

Background: NIPA is involved in timing mitotic entry and is inactivated by phosphorylation. The kinase responsible for NIPA phosphorylation at G2/M is unknown.
Results: We show that cell cycle-dependent phosphorylation of NIPA is mediated by ERK2.
Conclusion: NIPA is identified as one of the very few ERK2-specific substrates at the G2/M transition.
Significance: This demonstrates divergent functions of ERK1 and ERK2 in cell cycle regulation.
Keywords: Cancer Biology, Cell Cycle, MAP Kinases (MAPKs), Mitosis, Phosphorylation, Ubiquitin Ligase
Abstract
NIPA is an F-box-like protein that contributes to the timing of mitotic entry. It targets nuclear cyclin B1 for ubiquitination in interphase, whereas in G2/M phase, NIPA is inactivated by phosphorylation to allow for cyclin B1 accumulation, a critical event for proper G2/M transition. We recently specified three serine residues of NIPA and demonstrated a sequential phosphorylation at G2/M, where initial Ser-354 and Ser-359 phosphorylation is most crucial for SCFNIPA inactivation. In this study, we identified ERK2 as the kinase responsible for this critical initial phosphorylation step. Using in vitro kinase assays, we found that both ERK1 and ERK2 phosphorylated NIPA with high efficiency. Mutation of either Ser-354 or Ser-359 abolished ERK-dependent NIPA phosphorylation. Pharmacologic inhibition of ERK1/2 in cell lines resulted in decreased NIPA phosphorylation at G2/M. By combining cell cycle synchronization with stable expression of shRNA targeting either ERK1 or ERK2, we showed that ERK2 but not ERK1 mediated NIPA inactivation at G2/M. ERK2 knockdown led to a delay at the G2/M transition, a phenotype also observed in cells expressing a phospho-deficient mutant of NIPA. Thus, our data add to the recently described divergent functions of ERK1 and ERK2 in cell cycle regulation, which may be due in part to the differential ability of these kinases to phosphorylate and inactivate NIPA at G2/M.
Introduction
Timely degradation of proteins that control cell proliferation and apoptosis is an essential mechanism in keeping normal growth from turning into malignancy. We previously identified a novel mammalian E3 ubiquitin ligase that links ubiquitin-mediated protein destruction to the subcellular regulation of cyclin B1 (CCNB1) (1–5). SCFNIPA is defined by the F-box-like protein NIPA (nuclear interaction partner of anaplastic lymphoma kinase) and belongs to the SCF (SKP1/cullin/F-box) family of ubiquitin ligases. This family of ubiquitin ligases controls the abundance of key cell cycle regulatory proteins. The F-box protein is the only variable component of the SCF complex that mediates substrate binding and thus determines specificity of the respective SCF complex (6–9). The functions of the SCF-type ubiquitin ligases and F-box proteins in particular have been reviewed in detail (9). The prominent hallmark of the SCFNIPA complex is its ability to act as a cell cycle oscillator. The oscillating activity of the SCFNIPA complex is governed by the cell cycle-dependent inhibitory phosphorylation of NIPA in late G2 phase, which dissociates NIPA from the SCF core. This phosphorylation event was found to occur first on Ser-354 and Ser-359 and subsequently on Ser-395, suggesting that at least three distinct phosphorylation sites are involved in inactivation of NIPA at late G2 phase and mitosis (10). Previous studies demonstrated a sequential process in which the initial phosphorylation of NIPA at Ser-354 and Ser-359 disrupts and thereby inactivates the SCF complex. Then, CCNB1-CDK1 enhances phosphorylation at Ser-395 to ensure its own activity (10). Although we identified CCNB1-CDK1 as the kinase responsible for the phosphorylation of NIPA at Ser-395, the kinase(s) responsible for the initial phosphorylation at Ser-354 and Ser-359 remained enigmatic.
The MAPK cascade is required for the proliferation, embryogenesis, differentiation, and survival of somatic mammalian cells and is activated by a wide variety of mitogenic and oncogenic stimuli (reviewed in Refs. 11–14). Activation of the MAPK ERK1/2 has been linked to cell survival, growth, and differentiation, and the MAPKs JNK and p38 also control induction of apoptosis (15). More than 160 different substrates have been identified so far for the MAPK ERK1/2 (16), most of them with nuclear localization. ERK1/2 is part of the cell cycle machinery and is activated in early mitosis and inactivated before the exit from M phase (17–20). Enhanced ERK1/2 activation is associated with advanced cell cycle progression (21–23). It has been shown that mitogen-induced ERK1/2 activity can accelerate the G1/S transition (21, 24, 25). Recently, evidence has accumulated that ERK1/2 can also influence the G2/M transition (26–28). Several reports demonstrated activation of ERK1/2 in G2/M (29, 30) and also demonstrated a crucial role for these kinases in proper G2/M transition (31–33).
In Xenopus oocytes, activation of ERK1/2 is necessary for maturation from meiotic prophase I to metaphase II (34). After fertilization, inactivation of ERK1/2 is essential for the first G2/M progression (35, 36). Inappropriate activation of MAPK or its downstream kinases can cause cell cycle arrest at either G1/S or G2/M (17–20, 37, 38).
Here, we show that the F-box-like protein NIPA is a substrate of ERK2 and not ERK1 at the G2/M transition. We propose that the ERK2-mediated phosphorylation of NIPA is important for faithful cell cycle progression.
EXPERIMENTAL PROCEDURES
Plasmids, Inhibitors, Antibodies, and Immunological Procedures
pLMP-miR constructs (Open Biosystems) were designed as described previously (39).3 21-mer sequences for microRNA-30-based shRNA were as follows: ERK1, TTCCGCCATGAGAATGTTATA; ERK2, CAGGAAGATCTGAATTGTATA; and control, TCTCGCTTGGGCGAGAGTAAG. PD98059 (2′-amino-3′-methoxyflavone) and U0126 (1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene) were purchased from Cell Signaling. Mouse monoclonal antibodies were purchased from Sigma (anti-FLAG (M2) and anti-β-actin) and Santa Cruz Biotechnology (anti-CUL1, anti-cyclin B1 (GNS1), and anti-α-tubulin). Rabbit polyclonal and monoclonal antibodies were from Santa Cruz Biotechnology (anti-cyclin A and anti-SKP1), Cell Signaling (anti-ERK p42/44 MAPK and anti-phospho-ERK p42/44 MAPK (Thr-202/Tyr-204)), Upstate (anti-phospho-histone 3 (Ser-10)), and Abcam (anti-phospho-NIPA (Ser-354)). Anti-ALK4 antibody was a kind gift from Stephan W. Morris. Immunoblot analysis and immunoprecipitation were performed as described previously (39).
