suggest that PIAS1 can associate with PGC-1β and influence its co-activation ability without requiring SUMO modifications. Furthermore, luciferase reporter assays in co-transfected HepG2 cells also revealed a similar blocking effect of PIAS1 on PGC-1β co-activation of T0901317-stimulated LXRE3-Luc transcription (Fig. 7G), whereas PIAS1 exhibited no significant impact upon the ability of PGC-1β to co-activate TRβ1-dependent transcription of Pal-Luc upon T3 stimulation (Fig. 7G). This further indicates the specific action that PIAS1 exerted on the PGC-1-LXR pathway.
FIGURE 7.

PIAS1 interacts with PGC-1β and affects its SUMOylation and co-activation activity. A, PIAS1 could interact with both PGC-1β and PGC-1α but not with SRC-3. B, N-terminal portion of PIAS1 could associate with PGC-1β. 293T cells were co-transfected for 48 h with the indicated plasmids. Cell lysates were immunoprecipitated with anti-FLAG (A) or anti-Myc (B) antibody. Western immunoblotting was conducted using the tag or SRC-3 antibodies. C, PIAS1 could promote SUMOylation of PGC-1β. 293T cells were co-transfected for 48 h with the indicated plasmids. Cell lysates were prepared in the presence of 20 mm N-ethylmaleimide, and immunoblotting was performed using anti-PGC-1β for the detection of PGC-1β and the tag antibodies for PIAS1 and SUMO1. D and E, effect of PGC-1β sumoylation on its interaction with PIAS1 (D) or LXRβ (E). 293T cells were co-transfected for 48 h with the indicated plasmids. Cell lysates were immunoprecipitated with anti-FLAG (D) or anti-HA (E) antibody. Immunoblotting was conducted using anti-PGC-1β, anti-FLAG, anti-HA, or anti-Myc antibodies. Representative results in A–E are shown from at least two independent experiments. F and G, PIAS1 overexpression blunted the transcriptional co-activation activity of PGC-1β on LXRs but not on TRβ1. HepG2 cells were co-transfected for 48 h with SREBP1c-Pro-Luc (F) or the TK-LXRE3-Luc or TK-Pal-Luc reporter construct (G), together with the indicated plasmids. Cells were subsequently incubated with DMSO, 5 μm T0901317, or 100 nm T3 for 12 h before luciferase activity analysis. Data are presented as means ± S.E., with values normalized to DMSO-treated vector control without PGC-1β (set as 1). *, p < 0.05; #, p < 0.05 by two-way ANOVA. FL, full length. N.S., no significance.
PIAS1 Disrupts the Association between PGC-1 and LXRs
As PIAS1 could associate with both PGC-1 and LXR proteins, we tested if PIAS1 could affect the formation of the PGC-1·LXR complex using co-IP assays. When co-expressed in 293T cells, PIAS1 was able to be co-immunoprecipitated along with both PGC-1β and LXRβ in similar abundance (Fig. 8A), suggesting that PIAS1 has little preference for interacting with the two proteins. In contrast, co-IP of LXRβ indicated that PIAS1 co-expression could largely reduce the association of LXRβ with PGC-1β (Fig. 8B) but not with SRC-3 that was able to interact with LXRβ but not with PIAS1. In addition, co-IP of PGC-1 revealed that PIAS1 attenuated the association of LXRβ with PGC-1β as well as with PGC-1α (Fig. 8C). Furthermore, PIAS1 co-expression was also able to decrease the interaction of PGC-1β with LXRα (Fig. 8D), but it had little effect on its association with the thyroid hormone receptor 1β that can be co-activated by PGC-1β (42). Together, these data indicate that PIAS1 may disrupt the formation of the PGC-1·LXR complex by interacting with PGC-1 or LXR proteins, thereby diminishing the ability of PGC-1 to enhance LXR-dependent activation of gene transcription.
FIGURE 8.

PIAS1 attenuates the association between PGC-1 and LXRs. A, PIAS1 had no preference for interacting with PGC-1β and LXRβ. B, PIAS1 decreased the association of LXRβ with PGC-1β but not with SRC-3. C, PIAS1 affected the interaction of LXRβ with both PGC-1β and PGC-1α. D, PIAS1 attenuated the association of PGC-1β with LXRβ but not with TRβ1. 293T cells were co-transfected for 48 h with the indicated plasmids. Cell lysates were immunoprecipitated (IP) with the tag antibody as indicated, and immunoblotting was performed using anti-SRC-3 (B) or tag antibodies. Representative results are shown from at least two independent experiments.
