

Background: α1-Antitrypsin (α1AT) deficiency (α1ATD) is a consequence of defective folding, trafficking, and secretion of α1AT.
Results: SAHA restores the secretion of an active form of Z-α1AT in part through a calnexin- and HDAC7-sensitive dependent mechanism(s).
Conclusion: SAHA may represent a potential therapeutic approach for α1ATD.
Significance: SAHA is a regulator of the proteostasis biology of Z-α1AT, favoring export of a functional form to serum.
Keywords: ER Quality Control, Genetic Diseases, Histone Deacetylase, Histone Deacetylase Inhibitors, Protein Misfolding, α1-Antitrypsin, α1-Antitrypsin Deficiency, Calnexin, HDAC7, Proteostasis
Abstract
α1-Antitrypsin (α1AT) deficiency (α1ATD) is a consequence of defective folding, trafficking, and secretion of α1AT in response to a defect in its interaction with the endoplasmic reticulum proteostasis machineries. The most common and severe form of α1ATD is caused by the Z-variant and is characterized by the accumulation of α1AT polymers in the endoplasmic reticulum of the liver leading to a severe reduction (>85%) of α1AT in the serum and its anti-protease activity in the lung. In this organ α1AT is critical for ensuring tissue integrity by inhibiting neutrophil elastase, a protease that degrades elastin. Given the limited therapeutic options in α1ATD, a more detailed understanding of the folding and trafficking biology governing α1AT biogenesis and its response to small molecule regulators is required. Herein we report the correction of Z-α1AT secretion in response to treatment with the histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA), acting in part through HDAC7 silencing and involving a calnexin-sensitive mechanism. SAHA-mediated correction restores Z-α1AT secretion and serpin activity to a level 50% that observed for wild-type α1AT. These data suggest that HDAC activity can influence Z-α1AT protein traffic and that SAHA may represent a potential therapeutic approach for α1ATD and other protein misfolding diseases.
Introduction
Proper protein folding is essential to maintain cellular function and organismal health. The pathways that generate and protect the fold collectively make up the proteostasis network (PN)2 (1), a complex system of chaperones, folding enzymes, and degradative components (2–9). The PN is controlled by numerous signaling pathways such as the unfolded protein response (10, 11) and the heat shock response (12, 13) as well as Ca2+-sensing signaling pathways (5), autophagy (14, 15), integrated oxidative stress and inflammatory signaling pathways (16–21), and the acetylation proteostasis system (22). The PN includes components required for folding both cytosolic proteins and those found within the exocytic compartments, such as the endoplasmic reticulum (ER). Genetic disorders, including missense mutations, alter polypeptide chain folding energetics (4, 23), often putting protein folding out of reach of the folding capacity of the prevailing PN, triggering misfolding disease (2, 5, 6, 9, 24–27).
We now appreciate that α1AT deficiency (α1ATD) is a disorder of proteostasis-mediated protein folding and trafficking pathways (2, 14, 22, 27–32). α1AT is a soluble 52-kDa glycoprotein and the most abundant member of the serine protease inhibitor (SERPIN) family in plasma. It is delivered to the lung where α1AT is critical to prevent degradation of elastin, a key component ensuring the maintenance of lung elasticity. α1AT is considered as a metastable protein that is produced and secreted primarily by hepatocytes, with a poorly characterized contribution from lung macrophages and type II lung alveolar cells (14, 27, 33). α1AT acts as an inhibitor of serine proteases such as human neutrophil elastase (HNE), cathepsin G, and proteinase-3 by capturing these proteins in an irreversible, higher molecular weight suicide complex through loop-sheet insertion and proteolytic cleavage (33–35).
The reduced serum concentration of α1AT is a universal characteristic of α1ATD, reflecting the failure of folding and trafficking of α1AT through the secretory pathway (14, 33, 35, 36). The most common causes of inherited α1ATD are the Z- and S-variants of α1AT, which are caused by the E342K and E264V mutations, respectively, with the homozygous Z-α1AT being the most severe form of the disease (37, 38). The accumulation of Z-variant polymers in the ER can result in proteotoxic stress and associated chronic liver disease (27, 33, 39, 40). In contrast, a defect in ER export of all disease-associated α1AT variants leads to substantially reduced levels in the serum with severe consequences for anti-protease activity in the lung. As such, most α1AT variants found in α1ATD patients trigger lung symptoms with varying times of onset and sensitivity to the environment, particularly smoking, α1AT deficiency being one of the most important risk factor for onset of chronic obstructive pulmonary disease/emphysema (14, 27, 28, 33).
Histone acetyltransferases and deacetylases (HDACs) have recently been shown to play an important role in liver and lung physiology (16, 41–49). Histone acetyltransferase and HDAC mediate, respectively, the post-translational acetylation and deacetylation (50, 51) of histones as well as numerous other luminal and cytosolic proteins, including PN components (22, 52–56) and proteostasis regulatory pathways such as the key heat shock factor 1, involved in regulation of the cytosolic PN (57, 58). Recent studies have suggested that targeting HDAC activity with chemical HDAC inhibitors (HDACi) can provide substantial benefit for type II diabetes (50, 59, 60), cancer (61–63), rheumatoid arthritis (64), and chronic obstructive pulmonary disease/emphysema and asthma-associated airway inflammation (2, 27, 46, 65–67). Additionally, we have previously shown that HDACi can promote trafficking and function of the ΔF508 mutant of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, a variant that is responsible for the most common clinical presentation of cystic fibrosis (47, 68). HDACi have also been shown to correct the folding, trafficking, and function of ER-localized mutants of β-glucocerebrosidase, a lysosomal protein that when mutated is responsible for the onset of Gaucher disease, a lysosomal storage disorder (69). The combined studies have led us to propose that HDACi could strongly impact lung biology by altering the acetylation/deacetylation equilibrium (3) to improve trafficking and function (22) of the Z-variant.
Herein we identify an HDACi-sensitive pathway(s) involved in the correction of the defect(s) associated with Z-α1AT trafficking. We demonstrate that the suberoylanilide hydroxamic acid (SAHA)-dependent correction of Z-α1AT secretion operates, at least in part, through HDAC7 by modulating a calnexin-sensitive proteostasis pathway. Further characterization of SAHA-mediated correction of Z-α1AT revealed secretion of a functional Z-α1AT into the medium. The emerging evidence that HDACi can be effectively used to manage multiple misfolding phenotypes in cell and mouse model systems (47, 60, 68, 70–73) leads us to suggest that HDACs are proteostasis regulators (1, 4) that could have an important impact on α1ATD pathobiology and in the management of the misfolding disease in the clinic (22).
MATERIALS AND METHODS
Cell Culture
Wild-type (WT) and Z-variant IB3 cells (provided by T. Flotte, University of Massachusetts Medical School, Worcester, MA) were cultured in LHC-8 medium containing 10% (v/v) fetal bovine serum (FBS) and 200 μg/ml G418. HCT116 cells were purchased from ATCC (Manassas, VA) and cultured in DMEM containing 10% (v/v) FBS.
DNA Constructs and Generation of Stable HCT116 Cell Lines
Human WT and Z-variant (bases 73–1257) of α1AT were PCR-amplified using the following primers: forward (5′-TA CTC GAG GAG GAT CCC CAG GGA GAT-3′) and reverse (5′-GTC AGC GGC CGC TTA TTT TTG GGT GGG ATT-3′) and cloned into the XhoI and NotI sites of the pOZ bicistronic retroviral expression plasmid, introducing N-terminal FLAG and HA tags. The α1AT signal peptide (bases 1–72) was cloned upstream of the FLAG tag at the BglII site of the pOZ vector using annealed oligonucleotides. The HCT116 stable cell lines were generated as described previously (74–76).
siRNA-mediated Silencing
IB3 or HCT116 cells were plated in 12-well tissue culture dishes and grown to 40% confluency. Silencing of individual HDACs, EDEM3, ERO1L, and calnexin were performed by transfecting cells with RNAiMax (Invitrogen) and a final concentration of 50 nm siRNA according to the manufacturer's protocol and as previously described (47).
α1AT [35S]Methionine Pulse-labeling
HCT116 cells were incubated for 24 h in the absence or presence of 5 μm SAHA. The cells were washed with 3 changes of PBS and starved for 45 min in methionine-free MEM medium. Starvation medium was replaced with FBS-free growth medium containing 60 μCi of Easy Tag Express [35S]methionine (PerkinElmer Life Sciences), and cells were pulse-labeled for 15 min. The labeling media was subsequently replaced with FBS-free growth media and chased for the indicated times. Cell lysates and media were collected, α1AT was immunoprecipitated, and α1AT was identified by SDS-PAGE as described under “Immunoblotting” below.
α1AT Secretion Assays
2 h before measurement of α1AT secretion kinetics, cells were washed with PBS and incubated with 350 μl (12-well plate) or 700 μl (6-well plate) of FBS-free culture medium. After the 2-h incubation, cells were harvested, and the corresponding media were centrifuged at 1500 rpm for 30 min at 4 °C to separate cells and medium. After lysis of the cells, the amount of α1AT in the immature to mature forms in the lysate or secreted into the culture media was analyzed as described under “Immunoblotting” below.
α1AT-mediated HNE Inhibition Assay
To determine the activity of secreted α1AT, a fixed amount of media (20 μl) collected in each experiment was incubated with increasing amounts of human neutrophil elastase (0–10 ng) for 30 min at 37 °C. The binding reaction was terminated by the addition of SDS-PAGE buffer, and the samples were subsequently incubated at 95 °C for 5 min. The samples were then resolved by 8% (v/v) SDS-PAGE, transferred to nitrocellulose, and immunoblotted with anti-human α1AT antibody. The densitometry of the SDS-resistant complexes formed between either Z- or WT-α1AT and HNE (5 ng) were quantified using the software ImageJ in the linear region of the exposure.