Cell Culture and Cell Cycle Analysis
NIH/3T3 cells, primary Nipa-deficient MEFs (5), HEK293T cells, and Phoenix E cells were cultivated in DMEM containing 10% FCS, and KARPAS 299 cells were cultivated in RPMI 1640 medium containing 10% FCS. Transient transfections of HEK293T and Phoenix E cells were performed using FuGENE 6 (Roche Applied Science) or Lipofectamine 2000 (Invitrogen) transfection reagent. Cell synchronization at G2/M was performed by sequential culture with 2 mm thymidine and 40 ng/ml nocodazole, followed by mitotic shake off. Synchronization in G1/S was performed using double-thymidine block as described previously (1). For G0/G1 synchronization, cells were harvested in DMEM containing 0.5% FCS for 42 h. For BrdU incorporation and DNA content analysis, cells were pulsed with 10 μm BrdU (BD Biosciences) for 45 min and subsequently stained according to a standard protocol. Cell cycle distribution was determined by flow cytometry and subsequent analysis using FlowJo software (Tree Star Inc.). For mitotic index measurement, cells (HeLa) were grown on coverslips, and U0126 (5 μm)/Me2SO and nocodazole (40 ng/ml) were added at the respective time points and fixed in 3% paraformaldehyde. Immunofluorescence was performed as described previously (1).
Kinase Assays and GST Fusion Proteins
For GST fusion proteins, wild-type NIPA fragments (amino acids 352–402) and all NIPA point mutants (S354A, S359A, and S354A/S359A) were expressed in Escherichia coli using pGEX vectors (Amersham Biosciences). Protein expression and purification were performed as described (10).
For kinase assay using fully purified components, active forms of the kinases (ERK1 and ERK2) and purified GST substrates were transferred into the kinase reaction containing 250 mm Tris-HCl (pH 7.5), 50 mm MgCl2, 50 μm ATP, 1 μCi of [γ-32P]ATP (Amersham Biosciences), and 1 mm DTT. The kinase reaction was carried out at 30 °C for 5 min.
Retrovirus Production and Infection of NIH/3T3 Cells and MEFs
2 × 106 Phoenix E cells were plated on 60-mm dishes and transiently transfected using Lipofectamine 2000. Retroviral stocks were collected at 12-h intervals beginning 24 h after transfection. To generate stable retrovirus-infected cell lines of fibroblast origin, NIH/3T3 cells or MEFs were transduced by three rounds of infection every 12 h in retroviral supernatant supplemented with 4 μg/ml Polybrene.
RESULTS
p42/44 MAPK Phosphorylates NIPA in Vitro at Ser-354 and Ser-359
The phosphorylation of NIPA at G2/M involves a sequential process at three different phosphorylation sites. Previous studies demonstrated Ser-354 to be the important initial phosphorylation site dissociating NIPA from the SCF core complex (1). Whereas CCNB1-CDK1 has been identified as the kinase responsible for the final NIPA phosphorylation at Ser-395 (10), the kinase responsible for the first crucial phosphorylation at Ser-354 and Ser-359 remains unknown. Previously, several kinases were tested that are involved in the G2/M transition or predicted by consensus sequence analysis to be candidates for phosphorylation at Ser-354 and Ser-359, such as GSK3β, casein kinase 2, and Polo-like kinase 1. However, none of the tested kinases was able to specifically phosphorylate NIPA in vitro (10).
Recently, evidence has accumulated that MAPKs are involved in the regulation of the G2/M transition. In addition, NIPA Ser-359 has a p42/44 MAPK phosphorylation motif. To test the possible impact of MEK-activated kinases on the cell cycle-dependent phosphorylation of NIPA at the G2/M transition, we synchronized NIH/3T3 cells stably expressing FLAG-tagged NIPA in prometaphase and inhibited the Raf/MEK/ERK pathway with MEK inhibitors. PD98059 is a synthetic compound that inhibits noncompetitively the activation of MEK1 and, to a lesser extent, MEK2, thereby blocking ERK1/2 activity. In contrast, U0126 is an organic matter that inhibits MEK1 and MEK2 to the same extent. Moreover, in contrast with PD98059, U0126 blocks the already activated MEK isoforms, thereby inhibiting ERK1/2 activation in a more specific and potent way (40). The phosphorylation of NIPA was analyzed by the shift of the protein in electrophoretic mobility, which has previously been shown to be due to phosphorylation at Ser-354, Ser-359, and Ser-395 (10). As shown in Fig. 1 (A and B), both inhibitors abolished the phosphorylation of NIPA at G2/M. Cell cycle analyses demonstrated correct synchronization of the respective samples (Fig. 1, A and B, lower panels). This result suggested an involvement of MEK/ERK kinases in the phosphorylation of NIPA at G2/M. To further substantiate a potential role of the MAPKs ERK1 and ERK2 in the phosphorylation of NIPA in mitosis, we performed an in vitro kinase assay using purified ERK1 and ERK2 and a GST-NIPA fragment as a substrate. This NIPA fragment contains 51 amino acids (positions 352–402) around the relevant phosphorylation sites to minimize unspecific reactions. As shown in Fig. 1C, ERK1 and ERK2 were able to transfer 32P to the NIPA fragment. Next, we wanted to determine the relevant site for this in vitro phosphorylation. Because we previously demonstrated involvement of Ser-354 and Ser-359 in G2/M-specific NIPA phosphorylation (1, 10), we tested the respective NIPA mutants (S354A and S359A) and the double mutant (S354A/S359A) for phosphorylation by ERK1/2 in vitro. As shown in Fig. 1E (fifth lane), the double mutation of Ser-354 and Ser-359 totally abrogated the phosphorylation-dependent electrophoretic mobility shift of NIPA by ERK2. The same results could be observed for the single mutants S354A and S359A, indicating an involvement of both phosphorylation sites in the ERK2-dependent phosphorylation of NIPA in vitro (Fig. 1E). Similar results were obtained in vitro with ERK1 (Fig. 1D). Coomassie Blue staining confirmed comparable amounts of deployed substrate (Fig. 1F).
FIGURE 1.

p42/44 MAPK phosphorylates NIPA in vitro at Ser-354 and Ser-359. A and B, NIH/3T3 cells stably expressing FLAG-tagged NIPA were synchronized at G2/M by a 24-h thymidine block, followed by nocodazole arrest. After release of the cells from the thymidine block, the medium was supplemented with 5 μm MEK inhibitor U0126 (A), 50 μm MEK inhibitor PD98059 (B), or the equivalent amount of Me2SO. Immunoblot (IB) analyses to detect the phosphorylation-dependent mobility shift of FLAG-tagged NIPA (upper panels) and cell cycle analyses with propidium iodide (PI)-stained samples to ensure correct cell synchronization (lower panels) were performed. unsyn., unsynchronized. C–F, GST proteins of the NIPA protein binding region (amino acids 352–402), either WT (C) or point mutants of the indicated residues (D–F), were transferred to an in vitro kinase reaction to test for phosphorylation by purified active ERK1 (C and D) and ERK2 (C and E) in the presence of [32P]ATP. Phosphorylated proteins were visualized by autoradiography (D and E). GST protein loading was detected by Coomassie Blue staining (F).