PIAS1 Affects LXRβ Binding to Its Target DNA Sequence
To investigate the molecular mechanism that mediates the suppressive action of PIAS1 upon the transactivation activity of LXRβ, we performed ChIP assays to determine whether PIAS1 affects the DNA binding of LXRβ within the Srebp1c promoter containing two copies of LXRE (Fig. 9A). In co-transfected 293T cells, T0901317 stimulation increased the abundance of LXRβ bound to the Srebp1c promoter, and co-expressed PIAS1 apparently decreased it (Fig. 9A). To further examine whether PIAS1 is complexed with LXRβ on the target DNA sequence, we employed ChIP-reChIP assays. Consistently, LXRβ immunoprecipitation showed that PIAS1 reduced ligand-stimulated occupancy of LXRβ on the LXRE sequence of the Srebp1c promoter, even in the presence of co-expressed PGC-1β (Fig. 9B). However, secondary PIAS1 immunoprecipitation produced no detectable signals from the secondary ChIP experiments (Fig. 9B). In contrast, secondary PGC-1β immunoprecipitation revealed its association with LXRβ on the target LXRE sequence, which was also blunted by co-expression of PIAS1 (Fig. 9B). However, in 293T cells transfected with TK-Pal-Luc that harbors two copies of TRE, ChIP analysis showed that PIAS1 had no obvious effect on PGC-1β-enhanced TRβ1 binding to the TREs in the presence or absence of T3 stimulation (Fig. 9C). These data thus demonstrated that PIAS1 could selectively prevent LXRβ from binding to the LXRE sites, thereby suppressing its transactivation of target genes.
FIGURE 9.
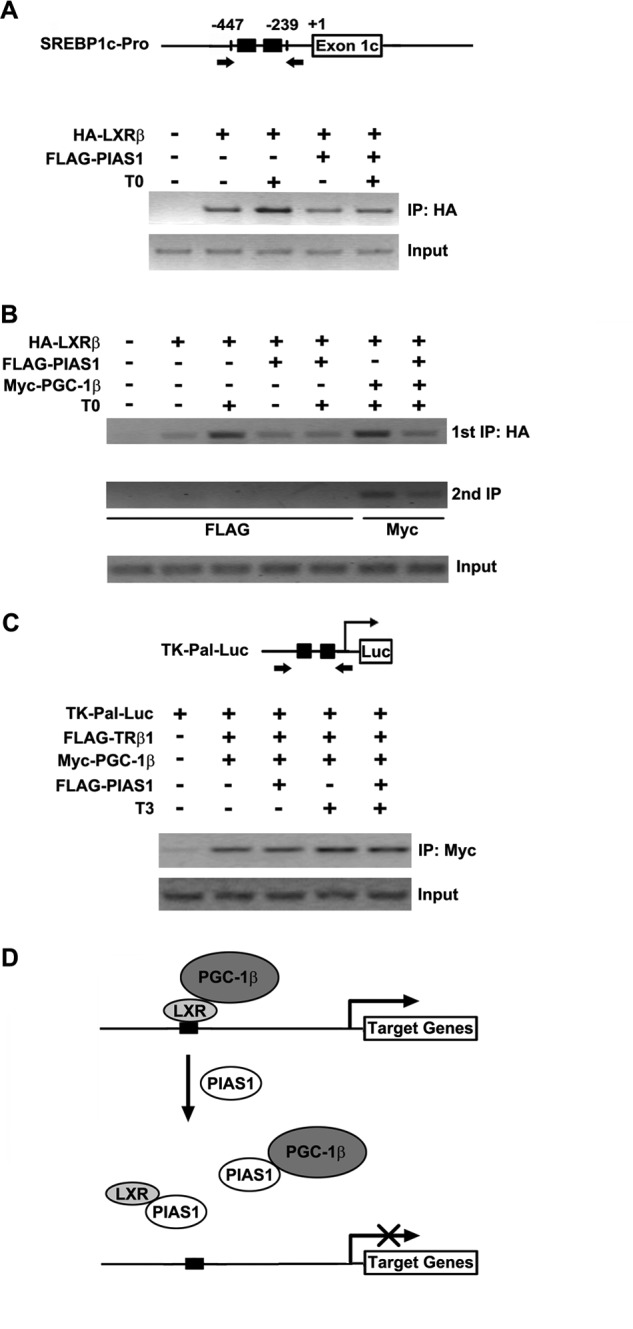
PIAS1 impairs the ability of LXRβ to bind to its target DNA sequence. A, schematic for the Srebp1c promoter containing the indicated LXREs. The arrows flanking the LXREs indicate the primers used for the ChIP assays in A and B. 293T cells were transfected for 48 h with empty vector (−) or the indicated plasmids and then treated with DMSO or 5 μm T0901317 for 12 h. Chromatin extracts were immunoprecipitated (IP) with anti-HA antibody, and the precipitated genomic DNA was subjected to PCR amplification. Cellular genomic DNA was used as the input control. B, 293T cells were co-transfected for 48 h with empty vector (−) or the indicated plasmids. Cells were likewise treated prior to chromatin immunoprecipitation with anti-HA antibody. The immunoprecipitates were then used for a secondary immunoprecipitation with anti-FLAG or anti-Myc antibody as indicated. The precipitated DNA was subjected to PCR amplification with cellular genomic DNA used as the input control. Representative results are shown from at least two independent experiments. C, schematic for the TK-Pal-Luc construct containing the indicated TREs. The arrows flanking the TREs indicate the primers used for the ChIP assay. 