Immunoblotting
Cell lysates were prepared in lysis buffer (50 mm Tris-HCl, 150 mm NaCl, 1% (v/v) Triton X-100, and protease inhibitors at 2 mg/ml of lysis buffer), and protein concentrations were determined by the Bradford protein assay (Thermo Scientific, Rockford, IL). Total protein and media were resuspended in 1× SDS sample buffer containing β-mercaptoethanol and incubated at 95 °C for 5 min. The samples 10–50 μg of total protein and 10–15 μl of media were then separated on a 10% (v/v) SDS-PAGE, transferred to nitrocellulose, and immunoblotted with anti-human α1AT antibody (Immunology Consultants Laboratory, Newberg, OR) or the indicated antibody. Detection was performed using chemiluminescence and the appropriate horseradish peroxidase-conjugated secondary antibodies. For native gel electrophoresis, 30 μl of cell media was separated on a 3–20% native gel according to the manufacturer's instructions (Expedeon Inc., San Diego, CA), and immunoblotting was performed as described above for SDS-PAGE. Hsp90 was used in general as loading control.
Analysis of Human Plasma Samples
Plasma samples from Z/Z (Z-α1AT) and M/M (WT-α1AT) individuals were separated on a 3–20% gradient native gel, and an immunoblot analysis of α1AT was performed as described above.
RESULTS
SAHA Corrects Secretion of Z-α1AT
Of the commonly available hepatic and lung cell lines, none endogenously express Z-α1AT but rather exhibit elevated levels of endogenous WT-α1AT, impeding our ability to analyze exogenously expressed Z-α1AT trafficking and secretion. To overcome this limitation, we generated cell culture models to study the contribution of proteostasis management on Z-α1AT trafficking. These include a human epithelial colorectal carcinoma cell line (HCT116) expressing N-terminal FLAG-tagged WT- or Z-α1AT, referred to as WT- and Z-HCT116, and a human bronchial epithelial cell line (IB3) expressing non-tagged WT- or Z-α1AT, referred to as WT- and Z-IB3. Traffic of the α1AT glycoprotein through the secretory pathway can be monitored by a change in its migration on SDS-PAGE in response to the processing of ER-acquired N-linked oligosaccharides (the immature form (I)) during trafficking through the Golgi to generate the slower migrating, mature glycoform (M). The latter is secreted into the serum (secreted form (S)) by the cell.
Both HCT116 and IB3 cell lines promoted maturation, trafficking, and constitutive secretion of WT-α1AT (supplemental Fig. S1, A and B), suggesting that the N-terminal FLAG tag has no impact on α1AT processing. Additionally, both cell lines recapitulated the ER retention of the Z-variant (supplemental Fig. S1, A and B), suggesting that both represent models for studying Z-α1AT deficiency.
HDACi have been shown to modulate multiple protein folding pathways and promote a significant level of correction to numerous protein misfolding diseases (22, 47, 61, 68, 69, 71, 77). Therefore, we examined the impact of HDACi treatment on the trafficking of the misfolded Z-α1AT variant. There are 18 human HDACs that cluster into four classes by sequence homology (16, 78, 79): Zn2+-dependent class I (HDAC1, HDAC2, HDAC3, and HDAC8), class II (HDAC4-HDAC7, HDAC9, and HDAC10) and class IV (HDAC11) enzymes, and the NAD+-dependent class III enzymes (sirtuins). We screened a panel of known pan HDACi that interferes with class I, II, and IV HDACs to assess their ability to alter the trafficking of Z-α1AT in Z-HCT116 cells. We detected an increase in the trafficking (Fig. 1A, left) and secretion (Fig. 1A, right) of Z-α1AT in response to the pan-HDACi: trichostatin A (TSA), Scriptaid, and SAHA as well as the class I-specific MS-275, with SAHA and Scriptaid exhibiting the highest level of benefit in restoring the secretion of Z-α1AT into the culture medium (Fig. 1A). Identical results were observed for SAHA and Scriptaid treatments in the Z-IB3 cell line expressing non-tagged α1AT (supplemental Fig. S1C), thereby indicating that the N-terminal FLAG tag used in HCT116 had no effect on the observed SAHA and Scriptaid-mediated correction. Because SAHA promoted efficient trafficking and is also an Food and Drug Administration-approved drug for management of metastatic disease, we focused our study on this compound.
FIGURE 1.

Correction of Z-α1AT trafficking in response to SAHA treatment. A, immunoblot analysis of α1AT and Hsp90 protein expression from cell lysates (I, immature; M, mature) (left) and culture media (S, secreted) (right) after treatment of Z-HCT116 cells with 5 μm SAHA, 5 μm MS-275, 0.1 μm trichostatin A (TSA), 1 μm Scriptaid, and DMSO is shown. Quantitative analysis of the mature α1AT (left) and secreted α1AT (right) is shown (mean ± S.D., n = 3). B, immunoblot analysis of Z-α1AT, HDAC7, and Hsp90 protein expression from cell lysates and culture media after SAHA treatment of Z-HCT116 cells at the indicated concentration is shown. Quantitative analysis of the -fold increase in secreted Z-α1AT relative to DMSO treatment (0 μm) is shown (mean ± S.D., n = 3). C, immunoblot analysis of α1AT, acetylated (Ac-H3), and total histone H3 (H3), and Hsp90 protein expression after treatment of Z-HCT116 cells with 5 μm SAHA for the indicated time (h) is shown. D, pulse-chase analysis of Z-HCT116 cells after treatment with DMSO (top) or 5 μm SAHA (bottom) from cell lysates (upper) and culture media (lower) for the indicated chase time (min) is shown. E, quantitative analysis of the pulse-chase experiments in D for the immature (left axis, closed symbols) and combined mature and secreted (right axis, open symbols) forms of Z-α1AT after treatment with DMSO (squares) or 5 μm SAHA (circles) is shown. Data shown denote the -fold change relative to t = 0 (mean ± S.D., n = 2). In all panels asterisks indicate p < 0.05 as determined by two-tailed t test using DMSO as the reference.
To better characterize the SAHA-mediated correction of Z-α1AT, both Z-HCT116 and Z-IB3 cells were treated with increasing concentrations of SAHA for 24 h (Fig. 1B and supplemental Fig. S1C). SAHA improved Z-α1AT trafficking and secretion in a dose-dependent manner, and correction was observed at concentrations as low as 0.5 μm, with maximal response observed at 5 μm (Fig. 1B and supplemental Fig. S1C). We also observed a dose-dependent increase in Z-α1AT mRNA starting at 0.5 μm (supplemental Fig. S1D), suggesting that the SAHA-mediated correction could, at least in part, be due to an increase in Z-α1AT transcription. WT-α1AT was equally responsive to SAHA, exhibiting an increased secretion at the 5 μm dosing (supplemental Fig. S1, E–F). Because both the WT and Z-variant exhibited increased trafficking and secretion in response SAHA treatment suggests that the SAHA-mediated correction of the Z-variant trafficking occurs via a conserved on-pathway mechanism.
To analyze the kinetics of the SAHA-mediated rescue of Z-α1AT in HCT116 cells, we performed a time course analysis using the optimal 5 μm dosing (Fig. 1C). We observed an increased level of the ER-localized immature (I) Z-α1AT after 8 h of treatment (Fig. 1C and supplemental Fig. S1G) and the appearance of both the mature (M) glycoform and secreted (S) Z-α1AT after 12 h of treatment (Fig. 1C and supplemental Fig. S1G). An apparent 1.5- and 1.8-fold increase in α1AT mRNA was seen at 8 and 12 h, respectively, as compared with DMSO (supplemental Fig. S1H). The difference in mRNA levels between these two time points was not statistically significant. However, an examination of the Z-α1AT protein levels in this same 4-h window revealed some important differences including 1) a statistically significant (∼50%) increase in the level of the ER-associated immature glycoform, 2) restoration of intracellular trafficking of Z-α1AT as evidenced by the appearance of the steady-state level of mature Golgi processed glycoform, and 3) a 7-fold increase in the secretion of the Z-variant relative to the basal level seen upon DMSO treatment (Fig. 1C and supplemental Fig. S1G). A significant increase in histone H3 acetylation was also observed as early as 2 h post-dosing with drug, consistent with the known epigenetic changes induced by SAHA (Fig. 1C). In contrast, no change in α1AT mRNA could be detected until 8 h post-treatment. These results suggest that, in addition to the apparent slight increase in expression or stability of Z-α1AT mRNA in response to SAHA, additional components could be transcriptionally and/or post-translationally impacted by SAHA through an HDAC-mediated mechanism(s), thereby contributing to the increased trafficking and secretion of Z-α1AT.
Given the elevated expression of the intracellular pool of Z-α1AT seen in response to SAHA treatment (Fig. 1A), we wanted to determine if the observed correction reflected a proportional “leak” of the misfolded ER pool into the secretory pathway or if we had achieved an increase in trafficking efficiency. An analysis of Z-α1AT trafficking when overexpressed at a level similar to that seen after SAHA treatment (supplemental Fig. S2A) revealed a level of maturation and secretion that was substantially lower than that observed in the presence of SAHA (supplemental Fig. S2A). To further characterize the differences in secretion between Z-α1AT overexpression and SAHA treatment, we analyzed the fractional distribution of Z-α1AT between the immature, mature, and secreted pools. Despite achieving a similar level of total Z-α1AT expression with both conditions (supplemental Fig. S2A), we observed that the mature and secreted pools of Z-α1AT were significantly higher in response to SAHA treatment, which was accompanied by a concomitant decrease in the immature glycoform (supplemental Fig. S2B). Conversely, the overexpression of Z-α1AT did not alter the fractional distribution of the various glycoforms as compared with that seen in the control conditions. These results indicate that the SAHA-mediated correction of Z-α1AT does not occur as a result of a proportional leak in response to Z-α1AT overexpression but rather from an alteration in the efficiency of Z-α1AT synthesis, folding, and/or trafficking, leading to productive maturation and secretion from the cell.
To confirm the above results, we performed a pulse-chase analysis using optimized SAHA dosing conditions (5 μm for 24 h) in Z-HCT116 cells (Fig. 1D). We observed that SAHA treatment caused an initial delay in the decay of the ER pool of Z-α1AT (Fig. 1E), supporting our hypothesis that this HDACi mediates an increased stability of the Z-variant. This initial increased stability was followed by a linear decay of the immature glycoform that was more rapid than that seen with vehicle treatment alone, suggesting either increased degradation or ER export. When the mature glycoform and secreted pool of Z-α1AT from these pulse-labeled cells were analyzed, a 4-fold increase in the rate of maturation and secretion of the Z-variant was observed after SAHA treatment as compared with that seen with DMSO (3.8 × 10−5 min−1 (SAHA) versus 0.9 × 10−5 min−1 (DMSO); Fig. 1E). This indicated that increased ER export largely contributes to the more rapid disappearance of the immature glycoform. Taken together, the above data (Fig. 1, A–E, supplemental Figs. S1, A–H, and S2, A and B) suggest that SAHA treatment corrects the trafficking defect associated with the Z-variant of α1AT and restores its secretion to the cell surface.