To further investigate the role of ERK1/2 in phosphorylating NIPA at the G2/M transition, we cloned retroviral vectors coding for two different shRNAs that specifically inhibit either ERK1 or ERK2. After retroviral infection and selection of NIH/3T3 cells stably expressing FLAG-tagged NIPA, we synchronized the cells in prometaphase. Fig. 2A shows the cell cycle-dependent phosphorylation of NIPA in control siRNA-transduced cells. ERK2 knockdown totally abolished this phosphorylation in G2/M-synchronized cells. In contrast, ERK1 knockdown was not able to block NIPA phosphorylation at G2/M (fourth lane). ERK immunoblots demonstrated the specific RNAi-mediated down-regulation of the two ERK isoforms. Cell cycle analyses (Fig. 2A, lower panel) showed appropriate synchronization of the cells at G2/M. To confirm this mechanism with endogenous NIPA protein, we infected parental NIH/3T3 cells with retrovirus encoding specific ERK2 shRNA (Fig. 2B) and showed that endogenous NIPA phosphorylation was abrogated at G2/M by ERK2 inhibition. Thus, whereas both ERK1 and ERK2 specifically phosphorylate NIPA in vitro, only ERK2 seems to mediate NIPA phosphorylation at the G2/M transition in synchronized cells.
FIGURE 2.

ERK2- but not ERK1-dependent phosphorylation of NIPA at G2/M leads to dissociation of the SCFNIPA complex. A, NIH/3T3 cells stably expressing FLAG-tagged NIPA were retrovirally infected with pLMP vectors encoding a microRNA-30-based shRNA directed against either ERK1 or ERK2 or a control sequence. 48 h after the last infection, cells were synchronized in prometaphase and monitored for NIPA phosphorylation at G2/M. In addition, immunoblot (IB) analyses of phospho-ERK1/2 and ERK1/2 were preformed to guarantee specific knockdown (upper panels), and propidium iodide (PI) cell cycle analyses were performed to ensure correct cell synchronization (lower panels). asyn., asynchronized; ko, knock-out. B, NIH/3T3 cells infected with pLMP vectors encoding either ERK2 or control shRNA were synchronized in prometaphase and monitored for endogenous NIPA phosphorylation at G2/M. Immunoblot analyses of ERK1/2 to guarantee specific knockdown and propidium iodide cell cycle analyses to ensure correct cell synchronization were preformed. C, for co-immunoprecipitation of components of the SCF core complex, NIH/3T3 cells stably expressing FLAG-tagged NIPA were infected with pLMP vectors encoding either ERK2 or control shRNA retrovirus. Thereafter, cells were synchronized at G2/M using a sequential thymidine/nocodazole treatment or left unsynchronized. FLAG-tagged NIPA was immunoprecipitated, and the bound protein fraction was subjected to immunoblot analyses for binding of CUL1. Cell cycle profiles of asynchronous and synchronized cells were analyzed by flow cytometry (lower panel). D, ERK2 is activated at the G2/M transition. NIH/3T3 cells were synchronized at G2/M (D) or G0/G1 (E) and subjected to immunoblot analyses with phospho-ERK and ERK. Phospho-histone 3 immunoblot and propidium iodide cell cycle analyses ensured correct cell synchronization. F, ERK activation during the cell cycle was monitored with immunoblotting of G0/G1-released NIH/3T3 cells. Propidium iodide cell cycle analyses ensured correct cell synchronization.
ERK2-dependent Phosphorylation of NIPA at G2/M Leads to Dissociation of the SCFNIPA Complex
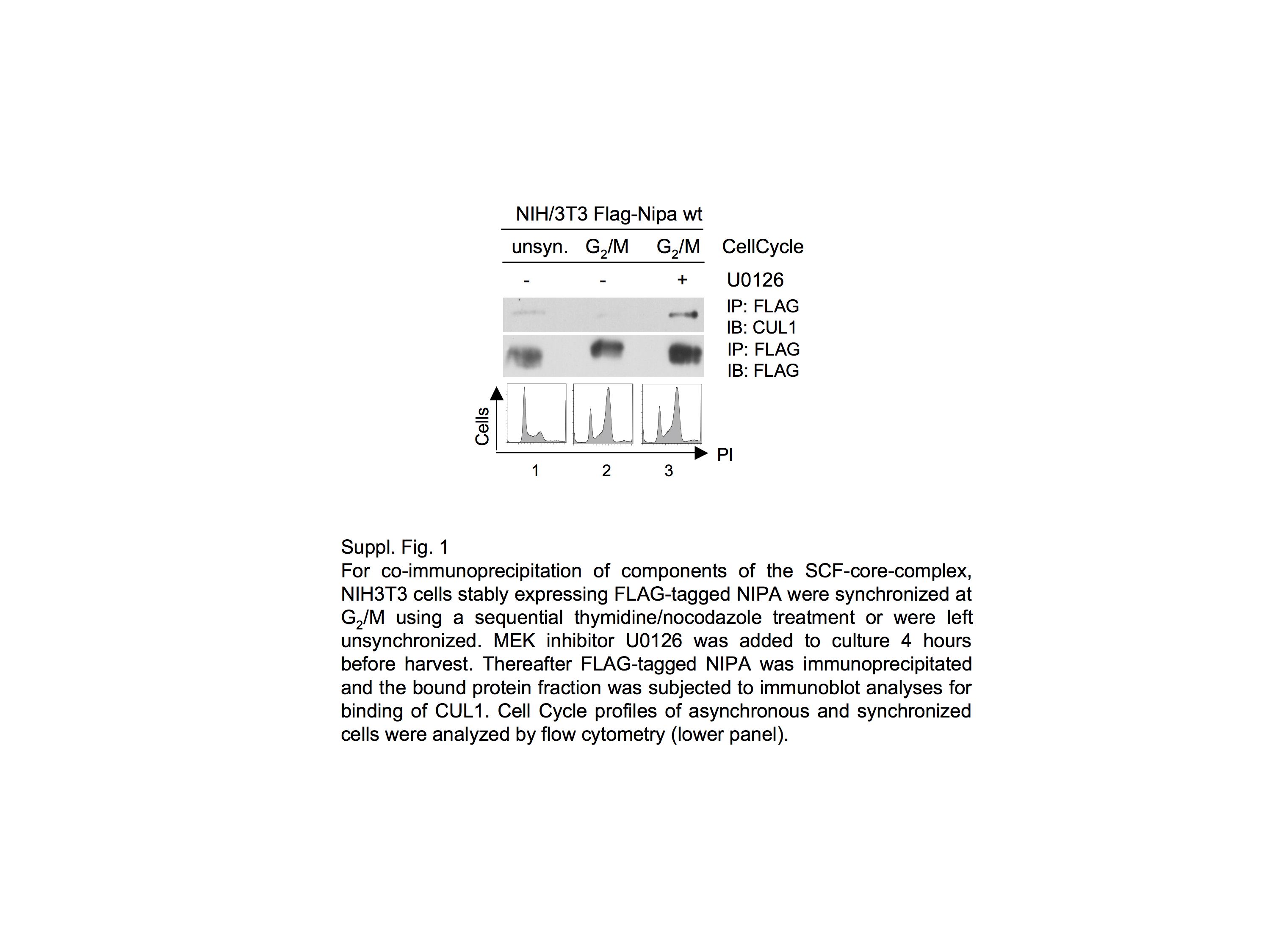
We next examined the functional consequences of NIPA phosphorylation by ERK2. The G2/M-specific phosphorylation of NIPA at Ser-354 has previously been shown to inhibit binding to components of the SCF core, thereby inactivating the SCFNIPA complex (1). We therefore analyzed the ability of NIPA to bind to the SCF core (CUL1) at G2/M when phosphorylated and thereby inactivated by ERK2. For this purpose, we utilized a MEK/ERK inhibitor as well as an ERK2-specific shRNA. NIH/3T3 cells stably expressing FLAG-tagged NIPA were synchronized at G2/M and treated with the MEK1/2 inhibitor U0126. FLAG-tagged NIPA was immunoprecipitated, and binding to endogenous CUL1 was evaluated (supplemental Fig. 1). Cells treated with the MEK inhibitor U0126 maintained binding to CUL1 in G2/M even to a higher extent than in unsynchronized cells. No binding was observed in untreated cells at G2/M. To specifically evaluate the effect of the ERK2-mediated phosphorylation of NIPA at the G2/M transition, the cells established in Fig. 2A were used for co-immunoprecipitation experiments. As shown in Fig. 2C, cells with specific ERK2 knockdown showed strong binding to CUL1 at G2/M, whereas the binding of cells transduced with control siRNA was markedly reduced. In both experiments (Fig. 2C and supplemental Fig. 1, lower panels), cell cycle analyses were performed to ensure synchronization in the specified cell cycle phase. Thus, the ERK2-specific phosphorylation of NIPA at Ser-354 and Ser-395 appears to be sufficient for its efficient dissociation from the SCF core complex at G2/M.