293T cells were transfected for 48 h with empty vector (−) or the indicated plasmids and then treated with DMSO or 100 nm T3 for 12 h. Chromatin immunoprecipitation was performed with anti-Myc antibody prior to PCR amplification. Cellular DNA was used as the input control. D, schematic model for PIAS1-directed repression of LXR-dependent gene transcription activation. PIAS1 can interact with LXRs as well as PGC-1 co-activators to disrupt their association. Without relying on the SUMO-modifications of LXRs, PIAS1 is able to block LXR binding to the promoters of their target genes (e.g. Srebp1c). Thus, PIAS1 acts to dampen ligand-activated, LXR-dependent gene expression programs such as lipogenesis.
DISCUSSION
Extensive studies have implicated PIAS proteins in regulation of a variety of cellular signaling events, such as STAT and NF-κB pathways in cytokine signaling (24, 43). This study revealed that the PIAS family member PIAS1 could act as a negative modulator of LXR-dependent up-regulation of lipogenic gene expression program, unmasking a potential regulatory role for PIAS proteins in lipid metabolism. As depicted by our schematic model (Fig. 9D), PIAS1 can associate with both LXR and PGC-1β proteins, attenuate the formation of the LXR·PGC-1β complex, and dampen LXR-dependent gene transcription by impairing the DNA binding ability of LXRs on target genes. Our results thus provide new insights into the physiological functions of PIAS proteins in addition to their actions in regulation of immune responses.
How PIAS proteins are regulated under normal or pathological conditions is largely unclear. Using diet-induced and genetic obesity mouse models, we found that the expression of both Pias1 and Pias3 in the liver was altered in association with obesity-induced metabolic stress conditions and exhibited an inverse correlation with that of lipogenic genes. Moreover, our results demonstrated that PIAS1 overexpression in primary hepatocytes could indeed down-regulate the expression of Srebp1c, a master lipogenic regulator (13), as well as critical enzymes involved in lipogenesis, leading to reduced fatty acid biosynthesis. Additionally, PIAS1 was able to interact with LXRs endogenously in a ligand-responsive fashion and block their ligand-stimulated transactivation activities. Although the precise mechanisms by which the transcription of Pias1 or Pias3 is metabolically regulated remain to be further understood, it also has yet to be defined whether hepatic PIAS3 can exert a similar regulatory effect on LXR-dependent lipogenic control as PIAS1. Selective gene knock-out mouse models would be of great value to decipher if PIAS1 and PIAS3 play redundant roles in regulation of lipid metabolism. Given the close relationship between immune signaling and metabolic homeostasis (44), it is tempting to speculate that PIAS proteins may exert a broader spectrum of metabolic actions by affecting not only nuclear receptors like LXRs, but also other classical PIAS-regulated pathways such as STAT signaling (24).