Chronic Low Dose Treatments Stabilize Z-α1AT Secretion after Removal of SAHA
We next tested whether a lower, daily chronic dosing schedule could sustain and/or enhance the observed increased trafficking and secretion in Z-HCT116 cells as previously shown for CFTR (47). To test this possibility, we first demonstrated a robust dose-dependent correction of Z-α1AT in response to 0.2 and 1 μm SAHA after a 120-h daily dosing regimen (Fig. 2A). Maximal levels of mature Z-α1AT were observed at the 24-h time point with a 1 μm dose of SAHA and remained constant over the entire 120-h period in both Z-HCT116 and Z-IB3 cells (Fig. 2B and supplemental Fig. S2C). Given that α1AT is constitutively secreted, the observed intracellular level of mature Z-α1AT suggests that a steady-state equilibrium of synthesis, degradation, and secretion was achieved in response to the chronic SAHA-dosing regimen after the first 24 h. In agreement with this interpretation, the level of Z-α1AT secreted in a 2-h window at each indicated time point revealed a steady-state level of secretion in the presence of SAHA in both Z-HCT116 and Z-IB3 cells (Fig. 2B and supplemental Fig. S2C).
FIGURE 2.

Chronic low dose SAHA treatment sustains correction of Z-α1AT. A, Z-α1AT expression in cell lysates (I, immature; M, mature) (upper panel) and culture media (S, secreted) (middle panel) after a chronic, daily (every 24 h) treatment with DMSO, 0.2 or 1 μm SAHA for 120 h. B, immunoblot analysis of Z-α1AT protein expression in cell lysates (upper panel) and culture media (middle panel) after a chronic daily dosing scheme (below) of Z-HCT116 cells with 1 μm SAHA or DMSO for the indicated times (h). C, immunoblot analysis of Z-α1AT protein expression in cell lysates and culture media after a chronic daily pretreatment with 1 μm SAHA for 1 or 5 days and subsequent drug washout for the indicated time (h). D, quantitative analysis of the amount of mature (left) and secreted (right) forms of α1AT after a 1 (closed squares)- and 5-day (closed circles) pretreatment with 1 μm SAHA as in C. Data are plotted as the -fold change of α1AT relative to the 5-day DMSO treatment control (open squares) (mean ± S.D., n = 3). The inset in panel D (right) represents an enlargement of the 24–72-h washout period. The asterisk indicates p < 0.05 as determined by two-tailed t test using DMSO as the reference.
To determine whether this new corrected, steady-state equilibrium of Z-α1AT synthesis, folding, trafficking, and secretion, achieved with the chronic low-dose protocol, would persist after drug withdrawal as previously observed for CFTR (47), we monitored the trafficking and secretion of Z-α1AT in Z-HCT116 cells following a washout after an initial treatment period of either 1 or 5 days with 1 μm SAHA (Fig. 2, C and D). After SAHA withdrawal, an overall decrease in the level of both the mature Golgi processed and secreted Z-α1AT was observed for both the 1 and 5 days pretreatment schemes. However, the decay of the SAHA-mediated correction exhibited dramatically different kinetics depending on the length of the initial treatment (Fig. 2, C and D). Specifically, when Z-HCT116 cells were pretreated with SAHA for only 1 day (1 μm), an exponential decrease in the accumulated mature glycoform and secreted pool of Z-α1AT was observed, leading to the levels seen in DMSO-treated cells after only a 24-h washout period (Fig. 2, C and D). In contrast, the disappearance of the intracellular pool of the mature glycoform of Z-α1AT exhibited linear decay kinetics after withdrawal of drug after the 5-day pretreatment scheme, suggesting a clear difference in how this pool is protected by the PN. Additionally, a statistically significant increase in the levels of both the mature glycoform and secreted Z-α1AT remained even after a 72-h washout when cells were exposed to the 1 μm chronic dosing regimen (Fig. 2, C and D). These data demonstrate that the SAHA-mediated correction of Z-α1AT persists even after compound withdrawal after a chronic treatment regimen, thereby suggesting that a SAHA-mediated correction of Z-α1AT secretion extends beyond simple transcriptional or translational regulation of α1AT expression. The latter effect was not expected to persist in the absence of the compound as observed in acute dosing regimens reflecting efficient washout of drug.
SAHA Partially Corrects Z-α1AT Trafficking Defect through HDAC7
The dose-dependent correction of Z-α1AT by SAHA in Z-HCT116 cells correlated with reduced levels of HDAC7 (Fig. 1B), a result consistent with previous reports on the effect of SAHA on HDAC7 expression (47, 80). To address the role of HDAC7 and possibly other HDACs (81) in the SAHA-mediated correction of Z-α1AT trafficking defect, we examined the effect of siRNA-mediated HDAC silencing in both the Z-HCT116 and Z-IB3 cell models. Upon examining the 11 members that make up the class I, II, and IV HDACs, only the silencing of HDAC7 (supplemental Fig. S3, A and B) resulted in correction of Z-α1AT in both Z-HCT116 (Fig. 3A) and Z-IB3 cells (Fig. 3B), yielding a 3- and 6-fold increase in Z-variant secretion, respectively (Fig. 3, A and B). The silencing of HDAC7 resulted in an ∼2.5-fold increase in α1AT mRNA expression (supplemental Fig. S3C) similar to the ∼3-fold increased observed after SAHA treatment (supplemental Fig. S3C).
FIGURE 3.
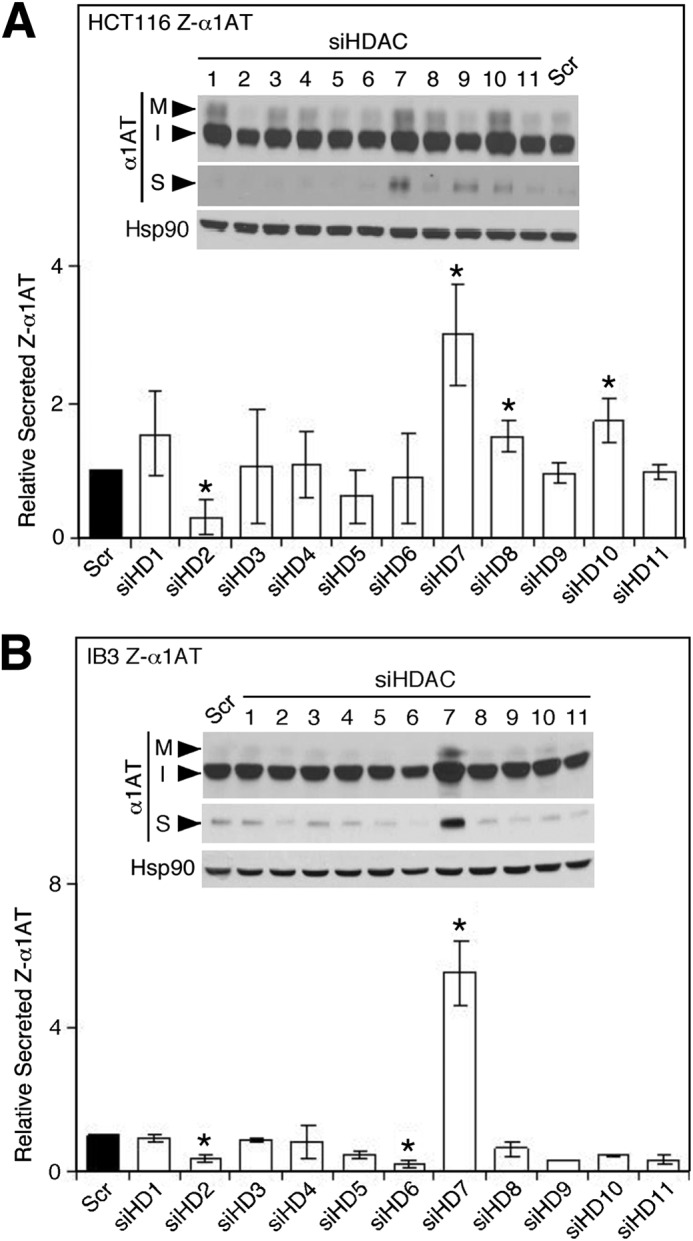
Silencing of HDAC7 corrects Z-α1AT maturation and secretion. Immunoblot analysis of Z-α1AT protein expression in cell lysates (upper panels: I, immature; M, mature) and culture media (middle panels: S, secreted) after siRNA-mediated silencing of HDACs 1–11 in Z-HCT116 cells (A) and Z-IB3 cells (B). Quantitative analysis of the level of secreted Z-α1AT in response to silencing of the indicated HDAC in Z-HCT116 (A) and Z-IB3 (B) cells. Data shown denote the -fold change in secreted Z-α1AT relative to scramble (Scr) control (mean ± S.D., n = 3). In all panels asterisks indicate p < 0.05 as determined by two-tailed t test using Scr control as the reference.
To provide additional support for our interpretation that the observed SAHA-mediated correction of Z-α1AT can act through HDAC7, we compared the effect of SAHA treatment alone or in combination with HDAC7 silencing (supplemental Fig. S4). Both siHDAC7 and SAHA treatments increased the stability of the immature pool of the Z-variant and restored maturation and secretion of Z-α1AT (supplemental Fig. S4). Combining the SAHA and siHDAC7 treatments did not further increase the stability of the ER-localized immature glycoform of the Z-α1AT compared with that seen with SAHA treatment alone (supplemental Fig. S4), raising the possibility that the increased stability of the Z-variant in the ER seen in response to SAHA acts through HDAC7. The combined treatment did result in an increase in maturation and secretion of Z-α1AT compared with either treatment alone (supplemental Fig. S4), but these effects were less then additive, suggesting that the SAHA-mediated trafficking correction also acts, at least in part, through an HDAC7-dependent mechanism. An analysis of the level of HDAC7 silencing seen in response to SAHA treatment alone or in combination with siHDAC7 treatment revealed a significant difference in the extent of HDAC7 knockdown (supplemental Fig. S4, upper) under these two conditions with the combined having a greater silencing (supplemental Fig. S4, upper). This differential HDAC7 silencing could explain the variable correction of Z-α1AT secretion seen with SAHA treatment alone or in combination with siHDAC7 treatment, with the latter having a more pronounced corrective benefit as well as an increased HDAC7 knockdown.