Because it is not clear that ERK kinases are activated in a cell cycle-dependent manner, we synchronized NIH/3T3 cells in different cell cycle phases and monitored activation of ERK1 and ERK2. Interestingly, whereas both ERK isoforms were inhibited at G0/G1 (Fig. 2E) and likewise activated throughout S phase (Fig. 2F), ERK2 seemed to be the prominent kinase at the G2/M transition (Fig. 2D).
Knockdown of ERK2 Leads to Delayed G2/M Progression
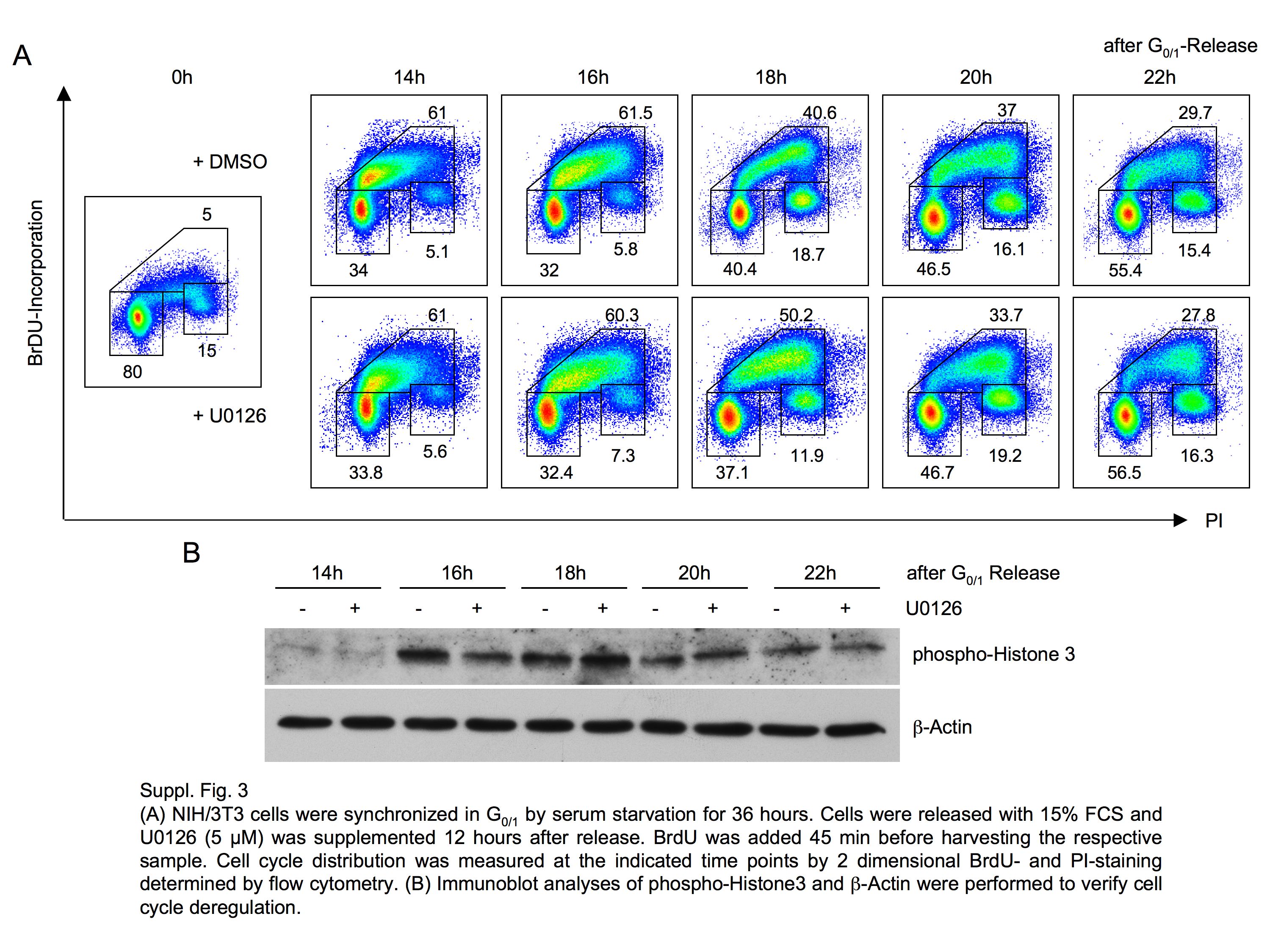
The functional consequence of an assembled SCFNIPA complex in mitosis is ongoing degradation of CCNB1 and thereby a delayed G2/M progression. We thus investigated the impact of ERK2 inhibition on mitotic entry. Again, we first used a broad MEK/ERK inhibitor (supplemental Fig. 2, A and B) and thereafter highly specific ERK2 down-regulation by RNAi (Fig. 3A). Cells were arrested in G1/S by double-thymidine block 48 h after RNAi transduction, and the block was subsequently removed to permit synchronized cell cycle progression. Experiments were repeated using G0/G1 release to ensure DNA replication checkpoint-independent activation of ERK kinases (supplemental Fig. 3, A and B). Using two-dimensional cell cycle analyses with BrdU labeling, an evident delay of mitotic entry was observed in cells in which NIPA phosphorylation was inhibited by ERK2 knockdown compared with control cells (Fig. 3A). A significant delay in mitotic entry by ERK2 inhibition started at 4 h after G1/S release (42% versus 26%) and remained up to 7 h (20% versus 5%). Notably, this cell cycle phenotype in ERK2 knockdown cells closely mimics the phenotype observed in cells expressing the phospho-deficient NIPA S354A mutant (1), arguing that ERK2-mediated reduced phosphorylation at this site may lead to the cell cycle delay. Western blot analysis confirmed efficient knockdown of ERK2, and the cell cycle effects obtained could also be demonstrated by staining for cell cycle-regulated proteins such as cyclin A and CCNB1 in these cells (supplemental Fig. 4).