Acting as critical regulators of transcription factors with the SUMO E3 ligase activity, PIAS proteins employ multiple mechanisms to modulate cellular gene expression programs. For instance, PIAS1 can inhibit the DNA binding activity of transcription factors, such as STAT1 and the p65 subunit of NF-κB (23, 45, 46). PIAS1 can also modulate gene expression by promoting SUMOylation of transcription factors, as in the case of nuclear receptors PPARγ and LXRβ (20, 21). In macrophages, SUMOylation can target PPARγ to nuclear receptor co-repressor complex, thereby preventing the repressive complex from being removed from the promoter of target genes (20). In astrocytes, PIAS1 can SUMOylate LXRβ in an LXR ligand-dependent manner, resulting in the formation of a complex with STAT1 and, consequently, diminishing the ability of STAT1 to bind to STAT1-responsive genes (21). In contrast, here we found that PIAS1 could interact with LXRs and blunt their transactivation activity without requiring PIAS1-promoted SUMOylation. Although our results showed that PIAS1 was able to impair the DNA binding ability of LXRβ for the Srebp1c promoter, it is currently unclear if PIAS1 suppression of LXR-regulated lipogenic genes involves the actions of co-repressors. Therefore, it is most likely that PIAS1 exerts its effects on lipogenic control by regulating the activity of LXRs in a distinct mechanism from that in PIAS1-mediated actions involved in LXR-dependent transrepression of inflammatory signaling.
Our co-IP experiments indicated that PIAS1 could also interact with PGC-1 proteins, the co-activators of LXRs, and negatively influenced the formation of PGC-1·LXR complex. This appeared to be selective because no apparent disrupting effect was seen on the SRC-3·LXRβ or PGC-1β·TRβ1 association, which might be at least in part ascribable to the inability of PIAS1 to interact with SRC-3 or TRβ1. Consequently, PIAS1 exerted a blunting effect on the ability of PGC-1β to co-activate LXRβ but not TRβ1 transcriptional activity. However, it is also conceivable that the action of PIAS1 on PGC1 may represent an additional layer of regulation, as PIAS1-mediated impairment of LXR binding to its target DNA most likely serves as a predominant mechanism for PIAS1's suppression of LXR-dependent gene expression. Although we were unable to determine whether there is a tripartite PGC-1β·PIAS1·LXRβ complex involved due to their interactions with each other, it is possible that PGC-1β and LXRβ may interact with the N-terminal region of PIAS1 in a competitive manner. In addition, it is worth noting that PIAS1 and PIAS3 proteins have been recently reported to enhance SUMO1 modification of PGC-1α, thus attenuating its functional activity (47). In this study, we found that PIAS1 could also promote PGC-1β SUMOylation without dramatically affecting its interaction with PIAS1 or LXR. Because we could not identify via mutational analysis the precise sites of PIAS1-dependent SUMOylation of PGC-1β (data not shown), it remains to be completely clarified whether PIAS1 affects the lipogenic activity of the PGC-1β-LXRβ pathway through PGC-1β SUMOylation. Nonetheless, it is likely that PIAS1 does not rely on the SUMO modification of PGC-1β to modulate its activity because the E3 ligase-deficient truncated PIAS1-N protein could also repress the co-activation of LXR-dependent gene transcription. Given the key role of PGC-1 in orchestrating metabolic programs in lipid homeostasis by regulating the transcription activities of LXRs as well as SREBP1c in response to physiological and nutritional cues (41), it warrants further investigations to define the exact regulatory interplays of PIAS proteins, PGC-1 and PGC-1-co-regulated partners.
In summary, our findings have revealed a suppressive action of PIAS1 on LXR-activated gene expression programs, particularly in lipogenesis. LXR ligands have been shown to bear beneficial effects in mice with metabolic disorders and cardiovascular diseases (16), and ligand-activated LXRs have also been suggested to mediate anti-inflammatory responses in macrophages or astrocytes (21, 48). A better understanding of the molecular mechanism by which PIAS proteins regulate the functional actions of LXRs, particularly under pathological conditions, may lead to new avenues to develop therapeutic strategies for the treatment of both immune and metabolic disorders.
Acknowledgments
We thank Dr. Liangyou Rui for the critical comments on the manuscript. We are grateful to Drs. Ke Shuai, Jihe Zhao, David J. Mangelsdorf, Jiandie Lin, and Jinqiu Zhou for their generosity in providing all the reagents.