SAHA/siHDAC7-mediated Correction of Z-α1AT Acts through the Calnexin Glycoprotein Cycle
A number of signaling pathways are activated by protein misfolding in the ER, including the unfolded protein response, which can be monitored by the phosphorylation state of eIF2α, the ER-overload response, which can be monitored by the phosphorylation state and degradation of IκB (p-IκB), and autophagy, which can be monitored by the amount of cleavage of LC3. Neither SAHA nor siHDAC7 treatments activated these pathways (supplemental Fig. S5A) nor did they alter the protein expression levels of calnexin, ERGIC53, protein disulfide isomerase, the ER-associated binding immunoglobulin protein (BiP), and valosin-containing protein (VCP/p97), known α1AT interacting partners thought to facilitate its folding, trafficking, and degradation (35, 82–87) (supplemental Fig. S5B).
Given that α1AT is a luminal glycoprotein, we focused our attention on PN pathways unique to the ER that manages glycoprotein folding. We first investigated the relationship between Z-α1AT folding and the calnexin cycle in response to SAHA because calnexin is a major regulator of glycoprotein folding in the ER (11), and it has recently been suggested to affect the trafficking of α1AT (82) as well as many other proteins including, for example, ΔF508-CFTR, the latter variant of which is corrected by both SAHA and siHDAC7 (47). Although we did not detect any change in the expression levels of calnexin in response to SAHA (supplemental Fig. S5B), we did detect a statistically significant 2- and 1.2-fold reduction in the recovery of calnexin with Z-α1AT by co-immunoprecipitation, in response to SAHA or siHDAC7 treatment, respectively (Fig. 4A and supplemental Fig. S5C). This decreased level of interaction was not due to a saturation of the available calnexin pool due to increased expression of Z-α1AT, as a significant (>99%) amount of the total calnexin remained in the unbound material, whereas 99% of the Z-α1AT was recovered in the immunoprecipitate (supplemental Fig. S5D). Given that SAHA and siHDAC7 are able to restore the trafficking of ΔF508 CFTR (47, 87) (supplemental Fig. S6, A and B), we monitored the binding of calnexin to ΔF508-CFTR. As for Z-α1AT, we also observed a decreased recovery of calnexin in response to both SAHA and siHDAC7 treatments (supplemental Fig. S6, A and B).
FIGURE 4.

SAHA treatment alters calnexin binding to Z-α1AT. A, immunoblot and quantitative analyses of Z-α1AT and calnexin (CANX) from an immunoprecipitation (IP) of Z-α1AT from Z-HCT116 cells treated with DMSO or 5 μm SAHA for 24 h are shown. B, immunoblot and quantitative analyses of the level of secreted Z-α1AT after treatment of Z-HCT116 cells with 100 μm miglustat (MIG), 2 μg/ml kifunensine (KIF), or vehicle (Veh) for 24 h in the presence (white bars) or absence (black bars) of 5 μm SAHA. The data represent the -fold change relative to vehicle control (mean ± S.D., n ≥ 2). C, immunoblot and quantitative analyses of Z-α1AT and CANX from an immunoprecipitation of α1AT from Z-HCT116 cells treated with vehicle (Veh), 2 μg/ml kifunensine, or kifunensine + 5 μm SAHA for 24 h. The data in A and C are shown as a ratio of recovered calnexin to immature (I) Z-α1AT (mean ± S.D., n = 3) relative to DMSO. In all panels, # and * indicate p < 0.05 as determined by two-tailed t test using SAHA + vehicle (#) or vehicle (*) as the reference.
To further address the role of calnexin in the SAHA-mediated correction of Z-α1AT, we examined the effect of two drugs known to alter the affinity of nascent proteins for calnexin: 1) miglustat, an inhibitor of glucosidases I and II, both of which are necessary for the proper cycling of calnexin from the nascent polypeptide chain during folding (88), and 2) kifunensine, an inhibitor of the ER α-1,2-mannosidase, whose activity is required to allow improperly folded proteins to exit the calnexin cycle and enter the degradation pathway (89, 90). As previously demonstrated (91–93), kifunensine treatment resulted in a modest increase in Z-α1AT secretion, whereas miglustat had no significant effect (Fig. 4B). When the SAHA and kifunensine treatments were combined, a decrease in Z-α1AT secretion was observed relative to that seen with SAHA alone, whereas miglustat again had no effect (Fig. 4B). Because the action of α-1,2-mannosidase is critical for the exit of client proteins from the calnexin cycle (82, 90), these results suggest that the SAHA-mediated correction proceeds through a similar pathway. These results are also consistent with the observation that SAHA-mediated disruption of the calnexin-α1AT interaction is inhibited in the presence of kifunensine but not in the presence of miglustat (Fig. 4C and supplemental Fig. S6C). Therefore, we raise the possibility that the calnexin/α1AT interaction represents a mechanistic target for the SAHA-mediated correction of Z-α1AT trafficking. For example, one possibility is that SAHA generates a more favorable ER folding environment that allows the Z-α1AT to acquire a stable fold and hence avoid the glycoprotein ERAD (GERAD) (93).
To provide further support for our interpretation that reducing the interaction of Z-α1AT with calnexin can restore the trafficking and secretion of the Z-variant, we tested the effect of silencing calnexin (siCANX) on Z-α1AT biogenesis. After calnexin silencing, we observed a 3-fold increase in the level of the immature form (Fig. 5, A and B), a 2-fold increase in the mature form (Fig. 5, A and C), and a 3-fold increase in the secreted pool (Fig. 5, A and D) of Z-α1AT relative to the siScramble (Scr)-transfected control. The level of correction seen upon calnexin silencing was similar to that seen with SAHA treatment alone (Fig. 5, A–D). When SAHA treatment was combined with calnexin silencing, we observed an additive effect (Fig. 5, A–D), suggesting that disruption of the calnexin-client interaction is not solely responsible for the observed effect of SAHA in correcting the defect associated with the Z-α1AT.
FIGURE 5.
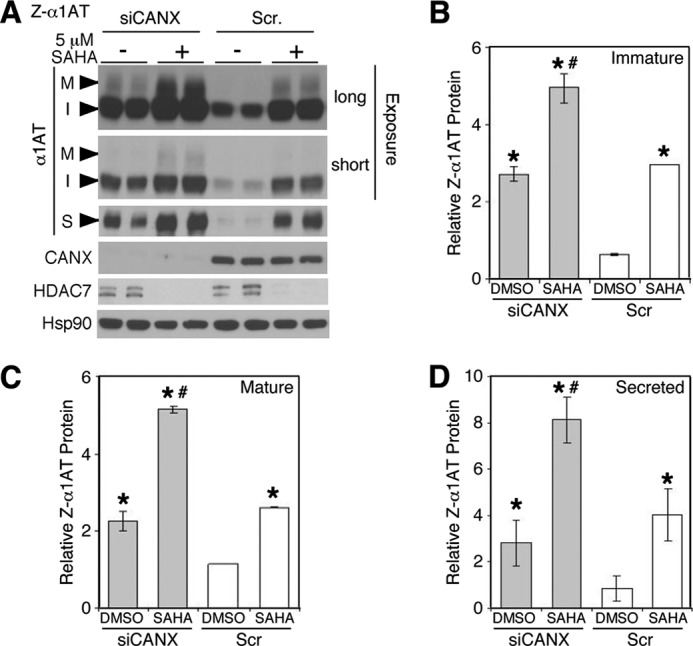
Calnexin silencing restores Z-αAT maturation and secretion. A, immunoblot analysis of Z-α1AT, CANX, and HDAC7 protein expression in cell lysates (M and I) and culture media (S) after siRNA-mediated calnexin silencing in the presence or absence of 5 μm SAHA in Z-HCT116 cells for 24 h. Two exposures are shown to highlight the separate effects of siCANX on the immature and mature bands. Quantitative analyses of the immature (B), mature (C), and secreted (D) forms of Z-α1AT after calnexin silencing (gray bar graph) relative to scrambled control (white bar graph) in the presence of a 5 μm SAHA or DMSO treatments of Z-HCT116 cells for 24 h. Data denote the -fold change in the protein expression of the indicated Z-α1AT form relative to DMSO + Scr control (mean ± S.D., n = 4). In all panels, the * and # indicate p < 0.05 as determined by two-tailed t test using DMSO + Scr (*) and SAHA + Scr (#) as reference, respectively.
Modification of the ER Proteostasis Environment upon SAHA Treatment
Given the above observations, we next attempted to identify additional components of the ER PN that could affect the trafficking of Z-α1AT. To identify changes in the ER PN, we investigated the effect of SAHA on the expression of 84 genes involved in ER-associated folding, degradation, post-translational modification, and signaling (supplemental Table S1). We observed that 7 genes exhibited a reduced expression, and 9 genes showed an increased expression (>2-fold) in response to SAHA treatment (Fig. 6A). An analysis of the functional relationship between these genes revealed that α1AT is tightly linked to the glycoprotein ERAD machinery through EDEM3, OS9, and SEL1L, whose expressions were increased upon SAHA treatment. This pathway has already been reported to be involved in the degradation of misfolded, ER-localized glycoprotein including another α1AT mutant, the Null Hong-Kong (NHK) species (94, 95). Thus, it is reasonable to conclude that EDEM3-OS9-SEL1L would also contribute to the degradation of the Z-variant. The results above reveal that although SAHA mediates an increased expression of Z-α1AT itself, it is also offset with an increased expression of the glycoprotein ERAD pathway charged with its degradation should it fail to fold properly. This suggests that the observed correction in response to SAHA is not a result of simply overwhelming the ER retention pathways typically associated with the Z-variant. Moreover, when EDEM3-silenced cells were treated with SAHA, we observed a synergistic effect on Z-α1AT correction (supplemental Fig. S7A), suggesting that SAHA and siEDEM3 target the Z-variant correction via parallel pathways. These results once again support a model of the SAHA-mediated correction of Z-α1AT without interfering or saturating the degradation pathway of the Z-variant.