FIGURE 3.

Knockdown of ERK2 leads to prolonged G2/M progression. A, NIH/3T3 cells stably expressing FLAG-tagged NIPA were retrovirally infected with pLMP vectors encoding ERK2-specific shRNA or a control sequence. 48 h after the last infection, cells were synchronized in G1/S by double-thymidine block. Cells were released from the block, and BrdU was added 45 min before harvesting the respective samples. Cell cycle distribution was measured at the indicated time points by two-dimensional BrdU and propidium iodide (PI) staining determined by flow cytometry. In addition, immunoblot analyses of phospho-ERK1/2 and ERK1/2 to guarantee specific ERK2 knockdown and of cyclins A and B1 to show prolonged mitotic progression were performed (supplemental Fig. 3). miR, microRNA. B, mitotic index of HeLa cells. Shown is representative phospho-histone 3 (FITC), α-tubulin (Texas Red), and DAPI (blue) staining of cells harvested 90 min after nocodazole arrest with or without MEK inhibitor U0126. DMSO, dimethyl sulfoxide. C, quantification of two independent experiments. 1500 cells/slide were counted for phospho-histone H3 positivity. D, immortalized NIPA knock-out (ko) MEFs were retrovirally infected with pLMP vectors encoding ERK2-specific shRNA or a control sequence. 48 h after the last infection, cells were synchronized in G1/S and released as described above. Cell cycle distribution was measured at the indicated time points by BrdU/propidium iodide/flow cytometry.
To demonstrate a direct relationship between ERK inhibition and delay in mitotic entry, we treated asynchronously growing HeLa cells with U0126 or Me2SO and nocodazole and calculated the mitotic index of the respective sample after 30, 60, 90, and 120 min by counting phospho-histone 3-positive cells. As shown in Fig. 3 (B and C), MEK inhibition led to a delay in mitotic entry with 4.22 ± 0.47 versus 6.89 ± 0.72 phospho-histone 3-positive cells after 60 min. To obtain further evidence that ERK2 inhibition delays mitotic entry in part by the reduced phosphorylation of NIPA, we utilized Nipa-deficient MEFs (5) and investigated the timing of mitotic entry with and without ERK2 knockdown. We hypothesized that if ERK2-dependent NIPA phosphorylation plays a crucial role in the timing of mitotic entry, the cell cycle delay mediated by ERK2 inhibition should be abolished in Nipa-deficient cells. Indeed, knockdown of ERK2 in Nipa-deficient MEFs showed no significant differences in mitotic entry compared with control microRNA knockdown cells (Fig. 3B). Together, these results indicate an involvement of ERK2-dependent NIPA phosphorylation at Ser-354 and Ser-359 at G2/M in the timing of mitotic entry.
Oncogene-mediated Phosphorylation of NIPA at Ser-354 Also Involves p42/44 MAPK
NIPA has been shown to be constitutively phosphorylated in NPM-ALK-expressing cells. The fusion protein NPM-ALK is an oncogenic tyrosine kinase, and the serine kinase responsible for the phosphorylation of NIPA at Ser-354 is not known. We thus asked whether ERK2 might also be involved in the oncogenic cell cycle-independent phosphorylation of NIPA in NPM-ALK-positive cells. To this end, we coexpressed NPM-ALK with FLAG-tagged NIPA in HEK293T cells and treated them with different concentrations of the MEK/ERK inhibitors U0126 and PD98059. The NPM-ALK-induced phosphorylation of NIPA could be completely abolished by inhibiting the MEK/ERK pathway with both compounds (Fig. 4, A and B). To further substantiate these data in human lymphoma cells, we analyzed the effects of ERK inhibition on NIPA phosphorylation in a human T-cell lymphoma cell line (KARPAS 299) that was established from the peripheral blood of a 25-year-old patient with NPM-ALK-positive anaplastic large cell lymphoma. Indeed, treatment of KARPAS 299 cells with 5 μm U0126 abolished the phosphorylation of NIPA at Ser-354 (Fig. 4C). These results indicate the involvement of ERK kinases not only in cell cycle-dependent NIPA phosphorylation but also in oncogene-mediated cell cycle-independent NIPA phosphorylation.
FIGURE 4.

NPM-ALK phosphorylates NIPA at Ser-354 via p42/44 MAPK. A and B, FLAG-tagged NIPA and NPM-ALK or MG (MSCV-IRES-GFP) empty vector was cotransfected in HEK293T cells. 48 h after transfection, the medium was supplemented with different concentrations of MEK inhibitor PD98059 (A), MEK inhibitor U0126 (B), or Me2SO for 12 h. Whole cell lysate extracts were prepared, and immunoblot (IB) analyses were subsequently performed using the indicated antibodies. C, KARPAS 299 cells were treated with 5 μm U0126 and harvested after 6 h. Whole cell lysate extracts were prepared, and immunoblot analyses were subsequently performed using the indicated antibodies. untr., untransfected.
Fig. 5 summarizes a model for SCFNIPA inactivation and faithful cell cycle progression based on the results shown here and in a previous report (10). The initial phosphorylation of NIPA at G2/M is mediated by ERK2 at Ser-354 and Ser-395. This leads to the dissociation of the SCFNIPA complex, allowing CCNB1 to accumulate in the nucleus. Subsequently, CCNB1-CDK1 phosphorylates NIPA in addition at Ser-395 to prevent any degradation by the SCFNIPA complex.
FIGURE 5.

Model of NIPA phosphorylation at G2/M. The initial phosphorylation at Ser-354 and Ser-359 by ERK2 dissociates and thereby inactivates the SCFNIPA complex in late G2, leading to increasing CCNB1-CDK1 kinase activity and subsequent mitotic entry. Thereafter, mitotic CCNB1-CDK1 kinase phosphorylates NIPA at Ser-395 to ensure its own activity.
DISCUSSION
It is known that ERK1/2 is part of the cell cycle machinery by regulating the G1/S (21, 24, 25) and G2/M (26–28) transitions. ERK1/2 activation leads to advanced cell cycle progression (21–23).