This work was supported by Ministry of Science and Technology Grants 2012CB524900 and 2011CB910900 from the 973 Program, National Natural Science Foundation Grants 81021002, 30988002, and 30830033, Chinese Academy of Sciences Knowledge Innovation Programs KSCX2-EW-R-09 and KSCX2-EW-Q-1-09, and Science and Technology Commission of Shanghai Municipality Grant 10XD1406400 (to Y. L.).
- LXR
- liver X receptor
- PIAS
- protein inhibitor of activated STAT
- PGC-1β
- peroxisome proliferator-activated receptor γ co-activator 1β
- SUMO
- small ubiquitin-like modifier
- LXRE
- LXR-responsive element
- SREBP1c
- sterol regulatory element-binding protein 1c
- FAS
- fatty-acid synthase
- SCD1
- stearoyl-CoA desaturase 1
- LFD
- low fat diet
- HFD
- high fat diet
- SRC-3
- steroid receptor co-activator-3
- ANOVA
- analysis of variance
- DBD
- DNA-binding domain
- LBD
- ligand-binding domain
- PPARγ
- peroxisome proliferator-activated receptor γ
- EGFP
- enhanced GFP
- TRE
- thyroid-responsive element
- T3
- triiodothyronine.
REFERENCES
- 1. Kalaany N. Y., Mangelsdorf D. J. (2006) LXRS and FXR. The yin and yang of cholesterol and fat metabolism. Annu. Rev. Physiol. 68, 159–191 [DOI] [PubMed] [Google Scholar]
- 2. Zelcer N., Tontonoz P. (2006) Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Invest. 116, 607–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lu T. T., Repa J. J., Mangelsdorf D. J. (2001) Orphan nuclear receptors as eLiXiRs and FiXeRs of sterol metabolism. J. Biol. Chem. 276, 37735–37738 [DOI] [PubMed] [Google Scholar]
- 4. Repa J. J., Mangelsdorf D. J. (2000) The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu. Rev. Cell Dev. Biol. 16, 459–481 [DOI] [PubMed] [Google Scholar]
- 5. Apfel R., Benbrook D., Lernhardt E., Ortiz M. A., Salbert G., Pfahl M. (1994) A novel orphan receptor specific for a subset of thyroid hormone-responsive elements and its interaction with the retinoid/thyroid hormone receptor subfamily. Mol. Cell. Biol. 14, 7025–7035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Willy P. J., Umesono K., Ong E. S., Evans R. M., Heyman R. A., Mangelsdorf D. J. (1995) LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 9, 1033–1045 [DOI] [PubMed] [Google Scholar]
- 7. Lehmann J. M., Kliewer S. A., Moore L. B., Smith-Oliver T. A., Oliver B. B., Su J. L., Sundseth S. S., Winegar D. A., Blanchard D. E., Spencer T. A., Willson T. M. (1997) Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J. Biol. Chem. 272, 3137–3140 [DOI] [PubMed] [Google Scholar]
- 8. Chawla A., Repa J. J., Evans R. M., Mangelsdorf D. J. (2001) Nuclear receptors and lipid physiology. Opening the X-files. Science 294, 1866–1870 [DOI] [PubMed] [Google Scholar]
- 9. Glass C. K., Rosenfeld M. G. (2000) The co-regulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 14, 121–141 [PubMed] [Google Scholar]
- 10. Alberti S., Schuster G., Parini P., Feltkamp D., Diczfalusy U., Rudling M., Angelin B., Björkhem I., Pettersson S., Gustafsson J. A. (2001) Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXRβ-deficient mice. J. Clin. Invest. 107, 565–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Janowski B. A., Willy P. J., Devi T. R., Falck J. R., Mangelsdorf D. J. (1996) An oxysterol signaling pathway mediated by the nuclear receptor LXRα. Nature 383, 728–731 [DOI] [PubMed] [Google Scholar]
- 12. Peet D. J., Turley S. D., Ma W., Janowski B. A., Lobaccaro J. M., Hammer R. E., Mangelsdorf D. J. (1998) Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXRα. Cell 93, 693–704 [DOI] [PubMed] [Google Scholar]