FIGURE 6.

Silencing of ERO1L restores Z-α1AT maturation and secretion. A, quantitative PCR analysis of the effect of a 5 μm SAHA treatment on the mRNA levels of 84 genes related to ER PN pathways in Z-HCT116 cells. The data are presented as -fold change in expression relative to DMSO treatment (mean ± S.D., n = 3), and genes with a statistically significant difference in expression (p < 0.05), as determined by Student's t test, are shown. B, immunoblot analysis of Z-α1AT, ERO1L, and HDAC7 protein expression in cells extracts and culture media (S) after the silencing of ERO1L (siERO1L) in the presence or absence of 5 μm SAHA in Z-HCT116 cells for 24 h. Quantitative analysis of the effect of ERO1L silencing (gray bar) on the immature, mature, and secreted forms of Z-α1AT relative to Scr control (white bar) in the presence of 5 μm SAHA or DMSO treatments of Z-HCT116 cells for 24 h is shown. Data are presented as -fold change in the expression of the indicated Z-α1AT form relative to DMSO + Scr control (mean ± S.D., n = 4). In all panels, the * and # indicate p < 0.05 as determined by two-tailed t test using DMSO + Scr (*) and SAHA + Scr (#) as reference.
To confirm a potential role for SAHA as a more general proteostasis regulator of α1AT biology, we further analyzed the contribution of ERO1L in the biogenesis of α1AT. We first analyzed the effect of SAHA on ERO1L expression levels (supplemental Fig. S7B). Both ERO1L mRNA and protein expression were reduced after SAHA treatment (Fig. 6A and supplemental Fig. S7B). To address whether the reduced ERO1L levels contribute to the observed SAHA-mediated rescue of Z-α1AT, we next monitored the trafficking ability of the Z-variant after siRNA-mediated silencing of ERO1L (siERO1L). The silencing of ERO1L caused an increase in the level of immature (1.8-fold), mature (2.3-fold), and secreted (1.8-fold) Z-α1AT relative to that seen in scramble control (Fig. 6B), which was further increased upon the addition of SAHA (Fig. 6B). This result indicates that SAHA acts, at least in part, through silencing of ERO1L and provides evidence that it represents a general proteostasis regulator capable of targeting key pathways mediating the biogenesis of Z-α1AT and correction of its trafficking defect.
SAHA Restores Z-α1AT Anti-protease Activity
Because we observed correction of Z-α1AT secretion in response to SAHA treatment, we tested whether the secreted Z-variant had anti-protease activity. To monitor this activity, we exploited the ability of α1AT to form an SDS-resistant complex with HNE, leading to a slower migration of the α1AT-HNE complex on SDS-PAGE (91, 96). Briefly, increasing quantities of HNE were incubated with a fixed amount of media recovered from WT-α1AT- or Z-α1AT-secreting cells. The resulting complexes were analyzed by SDS-PAGE for evidence of α1AT supershift to a higher molecular weight species representing the HNE complex. As expected, the WT-α1AT secreted by WT-HCT116 cells formed a higher molecular weight complex with HNE (Fig. 7A, top panel). Because SDS-resistant complexes were not observed when conditioned media from parental HCT116 cells was used, these complexes arise from the secretion of WT-α1AT (supplemental Fig. S8A). In contrast to the WT-α1AT result, no supershift was detected when the media from DMSO-treated Z-HCT116 cells were used (Fig. 7A, middle panel) or, surprisingly, siHDAC7-treated cells (supplemental Fig. S8B). However, given the sensitivity of the assay to HNE-mediated degradation of even WT-α1AT (Fig. 7A, top panel), we cannot rule out the possibility that the low level of secretion of the endogenous pool of Z-α1AT (DMSO vehicle) or that observed in the presence of siHDAC7 was degraded, precluding detection of the complexes. In contrast, in SAHA-treated cells the secreted Z-α1AT showed significant incorporation into SDS-resistant complexes when compared with that observed with the WT-α1AT (Fig. 7A, bottom panel). To identify the fraction of this SAHA-stimulated pool of Z-α1AT that retained SERPIN activity, we quantified the SDS-resistant complexes formed between Z- or WT-α1AT in the presence of similar levels of α1AT in the media and 5 ng HNE (Fig. 7B). We detected a 50% recovery of SDS-resistant Z-variant complexes relative to that seen with WT-α1AT (Fig. 7B), indicating that whereas the SAHA-mediated secreted pool does not restore the full level of activity seen with WT-α1AT, it retains sufficient activity to give a robust response in the HNE binding assay. This difference could be explained by the fact that Z-α1AT is secreted largely as a polymeric pool after SAHA treatment (Fig. 7C), consistent with the polymeric state found in plasma of Z-variant patients (Fig. 7D).
FIGURE 7.
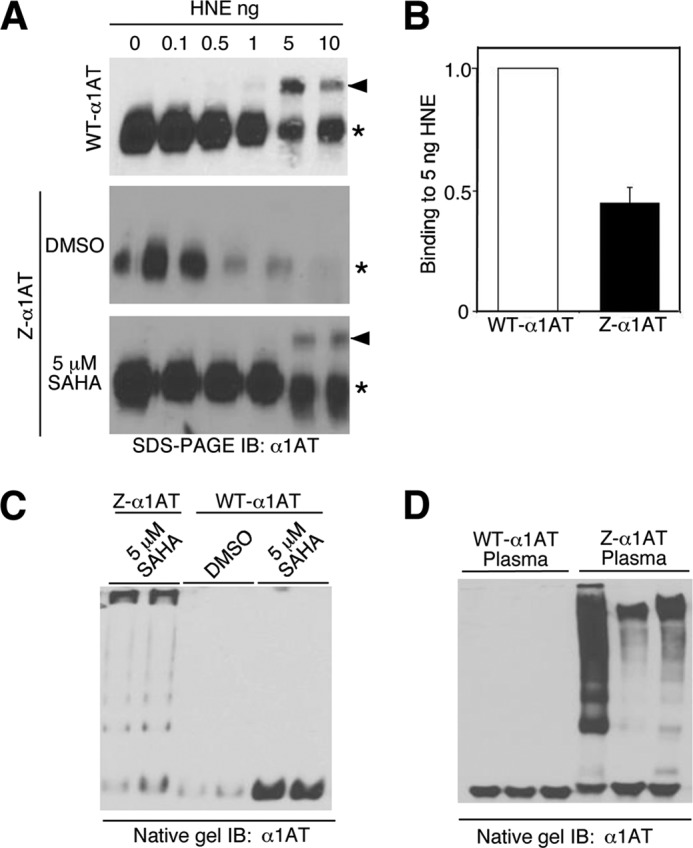
Effect of SAHA on Z-α1AT activity. A, immunoblot (IB) analysis of α1AT after incubation of HNE with secreted WT (top) or Z-α1AT from HCT116 cells treated with DMSO (middle) or 5 μm SAHA (bottom) for 24 h. Supershift of α1AT (arrowhead) relative to secreted α1AT (asterisk) indicates the presence of SDS-resistant complexes with HNE. B, quantitative analysis of -fold change in the amount of secreted α1AT bound to 5 ng of HNE for WT-α1AT and Z-α1AT after treatment with 5 μm SAHA (black) (mean ± S.D., n = 3). C, native gel analysis of secreted Z-α1AT and WT-α1AT after the treatment of HCT116 cells with DMSO or 5 μm SAHA for 24 h. D, native gel analysis of plasma samples from three individuals wild-type (WT-α1AT) and Z-variant (Z-α1AT) patients.
DISCUSSION
Herein, we report the improved secretion of an active species of Z-α1AT in response to SAHA. The mechanism of action for this HDACi-mediated correction is sensitive in part to the reduction of the levels of HDAC7 and to the modulation of the interaction of Z-α1AT with the ER glycoprotein chaperone calnexin that is likely linked to the activity of other ER PN components including ERO1L and the EDEM3-OS9-SEL1L complex.
Our results are in agreement with the increasing evidence that the cellular acetylome balance, managed through the opposing activities of histone acetyltransferases and HDACs, plays an important role as a key regulator of protein folding. Indeed, there is increasing evidence that HDAC pathways play a critical role in managing multiple protein structure relationships. HDACs are particularly well characterized for their impact on the quaternary structure of histones, controlling the dynamics of nucleosome assembly, leading to their ability to control access of DNA to the activity of transcription factors. Moreover, it is also now appreciated that HDACs have a strong influence on the activity of Hsp90 and the activity of heat shock factor 1 (6, 13, 57), the key transcription factor controlling the heat shock response favoring increased protection to protein misfolding. We have suggested that both histone and non-histone post-translational acetylation events represent only small part of the total cellular acetylome now recognizes to be managed by the acetylation proteostasis system (22, 97, 98).
Our characterization of the mechanism of action for the HDACi-mediated correction indicates that SAHA, at least in part, modulates the interaction between the N-glycans of Z-α1AT and the calnexin folding machinery. The latter is a key ER proteostasis component that recognizes glycoprotein-folding intermediates including α1AT and manages their folding (35, 82, 99–101). Thus, our data are consistent with the idea that SAHA targets a very early folding intermediate. Several different mechanisms could account for the ability of SAHA to alter the calnexin/client interaction and/or its interaction with additional ER PN components including degradative and/or redox-coupled components associated with folding steps. Calnexin is reported to be acetylated at Lys-137, a post-translation modification that localizes to the lectin domain of the protein (51). Therefore, a possible model whereby the acetylation state of calnexin alters the conformation of the lectin motif and inhibits its ability to associate with the maturing glycan chain of client proteins remains possible. This hypothesis is supported by the recent discovery of a class I HDAC(s) in the lumen of the ER (102), although a mechanism of their putative translocation into the ER lumen remains to be explained. Alternatively, and/or in addition, the trafficking of the Z-variant protein could be corrected in response to SAHA treatment through known proteostasis events discussed above and/or unknown HDACi-sensitive pathways. The latter could involve ER and/or cytosolic acetylation proteostasis system-sensitive trafficking components that must be aligned early in the folding process for effective ER export (7, 22, 47, 68). In agreement with this view, our preliminary systems level assessment of SAHA-mediated changes in gene expression of ER-associated PN components revealed altered expression of multiple proteins known to be functionally coupled to calnexin and that could contribute to the overall folding environment of the ER promoting WT folding and trafficking. Moreover, a model involving histone modification and correction through epigenetic pathways that affect trafficking is consistent with the persistence of the corrective effect of a low dose, chronic SAHA treatment regimen after compound withdrawal.