In this study, we aimed to identify the kinase responsible for the cell cycle-dependent phosphorylation of NIPA, leading to inactivation of the SCFNIPA complex. We found both ERK1 and ERK2 to be potential candidates because both isoforms were able to phosphorylate NIPA rapidly in an in vitro kinase assay. In vitro phosphorylation was greatly impaired in the NIPA mutants S354A and S359A. This finding was remarkable, as it has been shown that phosphorylation at Ser-354 and Ser-359 is most important for inactivation of the SCFNIPA complex (10). The phosphoacceptor site Ser-359 is followed by a proline, which is a motif for ERK1/2 phosphorylation (41–44), whereas at Ser-354, we were not able to identify any ERK substrate recognition motif (45). However, it is well known that ERK1/2 does not necessarily need any distinct consensus sequence for substrate recognition, as other kinases do (46, 47). Indeed, in further experiments, we obtained evidence that ERK2 is the kinase responsible for the phosphorylation of NIPA in a cell cycle-dependent manner. Importantly, this ERK2-mediated phosphorylation seems to lead to the functional inactivation of the SCFNIPA complex in mitosis. We were able to demonstrate that selective ERK2 knockdown leads to a delay in cell cycle progression and entry in mitosis. This phenotype of a prolonged G2 phase closely resembles the phenotype observed in cells expressing a constitutively active mutant of NIPA lacking the critical residue Ser-354 (1). Thus, we propose that ERK2 inhibition or mutation of Ser-354 leads to the permanent association of NIPA with the SCF core complex. Consequently, SCFNIPA continuously ubiquitinates CCNB1, thereby reducing mitosis-promoting factor activity (CCNB1-CDK1 complex) and delaying G2/M progression.
It has already been shown that ERK regulates mitosis-promoting factor activity at G2/M (23, 48, 49). Mitosis-promoting factor regulation is executed at the level of CDK1 activity and by the abundance of nuclear CCNB1. CDK1 activity is inhibited in interphase by WEE1- and MYT1-mediated phosphorylation (50–52). At G2/M, CDC25 dephosphorylates CDK1, leading to its activation (53–55). ERK1/2 plays a positive regulatory role in CDK1 activation by phosphorylating and thereby activating CDC25 (27). In addition, ERK1/2 is able to phosphorylate and inactivate CDK1 inhibitors such as MYT1 (56). At another regulatory level, ERK1/2 is also able to control the abundance of CCNB1 in the nucleus. ERK1/2 is able to phosphorylate CCNB1 at the cytoplasmic retention signal, thereby blocking the export of CCNB1 from the nucleus to the cytoplasm (57). In contrast to the above-described models of ERK-mediated regulation of the G2/M transition, the here-described mechanism involves an ERK isoform-specific phosphorylation. Enhancement of CCNB1 abundance at G2/M by the cell cycle-dependent phosphorylation of NIPA is achieved by ERK2, but not by ERK1.
This observation supports recent evidence for different functions of ERK1 and ERK2 in the regulation of the cell cycle. Initially, ERK1 and ERK2 were believed to be identical with regard to their function because of their highly similar structure (85% homology), synchronous activation, and identical subcellular localization in cells (11, 58, 59). However, recent studies support the hypothesis that ERK1 and ERK2 may have distinct functions, which cannot be compensated for by the other isoenzyme (60–63). With regard to cell cycle regulation, there are only a few publications describing a more important role for ERK2 than for ERK1 in mediating promitogenic cell cycle regulation (27, 64, 65). Our work fits well with this growing number of papers emphasizing an important role of specifically ERK2 for enhancing cell cycle progression. In addition, we have identified here one of the very few known ERK2-specific substrates mediating enhanced G2/M transition. The fact that cell cycle delay by blocking ERK2 could be observed only in NIPA-expressing cells, but not in Nipa-deficient cells, points to a potentially imperative role of this E3 ligase in ERK2-mediated cell cycle effects.
In summary, we propose a model (Fig. 5) in which the initial phosphorylation of NIPA by ERK2 at Ser-354 and Ser-359 dissociates the SCFNIPA complex. Subsequently, CCNB1-CDK1 accumulates and phosphorylates Ser-395 to further back up SCFNIPA inactivation and prevent its own degradation. Because checkpoint proteins such as NIPA are constitutively inactivated in tumor cells, ERK2 might represent an interesting target to reconstitute crucial cell cycle checkpoint control in malignant cells.
Supplementary Material
Acknowledgment
We thank Melanie Sickinger for excellent technical assistance.
This work was supported by Deutsche José Carreras Leukämie Stiftung (DJCLS) Grant R 08/02v and the Wilhelm Sander Stiftung (to J. D.), Technical University of Munich Research Grant KKF-B07-08 (to A. L. I.), German Research Foundation Grant DU 227/3-1 (to A. L. I. and J. D.), and German Research Foundation Grant BA 2851/3-1 and German Cancer Aid Grant 109543 (to F. B.).

This article contains supplemental Figs. 1–4.
Details of the construction of the various Nipa plasmids are available from the authors upon request.
- ALK
- anaplastic lymphoma kinase
- MEF
- mouse embryonic fibroblast
- NPM
- nucleophosmin.