- 13. Horton J. D., Goldstein J. L., Brown M. S. (2002) SREBPs. Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Repa J. J., Liang G., Ou J., Bashmakov Y., Lobaccaro J. M., Shimomura I., Shan B., Brown M. S., Goldstein J. L., Mangelsdorf D. J. (2000) Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev. 14, 2819–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Joseph S. B., Laffitte B. A., Patel P. H., Watson M. A., Matsukuma K. E., Walczak R., Collins J. L., Osborne T. F., Tontonoz P. (2002) Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J. Biol. Chem. 277, 11019–11025 [DOI] [PubMed] [Google Scholar]
- 16. Schultz J. R., Tu H., Luk A., Repa J. J., Medina J. C., Li L., Schwendner S., Wang S., Thoolen M., Mangelsdorf D. J., Lustig K. D., Shan B. (2000) Role of LXRs in control of lipogenesis. Genes Dev. 14, 2831–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cha J. Y., Repa J. J. (2007) The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 282, 743–751 [DOI] [PubMed] [Google Scholar]
- 18. Glass C. K., Ogawa S. (2006) Combinatorial roles of nuclear receptors in inflammation and immunity. Nat. Rev. Immunol. 6, 44–55 [DOI] [PubMed] [Google Scholar]
- 19. Joseph S. B., Castrillo A., Laffitte B. A., Mangelsdorf D. J., Tontonoz P. (2003) Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nature Medicine 9, 213–219 [DOI] [PubMed] [Google Scholar]
- 20. Pascual G., Fong A. L., Ogawa S., Gamliel A., Li A. C., Perissi V., Rose D. W., Willson T. M., Rosenfeld M. G., Glass C. K. (2005) A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 437, 759–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee J. H., Park S. M., Kim O. S., Lee C. S., Woo J. H., Park S. J., Joe E. H., Jou I. (2009) Differential SUMOylation of LXRα and LXRβ mediates transrepression of STAT1 inflammatory signaling in IFN-γ-stimulated brain astrocytes. Mol. Cell 35, 806–817 [DOI] [PubMed] [Google Scholar]
- 22. Chung C. D., Liao J., Liu B., Rao X., Jay P., Berta P., Shuai K. (1997) Specific inhibition of Stat3 signal transduction by PIAS3. Science 278, 1803–1805 [DOI] [PubMed] [Google Scholar]
- 23. Liu B., Liao J., Rao X., Kushner S. A., Chung C. D., Chang D. D., Shuai K. (1998) Inhibition of Stat1-mediated gene activation by PIAS1. Proc. Natl. Acad. Sci. U.S.A. 95, 10626–10631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shuai K., Liu B. (2005) Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 5, 593–605 [DOI] [PubMed] [Google Scholar]
- 25. Jackson P. K. (2001) A new RING for SUMO. Wrestling transcriptional responses into nuclear bodies with PIAS family E3 SUMO ligases. Genes Dev. 15, 3053–3058 [DOI] [PubMed] [Google Scholar]
- 26. Schmidt D., Müller S. (2003) PIAS/SUMO. New partners in transcriptional regulation. Cell. Mol. Life Sci. 60, 2561–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sharrocks A. D. (2006) PIAS proteins and transcriptional regulation. More than just SUMO E3 ligases? Genes Dev. 20, 754–758 [DOI] [PubMed] [Google Scholar]
- 28. Huang P., Li S., Shao M., Qi Q., Zhao F., You J., Mao T., Li W., Yan Z., Liu Y. (2010) Calorie restriction and endurance exercise share potent anti-inflammatory function in adipose tissues in ameliorating diet-induced obesity and insulin resistance in mice. Nutr. Metab. 7, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Song Y., Shan S., Zhang Y., Liu W., Ding W., Ren W., Xia H., Li X., Zhang Q., Zhao L., Li X., Yan J., Ying H. (2012) Ligand-dependent co-repressor acts as a novel co-repressor of thyroid hormone receptor and represses hepatic lipogenesis in mice. J. Hepatol. 56, 248–254 [DOI] [PubMed] [Google Scholar]
- 30. Godbey W. T., Wu K. K., Hirasaki G. J., Mikos A. G. (1999) Improved packing of poly(ethyleneimine)-DNA complexes increases transfection efficiency. Gene Ther. 6, 1380–1388 [DOI] [PubMed] [Google Scholar]