The role of SAHA as a regulator of PN biology affecting α1AT export is similar to our recent proposal for SAHA-mediated correction of ΔF508-CFTR (47), where alterations in the PN pathways contribute to the restoration of mutant function. Moreover, the need to energetically stabilize Z-α1AT for export through the PN is consistent with the recent use of the tetrapeptide chemical chaperone, TTAI, that binds the loop-sheet cleft defective in disease and that significantly improves the export of an HNE reactive Z-α1AT to the medium (103).
Our combined results lead us to suggest a model for the correction of Z-variant disease that reflects the role of multiple α1AT folding intermediates in the ER (Fig. 8) that contribute to degradative, polymer prone, and/or export pathways impacted by treatment with SAHA. In non-treated conditions (Fig. 8, left) Z-α1AT could in principle be found in up to four different biosynthetic pools, likely reflecting steps normally transiently occupied by WT α1AT. These include: 1) the pool being actively managed through the calnexin cycle (orange circles); 2) the major (>85%) pool targeted for degradation by the ERAD pathway (including its links to EDEM3-OS9-SEL1L) after hand-off from calnexin (blue circles); 3) a minor but cumulative toxic aggregated pool triggering liver disease (clustered black circles) (14, 29, 104–106); 4), an inefficiently secreted but potentially partially active polymeric Z-α1AT pool that can be found in serum of Z-variant patients (clustered orange circles in Golgi and extracellular pools) (107). After SAHA treatment (right), we suggest that Z-α1AT is redistributed between the various pools in a fashion that favors export. We observed a decrease in the soluble, ER-retained Z-α1AT pool, particularly its ability to populate the calnexin-sensitive pool (35, 90, 108, 109). Moreover, we observed a specific increase in the secretion efficiency of what was recovered as a functional Z-α1AT pool with a level approaching the activity WT-α1AT (50%) (Fig. 8, green circles). Thus, in the case of α1ATD, acetylation proteostasis system management by SAHA appears to promote a global remodeling of the ER and, potentially, supportive cytosolic PN pathways that promote an increase in functional serum levels (Fig. 8, post-translation modification) (22).
FIGURE 8.

A model for SAHA mediated effects on α1AT folding and trafficking. In non-treated conditions (left panel) Z-α1AT can be recovered in 4 pools. These include: 1) the pool being actively managed through the calnexin cycle (orange circles); 2) the major (>85%) pool targeted for degradation by the ERAD pathway (including its links to EDEM3-OS9-SEL1L) after hand-off from calnexin (blue circles); 3) a minor but cumulative toxic aggregated pool triggering liver disease (clustered black circles) (14, 29, 104–106); 4), an inefficiently secreted but partially active polymeric Z-α1AT pool that can be found in serum of Z-variant patients (clustered orange circles in Golgi and extracellular pools) (107). After SAHA treatment (right panel), we suggest that Z-α1AT is redistributed between the various pools to promote export. We observe a decrease in the ER retained Z-α1AT accompanied by an increase in the ERAD pathway as well as increased secretion of a functional form Z-α1AT (green circles). We suggest that the HDACi SAHA promotes remodeling of the ER proteostasis network either transcriptionally and/or post-translationally (PTM) to partially restore the secretion of functional Z-α1AT.
Curiously, targeting the pathway(s) that alters the cell capacity to correct trafficking defect of Z-α1AT by SAHA did not alleviate the aggregate pool nor activate autophagy pathways and thus may not reduce liver pathophysiology as occurs with carbamazepine (105). Rather, we would suggest that SAHA would have its main effect on the protection and/or restoration of lung function through the increased generation of a soluble secreted, functional pool (14, 105, 110). However, because HDACi have recently been reported to induce autophagy (111), the potential impact of select HDACi on aggregate load in the hepatocyte disease will require further investigation. Moreover, the impact of correction by SAHA on the propensity of Z-α1AT to generate a more monomeric versus the polymeric species that can be detected in serum of patients remains to be further explored (107) (Fig. 8, orange versus green circles (37)). Interestingly, combining EDEM3 silencing, which attenuates the glycoprotein ERAD pathway, with SAHA treatment, which modulates the calnexin-α1AT interaction favoring export, had a synergistic effect on the secretion of Z-α1AT. This observation suggests that the joint modulation of these pathways, both of which may limit the effective recovery of the Z-variant for export from ER, will provide a more significant therapeutic benefit for α1ATD than either alone.
In summary, our results show that SAHA and potentially other Zn2+-dependent class I, II, and/or IV pan HDACi can manage the activity of key pathways that influence the export of Z-variant α1AT in a fashion that can restore biological extracellular function (Fig. 8). Because SAHA and HDAC7 also overcome other disease-causing mutations including the correction of the trafficking and function of the ΔF508 CFTR variant responsible for cystic fibrosis (47), we propose that histone acetyltransferases/HDACs may function as more general proteostasis regulators (1).
Supplementary Material
Acknowledgments
We thank the α-1 Foundation and Dr. M. Brantly for generous help in the acquisition of Z-α1AT variant patient serum.
This work was supported, in whole or in part, by National Institutes of Health Grants GM33301, GM42336, and HL15674 (to W. E. B.).

This article contains supplemental Table S1 and Figs. S1–S8.
- PN
- proteostasis network
- ER
- endoplasmic reticulum
- α1ATD
- α1-antitrypsin (α1AT) deficiency
- HNE
- human neutrophil elastase
- HDAC
- histone deacetylase
- HDACi
- HDAC inhibitor
- SAHA
- suberoylanilide hydroxamic acid
- CANX
- calnexin
- siCANX
- silencing CANX
- Scr
- siScramble
- CFTR
- cystic fibrosis transmembrane conductance regulator
- ERAD
- endoplasmic reticulum-associated protein degradation.
REFERENCES
- 1. Balch W. E., Morimoto R. I., Dillin A., Kelly J. W. (2008) Adapting proteostasis for disease intervention. Science 319, 916–919 [DOI] [PubMed] [Google Scholar]
- 2. Bouchecareilh M., Balch W. E. (2011) Proteostasis. A new therapeutic paradigm for pulmonary disease. Proc. Am. Thorac. Soc. 8, 189–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hutt D., Balch W. E. (2010) Cell Biology. The proteome in balance. Science 329, 766–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Powers E. T., Morimoto R. I., Dillin A., Kelly J. W., Balch W. E. (2009) Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 78, 959–991 [DOI] [PubMed] [Google Scholar]
- 5. Ong D. S., Kelly J. W. (2011) Chemical and/or biological therapeutic strategies to ameliorate protein misfolding diseases. Curr. Opin. Cell Biol. 23, 231–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gidalevitz T., Prahlad V., Morimoto R. I. (2011) The stress of protein misfolding. From single cells to multicellular organisms. Cold Spring Harb. Perspect. Biol. 3, a009704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Balch W. E., Roth D. M., Hutt D. M. (2011) Emergent properties of proteostasis in managing cystic fibrosis. Cold Spring Harb. Perspect. Biol. 3, a004499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roth D. M., Balch W. E. (2011) Modeling general proteostasis. Proteome balance in health and disease. Curr. Opin. Cell Biol. 23, 126–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taylor R. C., Dillin A. (2011) Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 3, a004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Määttänen P., Gehring K., Bergeron J. J., Thomas D. Y. (2010) Protein quality control in the ER. The recognition of misfolded proteins. Semin. Cell Dev. Biol. 21, 500–511 [DOI] [PubMed] [Google Scholar]
- 11. Walter P., Ron D. (2011) The unfolded protein response. From stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 12. Morimoto R. I. (2008) Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 22, 1427–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Akerfelt M., Morimoto R. I., Sistonen L. (2010) Heat shock factors. Integrators of cell stress, development, and lifespan. Nat. Rev. Mol. Cell Biol. 11, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perlmutter D. H. (2011) α1-Antitrypsin deficiency. Importance of proteasomal and autophagic degradative pathways in disposal of liver disease-associated protein aggregates. Annu. Rev. Med. 62, 333–345 [DOI] [PubMed] [Google Scholar]
- 15. Kroemer G., Mariño G., Levine B. (2010) Autophagy and the integrated stress response. Mol. Cell 40, 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shakespear M. R., Halili M. A., Irvine K. M., Fairlie D. P., Sweet M. J. (2011) Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 32, 335–343 [DOI] [PubMed] [Google Scholar]
- 17. Malhi H., Kaufman R. J. (2011) Endoplasmic reticulum stress in liver disease. J. Hepatol. 54, 795–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kitamura M. (2011) Control of NF-κB and inflammation by the unfolded protein response. Int. Rev. Immunol. 30, 4–15 [DOI] [PubMed] [Google Scholar]
- 19. Grabiec A. M., Tak P. P., Reedquist K. A. (2011) Function of histone deacetylase inhibitors in inflammation. Crit. Rev. Immunol. 31, 233–263 [DOI] [PubMed] [Google Scholar]
- 20. Belorgey D., Irving J. A., Ekeowa U. I., Freeke J., Roussel B. D., Miranda E., Pérez J., Robinson C. V., Marciniak S. J., Crowther D. C., Michel C. H., Lomas D. A. (2011) Characterization of serpin polymers in vitro and in vivo. Methods 53, 255–266 [DOI] [PubMed] [Google Scholar]
- 21. Singh S., Vrishni S., Singh B. K., Rahman I., Kakkar P. (2010) Nrf2-ARE stress response mechanism. A control point in oxidative stress-mediated dysfunctions and chronic inflammatory diseases. Free Radic. Res. 44, 1267–1288 [DOI] [PubMed] [Google Scholar]