REFERENCES
- 1. Bassermann F., von Klitzing C., Münch S., Bai R. Y., Kawaguchi H., Morris S. W., Peschel C., Duyster J. (2005) NIPA defines an SCF-type mammalian E3 ligase that regulates mitotic entry. Cell 122, 45–57 [DOI] [PubMed] [Google Scholar]
- 2. Ouyang T., Bai R. Y., Bassermann F., von Klitzing C., Klumpen S., Miething C., Morris S. W., Peschel C., Duyster J. (2003) Identification and characterization of a nuclear interacting partner of anaplastic lymphoma kinase (NIPA). J. Biol. Chem. 278, 30028–30036 [DOI] [PubMed] [Google Scholar]
- 3. Bassermann F., Peschel C., Duyster J. (2005) Mitotic entry: a matter of oscillating destruction. Cell Cycle 4, 1515–1517 [DOI] [PubMed] [Google Scholar]
- 4. von Klitzing C., Huss R., Illert A. L., Fröschl A., Wötzel S., Peschel C., Bassermann F., Duyster J. (2011) APC/CCdh1-mediated degradation of the F-box protein NIPA is regulated by its association with Skp1. PLoS ONE 6, e28998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Illert A. L., Kawaguchi H., Antinozzi C., Bassermann F., Quintanilla-Martinez L., von Klitzing C., Hiwatari M., Peschel C., de Rooij D. G., Morris S. W., Barchi M., Duyster J. (2012) Targeted inactivation of nuclear interaction partner of ALK disrupts meiotic prophase. Development 139, 2523–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bai C., Sen P., Hofmann K., Ma L., Goebl M., Harper J. W., Elledge S. J. (1996) SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 86, 263–274 [DOI] [PubMed] [Google Scholar]
- 7. Feldman R. M., Correll C. C., Kaplan K. B., Deshaies R. J. (1997) A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 91, 221–230 [DOI] [PubMed] [Google Scholar]
- 8. Skowyra D., Craig K. L., Tyers M., Elledge S. J., Harper J. W. (1997) F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell 91, 209–219 [DOI] [PubMed] [Google Scholar]
- 9. Cardozo T., Pagano M. (2004) The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 5, 739–751 [DOI] [PubMed] [Google Scholar]
- 10. Bassermann F., von Klitzing C., Illert A. L., Münch S., Morris S. W., Pagano M., Peschel C., Duyster J. (2007) Multisite phosphorylation of nuclear interaction partner of ALK (NIPA) at G2/M involves cyclin B1/Cdk1. J. Biol. Chem. 282, 15965–15972 [DOI] [PubMed] [Google Scholar]
- 11. Chang L., Karin M. (2001) Mammalian MAP kinase signaling cascades. Nature 410, 37–40 [DOI] [PubMed] [Google Scholar]
- 12. Hindley A., Kolch W. (2002) Extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK)-independent functions of Raf kinases. J. Cell Sci. 115, 1575–1581 [DOI] [PubMed] [Google Scholar]
- 13. Kolch W. (2005) Coordinating ERK/MAPK signaling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 6, 827–837 [DOI] [PubMed] [Google Scholar]
- 14. Dhillon A. S., Hagan S., Rath O., Kolch W. (2007) MAP kinase signaling pathways in cancer. Oncogene 26, 3279–3290 [DOI] [PubMed] [Google Scholar]
- 15. Xia Z., Dickens M., Raingeaud J., Davis R. J., Greenberg M. E. (1995) Opposing effects of ERK and JNK p38 MAP kinases on apoptosis. Science 270, 1326–1331 [DOI] [PubMed] [Google Scholar]
- 16. Yoon S., Seger R. (2006) The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24, 21–44 [DOI] [PubMed] [Google Scholar]
- 17. MacNicol A. M., Muslin A. J., Howard E. L., Kikuchi A., MacNicol M. C., Williams L. T. (1995) Regulation of Raf-1-dependent signaling during early Xenopus development. Mol. Cell. Biol. 15, 6686–6693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murakami M. S., Vande Woude G. F. (1998) Analysis of the early embryonic cell cycles of Xenopus; regulation of cell cycle length by Xe-wee1 and Mos. Development 125, 237–248 [DOI] [PubMed] [Google Scholar]
- 19. Guadagno T. M., Ferrell J. E., Jr. (1998) Requirement for MAPK activation for normal mitotic progression in Xenopus egg extracts. Science 282, 1312–1315 [DOI] [PubMed] [Google Scholar]
- 20. Chau A. S., Shibuya E. K. (1999) Inactivation of p42 mitogen-activated protein kinase is required for exit from M phase after cyclin destruction. J. Biol. Chem. 274, 32085–32090 [DOI] [PubMed] [Google Scholar]
- 21. Yamamoto T., Ebisuya M., Ashida F., Okamoto K., Yonehara S., Nishida E. (2006) Continuous ERK activation down-regulates antiproliferative genes throughout G1 phase to allow cell cycle progression. Curr. Biol. 16, 1171–1182 [DOI] [PubMed] [Google Scholar]
- 22. Roberts E. C., Shapiro P. S., Nahreini T. S., Pages G., Pouyssegur J., Ahn N. G. (2002) Distinct cell cycle timing requirements for extracellular signal-regulated kinase and phosphoinositide 3-kinase signaling pathways in somatic cell mitosis. Mol. Cell. Biol. 22, 7226–7241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chambard J. C., Lefloch R., Pouysségur J., Lenormand P. (2007) ERK implication in cell cycle regulation. Biochim. Biophys. Acta 1773, 1299–1310 [DOI] [PubMed] [Google Scholar]
- 24. Roovers K., Assoian R. K. (2000) Integrating the MAP kinase signal into the G1 phase cell cycle machinery. BioEssays 22, 818–826 [DOI] [PubMed] [Google Scholar]
- 25. Meloche S., Pouysségur J. (2007) The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1-to-S phase transition. Oncogene 26, 3227–3239 [DOI] [PubMed] [Google Scholar]
- 26. Dumesic P. A., Scholl F. A., Barragan D. I., Khavari P. A. (2009) Erk1/2 MAP kinases are required for epidermal G2/M progression. J. Cell Biol. 185, 409–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang R., He G., Nelman-Gonzalez M., Ashorn C. L., Gallick G. E., Stukenberg P. T., Kirschner M. W., Kuang J. (2007) Regulation of Cdc25C by ERK-MAP kinases during the G2/M transition. Cell 128, 1119–1132 [DOI] [PubMed] [Google Scholar]
- 28. Liu X., Yan S., Zhou T., Terada Y., Erikson R. L. (2004) The MAP kinase pathway is required for entry into mitosis and cell survival. Oncogene 23, 763–776 [DOI] [PubMed] [Google Scholar]
- 29. Lavoie J. N., L'Allemain G., Brunet A., Müller R., Pouysségur J. (1996) Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 271, 20608–20616 [DOI] [PubMed] [Google Scholar]
- 30. Balmanno K., Cook S. J. (1999) Sustained MAP kinase activation is required for the expression of cyclin D1, p21Cip1, and a subset of AP-1 proteins in CCL39 cells. Oncogene 18, 3085–3097 [DOI] [PubMed] [Google Scholar]
- 31. Hayne C., Tzivion G., Luo Z. (2000) Raf-1/MEK/MAPK pathway is necessary for the G2/M transition induced by nocodazole. J. Biol. Chem. 275, 31876–31882 [DOI] [PubMed] [Google Scholar]
- 32. Tolwinski N. S., Shapiro P. S., Goueli S., Ahn N. G. (1999) Nuclear localization of mitogen-activated protein kinase kinase 1 (MKK1) is promoted by serum stimulation and G2/M progression. Requirement for phosphorylation at the activation lip and signaling downstream of MKK. J. Biol. Chem. 274, 6168–6174 [DOI] [PubMed] [Google Scholar]