- 31. Qiu Y., Mao T., Zhang Y., Shao M., You J., Ding Q., Chen Y., Wu D., Xie D., Lin X., Gao X., Kaufman R. J., Li W., Liu Y. (2010) A crucial role for RACK1 in the regulation of glucose-stimulated IRE1α activation in pancreatic beta cells. Sci. Signal. 3, ra7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mao T., Shao M., Qiu Y., Huang J., Zhang Y., Song B., Wang Q., Jiang L., Liu Y., Han J. D., Cao P., Li J., Gao X., Rui L., Qi L., Li W., Liu Y. (2011) PKA phosphorylation couples hepatic inositol-requiring enzyme 1α to glucagon signaling in glucose metabolism. Proc. Natl. Acad. Sci. U.S.A. 108, 15852–15857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McGarry J. D., Takabayashi Y., Foster D. W. (1978) The role of malonyl-CoA in the coordination of fatty acid synthesis and oxidation in isolated rat hepatocytes. J. Biol. Chem. 253, 8294–8300 [PubMed] [Google Scholar]
- 34. Jiang L., Wang Q., Yu Y., Zhao F., Huang P., Zeng R., Qi R. Z., Li W., Liu Y. (2009) Leptin contributes to the adaptive responses of mice to high-fat diet intake through suppressing the lipogenic pathway. PloS ONE 4, e6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Collins J. L., Fivush A. M., Watson M. A., Galardi C. M., Lewis M. C., Moore L. B., Parks D. J., Wilson J. G., Tippin T. K., Binz J. G., Plunket K. D., Morgan D. G., Beaudet E. J., Whitney K. D., Kliewer S. A., Willson T. M. (2002) Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J. Med. Chem. 45, 1963–1966 [DOI] [PubMed] [Google Scholar]
- 36. Kotaja N., Karvonen U., Jänne O. A., Palvimo J. J. (2002) PIAS proteins modulate transcription factors by functioning as SUMO-1 ligases. Mol. Cell. Biol. 22, 5222–5234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Minty A., Dumont X., Kaghad M., Caput D. (2000) Covalent modification of p73α by SUMO-1. Two-hybrid screening with p73 identifies novel SUMO-1-interacting proteins and a SUMO-1 interaction motif. J. Biol. Chem. 275, 36316–36323 [DOI] [PubMed] [Google Scholar]
- 38. Rodriguez M. S., Dargemont C., Hay R. T. (2001) SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 276, 12654–12659 [DOI] [PubMed] [Google Scholar]
- 39. Finck B. N., Kelly D. P. (2006) PGC-1 co-activators. Inducible regulators of energy metabolism in health and disease. J. Clin. Invest. 116, 615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin J., Handschin C., Spiegelman B. M. (2005) Metabolic control through the PGC-1 family of transcription co-activators. Cell Metab. 1, 361–370 [DOI] [PubMed] [Google Scholar]
- 41. Lin J., Yang R., Tarr P. T., Wu P. H., Handschin C., Li S., Yang W., Pei L., Uldry M., Tontonoz P., Newgard C. B., Spiegelman B. M. (2005) Hyperlipidemic effects of dietary saturated fats mediated through PGC-1β co-activation of SREBP. Cell 120, 261–273 [DOI] [PubMed] [Google Scholar]
- 42. Wu Y., Delerive P., Chin W. W., Burris T. P. (2002) Requirement of helix 1 and the AF-2 domain of the thyroid hormone receptor for co-activation by PGC-1. J. Biol. Chem. 277, 8898–8905 [DOI] [PubMed] [Google Scholar]
- 43. Shuai K. (2006) Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 16, 196–202 [DOI] [PubMed] [Google Scholar]
- 44. Hotamisligil G. S., Erbay E. (2008) Nutrient sensing and inflammation in metabolic diseases. Nat. Rev. Immunol. 8, 923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu B., Mink S., Wong K. A., Stein N., Getman C., Dempsey P. W., Wu H., Shuai K. (2004) PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nat. Immunol. 5, 891–898 [DOI] [PubMed] [Google Scholar]
- 46. Liu B., Yang R., Wong K. A., Getman C., Stein N., Teitell M. A., Cheng G., Wu H., Shuai K. (2005) Negative regulation of NF-κB signaling by PIAS1. Mol. Cell. Biol. 25, 1113–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rytinki M. M., Palvimo J. J. (2009) SUMOylation attenuates the function of PGC-1alpha. J. Biol. Chem. 284, 26184–26193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ghisletti S., Huang W., Ogawa S., Pascual G., Lin M. E., Willson T. M., Rosenfeld M. G., Glass C. K. (2007) Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARγ. Mol. Cell 25, 57–70 [DOI] [PMC free article] [PubMed] [Google Scholar]