- 22. Hutt D. M., Balch W. E. (2012) Cold Spring Harb. Perspect. Biol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oliveberg M., Wolynes P. G. (2005) The experimental survey of protein-folding energy landscapes. Q. Rev. Biophys. 38, 245–288 [DOI] [PubMed] [Google Scholar]
- 24. Eichner T., Radford S. E. (2011) A diversity of assembly mechanisms of a generic amyloid fold. Mol. Cell 43, 8–18 [DOI] [PubMed] [Google Scholar]
- 25. Lindquist S. L., Kelly J. W. (2011) Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases. Progress and prognosis. Cold Spring Harb. Perspect. Biol. 3, a004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kikis E. A., Gidalevitz T., Morimoto R. I. (2010) Protein homeostasis in models of aging and age-related conformational disease. Adv. Exp. Med. Biol. 694, 138–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bouchecareilh M., Balch W. E. (2012) Proteostasis, an emerging therapeutic paradigm for managing inflammatory airway stress disease. Curr. Mol. Med. 12, 815–826 [DOI] [PubMed] [Google Scholar]
- 28. Sifers R. N. (2010) Medicine. Clearing conformational disease. Science 329, 154–155 [DOI] [PubMed] [Google Scholar]
- 29. Perlmutter D. H., Silverman G. A. (2011) Hepatic fibrosis and carcinogenesis in α1-antitrypsin deficiency. A prototype for chronic tissue damage in gain-of-function disorders. Cold Spring Harb. Perspect. Biol. 3, a005801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Flotte T. R., Mueller C. (2011) Gene therapy for α1-antitrypsin deficiency. Hum. Mol. Genet. 20, R87–R92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gooptu B., Ekeowa U. I., Lomas D. A. (2009) Mechanisms of emphysema in α1-antitrypsin deficiency. Molecular and cellular insights. Eur. Respir. J. 34, 475–488 [DOI] [PubMed] [Google Scholar]
- 32. Ekeowa U. I., Freeke J., Miranda E., Gooptu B., Bush M. F., Pérez J., Teckman J., Robinson C. V., Lomas D. A. (2010) Defining the mechanism of polymerization in the serpinopathies. Proc. Natl. Acad. Sci. U.S.A. 107, 17146–17151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ekeowa U. I., Marciniak S. J., Lomas D. A. (2011) α1-antitrypsin deficiency and inflammation. Expert Rev. Clin. Immunol. 7, 243–252 [DOI] [PubMed] [Google Scholar]
- 34. Gooptu B., Lomas D. A. (2009) Conformational pathology of the serpins. Themes, variations, and therapeutic strategies. Annu. Rev. Biochem. 78, 147–176 [DOI] [PubMed] [Google Scholar]
- 35. Sifers R. N. (2010) Intracellular processing of α1-antitrypsin. Proc. Am. Thorac. Soc. 7, 376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yamasaki M., Sendall T. J., Pearce M. C., Whisstock J. C., Huntington J. A. (2011) Molecular basis of α1-antitrypsin deficiency revealed by the structure of a domain-swapped trimer. EMBO Rep. 12, 1011–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stoller J. K., Aboussouan L. S. (2012) A review of α1-antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 185, 246–259 [DOI] [PubMed] [Google Scholar]
- 38. Kelly E., Greene C. M., Carroll T. P., McElvaney N. G., O'Neill S. J. (2010) α1-antitrypsin deficiency. Respir. Med. 104, 763–772 [DOI] [PubMed] [Google Scholar]
- 39. Topic A., Prokic D., Stankovic I. (2011) α1-Antitrypsin deficiency in early childhood. Fetal Pediatr. Pathol. 30, 312–319 [DOI] [PubMed] [Google Scholar]
- 40. Marciniak S. J., Lomas D. A. (2010) α1-Antitrypsin deficiency and autophagy. N. Engl. J. Med. 363, 1863–1864 [DOI] [PubMed] [Google Scholar]
- 41. To M., Yamamura S., Akashi K., Charron C. E., Barnes P. J., Ito K. (2012) Defect of adaptation to hypoxia in patients with COPD due to reduction of histone deacetylase 7. Chest 141, 1233–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mizuno S., Yasuo M., Bogaard H. J., Kraskauskas D., Natarajan R., Voelkel N. F. (2011) Inhibition of histone deacetylase causes emphysema. Am. J. Physiol. Lung Cell Mol. Physiol. 300, L402–L413 [DOI] [PubMed] [Google Scholar]
- 43. Min T., Bodas M., Mazur S., Vij N. (2011) Critical role of proteostasis. Imbalance in pathogenesis of COPD and severe emphysema. J. Mol. Med. 89, 577–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mercado N., Thimmulappa R., Thomas C. M., Fenwick P. S., Chana K. K., Donnelly L. E., Biswal S., Ito K., Barnes P. J. (2011) Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem. Biophys. Res. Commun. 406, 292–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Isajevs S., Taivans I., Svirina D., Strazda G., Kopeika U. (2011) Patterns of inflammatory responses in large and small airways in smokers with and without chronic obstructive pulmonary disease. Respiration 81, 362–371 [DOI] [PubMed] [Google Scholar]
- 46. Banerjee A., Trivedi C. M., Damera G., Jiang M., Jester W., Hoshi T., Epstein J. A., Panettieri R. A., Jr. (2012) Trichostatin A abrogates airway constriction, but not inflammation, in murine and human asthma models. Am. J. Respir. Cell Mol. Biol. 46, 132–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hutt D. M., Herman D., Rodrigues A. P., Noel S., Pilewski J. M., Matteson J., Hoch B., Kellner W., Kelly J. W., Schmidt A., Thomas P. J., Matsumura Y., Skach W. R., Gentzsch M., Riordan J. R., Sorscher E. J., Okiyoneda T., Yates J. R., 3rd, Lukacs G. L., Frizzell R. A., Manning G., Gottesfeld J. M., Balch W. E. (2010) Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol. 6, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chung K. F., Marwick J. A. (2010) Molecular mechanisms of oxidative stress in airways and lungs with reference to asthma and chronic obstructive pulmonary disease. Ann. N.Y. Acad. Sci. 1203, 85–91 [DOI] [PubMed] [Google Scholar]
- 49. Rajendrasozhan S., Yao H., Rahman I. (2009) Current perspectives on role of chromatin modifications and deacetylases in lung inflammation in COPD. COPD 6, 291–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Peserico A., Simone C. (2011) Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J Biomed. Biotechnol. 2011, 371832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 52. Mollapour M., Neckers L. (2012) Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta 1823, 648–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cook C., Petrucelli L. (2012) Tau triage decisions mediated by the chaperone network. J. Alzheimers Dis., in press [DOI] [PubMed] [Google Scholar]
- 54. Kekatpure V. D., Dannenberg A. J., Subbaramaiah K. (2009) HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J. Biol. Chem. 284, 7436–7445 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55. Scroggins B. T., Robzyk K., Wang D., Marcu M. G., Tsutsumi S., Beebe K., Cotter R. J., Felts S., Toft D., Karnitz L., Rosen N., Neckers L. (2007) An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell 25, 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Murphy P. J., Morishima Y., Kovacs J. J., Yao T. P., Pratt W. B. (2005) Regulation of the dynamics of hsp90 action on the glucocorticoid receptor by acetylation/deacetylation of the chaperone. J. Biol. Chem. 280, 33792–33799 [DOI] [PubMed] [Google Scholar]
- 57. Zeng L., Chen R., Liang F., Tsuchiya H., Murai H., Nakahashi T., Iwai K., Takahashi T., Kanda T., Morimoto S. (2009) Silent information regulator, Sirtuin 1, and age-related diseases. Geriatr. Gerontol. Int. 9, 7–15 [DOI] [PubMed] [Google Scholar]
- 58. Westerheide S. D., Anckar J., Stevens S. M., Jr., Sistonen L., Morimoto R. I. (2009) Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323, 1063–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ozcan U., Yilmaz E., Ozcan L., Furuhashi M., Vaillancourt E., Smith R. O., Görgün C. Z., Hotamisligil G. S. (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Christensen D. P., Dahllöf M., Lundh M., Rasmussen D. N., Nielsen M. D., Billestrup N., Grunnet L. G., Mandrup-Poulsen T. (2011) Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol. Med. 17, 378–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Leggatt G. R., Gabrielli B. (2012) Histone deacetylase inhibitors in the generation of the anti-tumor immune response. Immunol. Cell Biol. 90, 33–38 [DOI] [PubMed] [Google Scholar]
- 62. Marks P. A. (2010) The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin. Investig. Drugs 19, 1049–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jagannath S., Dimopoulos M. A., Lonial S. (2010) Combined proteasome and histone deacetylase inhibition. A promising synergy for patients with relapsed/refractory multiple myeloma. Leuk. Res. 34, 1111–1118 [DOI] [PubMed] [Google Scholar]
- 64. Buckland J. (2011) Rheumatoid arthritis, HDAC and HDACi, pathogenetic and mechanistic insights. Nat. Rev. Rheumatol. 7, 682. [DOI] [PubMed] [Google Scholar]
- 65. Marwick J. A., Ito K., Adcock I. M., Kirkham P. A. (2007) Oxidative stress and steroid resistance in asthma and COPD. pharmacological manipulation of HDAC-2 as a therapeutic strategy. Expert Opin. Ther. Targets 11, 745–755 [DOI] [PubMed] [Google Scholar]
- 66. Halili M. A., Andrews M. R., Sweet M. J., Fairlie D. P. (2009) Histone deacetylase inhibitors in inflammatory disease. Curr. Top. Med. Chem. 9, 309–319 [DOI] [PubMed] [Google Scholar]
- 67. Marwick J. A., Adcock I. M., Chung K. F. (2010) Overcoming reduced glucocorticoid sensitivity in airway disease. Molecular mechanisms and therapeutic approaches. Drugs 70, 929–948 [DOI] [PubMed] [Google Scholar]
- 68. Hutt D. M., Olsen C. A., Vickers C. J., Herman D., Chalfant M., Montero A., Leman L. J., Burkle R., Maryanoff B. E., Balch W. E., Ghadiri M. R. (2011) Potential agents for treating cystic fibrosis. Cyclic tetrapeptides that restore trafficking and activity of ΔF508-CFTR. ACS Med. Chem. Lett. 2, 703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lu J., Yang C., Chen M., Ye D. Y., Lonser R. R., Brady R. O., Zhuang Z. (2011) Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease. Proc. Natl. Acad. Sci. U.S.A. 108, 21200–21205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hageman J., van Waarde M. A., Zylicz A., Walerych D., Kampinga H. H. (2011) The diverse members of the mammalian HSP70 machine show distinct chaperone-like activities. Biochem. J. 435, 127–142 [DOI] [PubMed] [Google Scholar]