- 33. Zecevic M., Catling A. D., Eblen S. T., Renzi L., Hittle J. C., Yen T. J., Gorbsky G. J., Weber M. J. (1998) Active MAP kinase in mitosis: localization at kinetochores and association with the motor protein CENP-E. J. Cell Biol. 142, 1547–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kosako H., Gotoh Y., Nishida E. (1994) Requirement for the MAP kinase kinase/MAP kinase cascade in Xenopus oocyte maturation. EMBO J. 13, 2131–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Abrieu A., Fisher D., Simon M. N., Dorée M., Picard A. (1997) MAPK inactivation is required for the G2-to-M phase transition of the first mitotic cell cycle. EMBO J. 16, 6407–6413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bitangcol J. C., Chau A. S., Stadnick E., Lohka M. J., Dicken B., Shibuya E. K. (1998) Activation of the p42 mitogen-activated protein kinase pathway inhibits Cdc2 activation and entry into M phase in cycling Xenopus egg extracts. Mol. Biol. Cell 9, 451–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bhatt R. R., Ferrell J. E., Jr. (1999) The protein kinase p90 Rsk as an essential mediator of cytostatic factor activity. Science 286, 1362–1365 [DOI] [PubMed] [Google Scholar]
- 38. Gross S. D., Schwab M. S., Lewellyn A. L., Maller J. L. (1999) Induction of metaphase arrest in cleaving Xenopus embryos by the protein kinase p90Rsk. Science 286, 1365–1367 [DOI] [PubMed] [Google Scholar]
- 39. Albers C., Illert A. L., Miething C., Leischner H., Thiede M., Peschel C., Duyster J. (2011) An RNAi-based system for loss-of-function analysis identifies Raf-1 as a crucial mediator of BCR-ABL-driven leukemogenesis. Blood 118, 2200–2210 [DOI] [PubMed] [Google Scholar]
- 40. Favata M. F., Horiuchi K. Y., Manos E. J., Daulerio A. J., Stradley D. A., Feeser W. S., Van Dyk D. E., Pitts W. J., Earl R. A., Hobbs F., Copeland R. A., Magolda R. L., Scherle P. A., Trzaskos J. M. (1998) Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 273, 18623–18632 [DOI] [PubMed] [Google Scholar]
- 41. Brummer T., Naegele H., Reth M., Misawa Y. (2003) Identification of novel ERK-mediated feedback phosphorylation sites at the C terminus of B-Raf. Oncogene 22, 8823–8834 [DOI] [PubMed] [Google Scholar]
- 42. Pearson G., Robinson F., Beers Gibson T., Xu B. E., Karandikar M., Berman K., Cobb M. H. (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 22, 153–183 [DOI] [PubMed] [Google Scholar]
- 43. Ramos J. W. (2008) The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int. J. Biochem. Cell Biol. 40, 2707–2719 [DOI] [PubMed] [Google Scholar]
- 44. Shaul Y. D., Seger R. (2007) The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim. Biophys. Acta 1773, 1213–1226 [DOI] [PubMed] [Google Scholar]
- 45. Gonzalez F. A., Raden D. L., Davis R. J. (1991) Identification of substrate recognition determinants for human ERK1 and ERK2 protein kinases. J. Biol. Chem. 266, 22159–22163 [PubMed] [Google Scholar]
- 46. Chuderland D., Seger R. (2005) Protein-protein interactions in the regulation of the extracellular signal-regulated kinase. Mol. Biotechnol. 29, 57–74 [DOI] [PubMed] [Google Scholar]
- 47. Corbalan-Garcia S., Yang S. S., Degenhardt K. R., Bar-Sagi D. (1996) Identification of the mitogen-activated protein kinase phosphorylation sites on human Sos1 that regulate interaction with Grb2. Mol. Cell. Biol. 16, 5674–5682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Abrieu A., Dorée M., Fisher D. (2001) The interplay between cyclin B-Cdc2 kinase (MPF) and MAP kinase during maturation of oocytes. J. Cell Sci. 114, 257–267 [DOI] [PubMed] [Google Scholar]
- 49. Abrieu A., Dorée M., Picard A. (1997) Mitogen-activated protein kinase activation down-regulates a mechanism that inactivates cyclin B-Cdc2 kinase in G2-arrested oocytes. Mol. Biol. Cell 8, 249–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Heald R., McLoughlin M., McKeon F. (1993) Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell 74, 463–474 [DOI] [PubMed] [Google Scholar]
- 51. King R. W., Jackson P. K., Kirschner M. W. (1994) Mitosis in transition. Cell 79, 563–571 [DOI] [PubMed] [Google Scholar]
- 52. Liu F., Stanton J. J., Wu Z., Piwnica-Worms H. (1997) The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol. Cell. Biol. 17, 571–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Borgne A., Meijer L. (1996) Sequential dephosphorylation of p34cdc2 on Thr-14 and Tyr-15 at the prophase/metaphase transition. J. Biol. Chem. 271, 27847–27854 [DOI] [PubMed] [Google Scholar]
- 54. Gautier J., Solomon M. J., Booher R. N., Bazan J. F., Kirschner M. W. (1991) Cdc25 is a specific tyrosine phosphatase that directly activates p34cdc2. Cell 67, 197–211 [DOI] [PubMed] [Google Scholar]
- 55. Krek W., Nigg E. A. (1991) Mutations of p34cdc2 phosphorylation sites induce premature mitotic events in HeLa cells: evidence for a double block to p34cdc2 kinase activation in vertebrates. EMBO J. 10, 3331–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Palmer A., Gavin A. C., Nebreda A. R. (1998) A link between MAP kinase and p34cdc2/cyclin B during oocyte maturation: p90rsk phosphorylates and inactivates the p34cdc2 inhibitory kinase Myt1. EMBO J. 17, 5037–5047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Walsh S., Margolis S. S., Kornbluth S. (2003) Phosphorylation of the cyclin B1 cytoplasmic retention sequence by mitogen-activated protein kinase and Plx. Mol. Cancer Res. 1, 280–289 [PubMed] [Google Scholar]
- 58. Boulton T. G., Nye S. H., Robbins D. J., Ip N. Y., Radziejewska E., Morgenbesser S. D., DePinho R. A., Panayotatos N., Cobb M. H., Yancopoulos G. D. (1991) ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine-phosphorylated in response to insulin and NGF. Cell 65, 663–675 [DOI] [PubMed] [Google Scholar]
- 59. Seger R., Krebs E. G. (1995) The MAPK signaling cascade. FASEB J. 9, 726–735 [PubMed] [Google Scholar]
- 60. Hatano N., Mori Y., Oh-hora M., Kosugi A., Fujikawa T., Nakai N., Niwa H., Miyazaki J., Hamaoka T., Ogata M. (2003) Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells 8, 847–856 [DOI] [PubMed] [Google Scholar]
- 61. Saba-El-Leil M. K., Vella F. D., Vernay B., Voisin L., Chen L., Labrecque N., Ang S. L., Meloche S. (2003) An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep. 4, 964–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bessard A., Frémin C., Ezan F., Fautrel A., Gailhouste L., Baffet G. (2008) RNAi-mediated ERK2 knockdown inhibits growth of tumor cells in vitro and in vivo. Oncogene 27, 5315–5325 [DOI] [PubMed] [Google Scholar]
- 63. Frémin C., Ezan F., Boisselier P., Bessard A., Pagès G., Pouysségur J., Baffet G. (2007) ERK2 but not ERK1 plays a key role in hepatocyte replication: an RNAi-mediated ERK2 knockdown approach in wild-type and ERK1 null hepatocytes. Hepatology 45, 1035–1045 [DOI] [PubMed] [Google Scholar]
- 64. Lloyd A. C. (2006) Distinct functions for ERKs? J. Biol. 5, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vantaggiato C., Formentini I., Bondanza A., Bonini C., Naldini L., Brambilla R. (2006) ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J. Biol. 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}