- 71. Pipalia N. H., Cosner C. C., Huang A., Chatterjee A., Bourbon P., Farley N., Helquist P., Wiest O., Maxfield F. R. (2011) Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 108, 5620–5625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Thomas E. A., Coppola G., Desplats P. A., Tang B., Soragni E., Burnett R., Gao F., Fitzgerald K. M., Borok J. F., Herman D., Geschwind D. H., Gottesfeld J. M. (2008) The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington disease transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 105, 15564–15569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chuang D. M., Leng Y., Marinova Z., Kim H. J., Chiu C. T. (2009) Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 32, 591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang L. J., Liu X., Gafken P. R., Kioussi C., Leid M. (2009) A chicken ovalbumin upstream promoter transcription factor I (COUP-TFI) complex represses expression of the gene encoding tumor necrosis factor α-induced protein 8 (TNFAIP8). J. Biol. Chem. 284, 6156–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shi Y., Sawada J., Sui G., Affar el B., Whetstine J. R., Lan F., Ogawa H., Luke M. P., Nakatani Y. (2003) Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422, 735–738 [DOI] [PubMed] [Google Scholar]
- 76. Nakatani Y., Ogryzko V. (2003) Immunoaffinity purification of mammalian protein complexes. Methods Enzymol. 370, 430–444 [DOI] [PubMed] [Google Scholar]
- 77. Hathorn T., Snyder-Keller A., Messer A. (2011) Nicotinamide improves motor deficits and up-regulates PGC-1α and BDNF gene expression in a mouse model of Huntington disease. Neurobiol. Dis. 41, 43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Marks P. A., Dokmanovic M. (2005) Histone deacetylase inhibitors. Discovery and development as anticancer agents. Expert Opin. Investig. Drugs 14, 1497–1511 [DOI] [PubMed] [Google Scholar]
- 79. Blander G., Guarente L. (2004) The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 73, 417–435 [DOI] [PubMed] [Google Scholar]
- 80. Dokmanovic M., Perez G., Xu W., Ngo L., Clarke C., Parmigiani R. B., Marks P. A. (2007) Histone deacetylase inhibitors selectively suppress expression of HDAC7. Mol. Cancer Ther. 6, 2525–2534 [DOI] [PubMed] [Google Scholar]
- 81. Mielcarek M., Benn C. L., Franklin S. A., Smith D. L., Woodman B., Marks P. A., Bates G. P. (2011) SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington disease. PLoS One 6, e27746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cameron P. H., Chevet E., Pluquet O., Thomas D. Y., Bergeron J. J. (2009) Calnexin phosphorylation attenuates the release of partially misfolded α1-antitrypsin to the secretory pathway. J. Biol. Chem. 284, 34570–34579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nyfeler B., Reiterer V., Wendeler M. W., Stefan E., Zhang B., Michnick S. W., Hauri H. P. (2008) Identification of ERGIC-53 as an intracellular transport receptor of α1-antitrypsin. J. Cell Biol. 180, 705–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Papp E., Száraz P., Korcsmáros T., Csermely P. (2006) Changes of endoplasmic reticulum chaperone complexes, redox state, and impaired protein disulfide reductase activity in misfolding α1-antitrypsin transgenic mice. FASEB J. 20, 1018–1020 [DOI] [PubMed] [Google Scholar]
- 85. Graham K. S., Le A., Sifers R. N. (1990) Accumulation of the insoluble PiZ-variant of human α1-antitrypsin within the hepatic endoplasmic reticulum does not elevate the steady-state level of grp78/BiP. J. Biol. Chem. 265, 20463–20468 [PubMed] [Google Scholar]
- 86. Shen Y., Ballar P., Fang S. (2006) Ubiquitin ligase gp78 increases solubility and facilitates degradation of the Z-variant of α1-antitrypsin. Biochem. Biophys. Res. Commun. 349, 1285–1293 [DOI] [PubMed] [Google Scholar]
- 87. Schmidt B. Z., Perlmutter D. H. (2005) Grp78, Grp94, and Grp170 interact with α1-antitrypsin mutants that are retained in the endoplasmic reticulum. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G444–G455 [DOI] [PubMed] [Google Scholar]
- 88. Norez C., Noel S., Wilke M., Bijvelds M., Jorna H., Melin P., DeJonge H., Becq F. (2006) Rescue of functional delF508-CFTR channels in cystic fibrosis epithelial cells by the α-glucosidase inhibitor miglustat. FEBS Lett. 580, 2081–2086 [DOI] [PubMed] [Google Scholar]
- 89. Cormier J. H., Tamura T., Sunryd J. C., Hebert D. N. (2009) EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol. Cell 34, 627–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Pan S., Wang S., Utama B., Huang L., Blok N., Estes M. K., Moremen K. W., Sifers R. N. (2011) Golgi localization of ERManI defines spatial separation of the mammalian glycoprotein quality control system. Mol. Biol. Cell 22, 2810–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Marcus N. Y., Perlmutter D. H. (2000) Glucosidase and mannosidase inhibitors mediate increased secretion of mutant α1-antitrypsin Z. J. Biol. Chem. 275, 1987–1992 [DOI] [PubMed] [Google Scholar]
- 92. Reiterer V., Nyfeler B., Hauri H. P. (2010) Role of the lectin VIP36 in post-ER quality control of human α1-antitrypsin. Traffic 11, 1044–1055 [DOI] [PubMed] [Google Scholar]
- 93. Cabral C. M., Liu Y., Moremen K. W., Sifers R. N. (2002) Organizational diversity among distinct glycoprotein endoplasmic reticulum-associated degradation programs. Mol. Biol. Cell 13, 2639–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hirao K., Natsuka Y., Tamura T., Wada I., Morito D., Natsuka S., Romero P., Sleno B., Tremblay L. O., Herscovics A., Nagata K., Hosokawa N. (2006) EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 281, 9650–9658 [DOI] [PubMed] [Google Scholar]
- 95. Christianson J. C., Shaler T. A., Tyler R. E., Kopito R. R. (2008) OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 10, 272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Burrows J. A., Willis L. K., Perlmutter D. H. (2000) Chemical chaperones mediate increased secretion of mutant α 1-antitrypsin (α1-AT) Z. A potential pharmacological strategy for prevention of liver injury and emphysema in α1-AT deficiency. Proc. Natl. Acad. Sci. U.S.A. 97, 1796–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Aka J. A., Kim G. W., Yang X. J. (2011) K-acetylation and its enzymes. Overview and new developments. Handb. Exp. Pharmacol. 206, 1–12 [DOI] [PubMed] [Google Scholar]
- 98. Gnad F., Ren S., Choudhary C., Cox J., Mann M. (2010) Predicting post-translational lysine acetylation using support vector machines. Bioinformatics 26, 1666–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Galli C., Bernasconi R., Soldà T., Calanca V., Molinari M. (2011) Malectin participates in a backup glycoprotein quality control pathway in the mammalian ER. PLoS One 6, e16304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hosokawa N., Wada I., Natsuka Y., Nagata K. (2006) EDEM accelerates ERAD by preventing aberrant dimer formation of misfolded α1-antitrypsin. Genes Cells 11, 465–476 [DOI] [PubMed] [Google Scholar]
- 101. Oda Y., Hosokawa N., Wada I., Nagata K. (2003) EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299, 1394–1397 [DOI] [PubMed] [Google Scholar]
- 102. Kahali S., Sarcar B., Prabhu A., Seto E., Chinnaiyan P. (2012) Class I histone deacetylases localize to the endoplasmic reticulum and modulate the unfolded protein response. FASEB J. 26, 2437–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Alam S., Wang J., Janciauskiene S., Mahadeva R. (2012) Preventing and reversing the cellular consequences of Z α1-antitrypsin accumulation by targeting s4A. J. Hepatol. 57, 116–124 [DOI] [PubMed] [Google Scholar]
- 104. Hidvegi T., Mukherjee A., Ewing M., Kemp C., Perlmutter D. H. (2011) The role of autophagy in α1-antitrypsin deficiency. Methods Enzymol. 499, 33–54 [DOI] [PubMed] [Google Scholar]
- 105. Hidvegi T., Ewing M., Hale P., Dippold C., Beckett C., Kemp C., Maurice N., Mukherjee A., Goldbach C., Watkins S., Michalopoulos G., Perlmutter D. H. (2010) An autophagy-enhancing drug promotes degradation of mutant α1-antitrypsin Z and reduces hepatic fibrosis. Science 329, 229–232 [DOI] [PubMed] [Google Scholar]
- 106. Kaushal S., Annamali M., Blomenkamp K., Rudnick D., Halloran D., Brunt E. M., Teckman J. H. (2010) Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp. Biol. Med. (Maywood) 235, 700–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ogushi F., Fells G. A., Hubbard R. C., Straus S. D., Crystal R. G. (1987) Z-type α1-antitrypsin is less competent than M1-type α1-antitrypsin as an inhibitor of neutrophil elastase. J. Clin. Invest. 80, 1366–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Pan S., Huang L., McPherson J., Muzny D., Rouhani F., Brantly M., Gibbs R., Sifers R. N. (2009) Single nucleotide polymorphism-mediated translational suppression of endoplasmic reticulum mannosidase I modifies the onset of end-stage liver disease in α1-antitrypsin deficiency. Hepatology 50, 275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Mast S. W., Diekman K., Karaveg K., Davis A., Sifers R. N., Moremen K. W. (2005) Human EDEM2, a novel homolog of family 47 glycosidases, is involved in ER-associated degradation of glycoproteins. Glycobiology 15, 421–436 [DOI] [PubMed] [Google Scholar]
- 110. Gosai S. J., Kwak J. H., Luke C. J., Long O. S., King D. E., Kovatch K. J., Johnston P. A., Shun T. Y., Lazo J. S., Perlmutter D. H., Silverman G. A., Pak S. C. (2010) Automated high-content live animal drug screening using C. elegans expressing the aggregation prone serpin α1-antitrypsin Z. PLoS One 5, e15460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yi C., Ma M., Ran L., Zheng J., Tong J., Zhu J., Ma C., Sun Y., Zhang S., Feng W., Zhu L., Le Y., Gong X., Yan X., Hong B., Jiang F. J., Xie Z., Miao D., Deng H., Yu L. (2012) Function and molecular mechanism of acetylation in autophagy regulation. Science 336, 474–477 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.