

Background: TLR2 SNPs are linked to tuberculosis, but the mechanisms by which they alter TLR signaling are unclear.
Results: R753Q TLR2 showed impaired tyrosine phosphorylation, dimerization with TLR6, MyD88 recruitment, and induction of NF-κB and cytokines upon mycobacterial challenge.
Conclusion: R753Q polymorphism blocks TLR2 tyrosine phosphorylation and signalosome assembly.
Significance: Deciphering how SNPs alter TLR signaling advances TLR immunobiology and facilitates design of new therapeutic strategies.
Keywords: Adaptor Proteins, Mutant, Receptor Structure-Function, Signal Transduction, Toll-like Receptors (TLR), Single Nucleotide Polymorphism
Abstract
The R753Q polymorphism in the Toll-IL-1 receptor domain of Toll-like receptor 2 (TLR2) has been linked to increased incidence of tuberculosis and other infectious diseases, but the mechanisms by which it affects TLR2 functions are unclear. Here, we studied the impact of the R753Q polymorphism on TLR2 expression, hetero-dimerization with TLR6, tyrosine phosphorylation, and recruitment of myeloid differentiation primary response protein (MyD) 88 and MyD88 adapter-like (Mal). Complementation of HEK293 cells with transfected WT or R753Q TLR2 revealed their comparable total levels and only minimal changes in cell surface expression of the mutant species. Notably, even a 100-fold increase in amounts of transfected R753Q TLR2 versus WT variant did not overcome the compromised ability of the mutant TLR2 to activate nuclear factor κB (NF-κB), indicating that a minimal decrease in cell surface levels of the R753Q TLR2 cannot account for the signaling deficiency. Molecular modeling studies suggested that the R753Q mutation changes the electrostatic potential of the DD loop and results in a discrete movement of the residues critical for protein-protein interactions. Confirming these predictions, biochemical assays demonstrated that R753Q TLR2 exhibits deficient agonist-induced tyrosine phosphorylation, hetero-dimerization with TLR6, and recruitment of Mal and MyD88. These proximal signaling deficiencies correlated with impaired capacities of the R753Q TLR2 to mediate p38 phosphorylation, NF-κB activation, and induction of IL-8 mRNA in transfected HEK293 cells challenged with inactivated Mycobacterium tuberculosis or mycobacterial components. Thus, the R753Q polymorphism renders TLR2 signaling-incompetent by impairing its tyrosine phosphorylation, dimerization with TLR6, and recruitment of Mal and MyD88.
Introduction
Innate immune cells, such as macrophages, dendritic cells, and neutrophils, detect conserved structures of microbial pathogens via pattern recognition receptors, including membrane-associated Toll-like receptors (TLRs)2 (1–3). TLRs are type I glycoproteins expressed on the cell surface (TLR1, TLR2, TLR4–6, TLR11) or intracellularly in the endoplasmic reticulum and endosomes (TLR3, TLR7–9) (4). They contain an ectodomain involved in ligand recognition and co-receptor interactions, a transmembrane region, and an intracellular signaling Toll-IL-1R (TIR) domain (5). TLRs expressed on the cell surface recognize bacterial lipids (e.g. LPS detection by TLR4) (2) and proteins (flagellin detection by TLR5) (6), whereas endosomal TLRs sense viral dsRNA (TLR3) (7), ssRNA (TLR7/8) (8, 9), and hypomethylated CpG motifs present in microbial DNA (TLR9) (10–14). TLR2 cooperates with TLR1 or TLR6 to sense tri- or diacylated lipoproteins, respectively, expressed by Gram-positive bacteria, mycobacteria, mycoplasma, and fungi (15–18).
Ligand recognition initiates TLR2 dimerization with TLR1 or TLR6 that brings together their TIR domains and triggers TLR2 tyrosine phosphorylation (15, 17, 19, 20), forming docking platforms to enable recruitment of myeloid differentiation primary response protein (MyD) 88 (4, 5). MyD88 interacts with MyD88 adapter-like (Mal) and TLR2 via TIR-TIR domain interactions (21, 22), forming a scaffold to recruit interleukin-1 (IL-1) receptor-associated kinases (IRAK) 4, IRAK1, and IRAK2 (23, 24) that associate with the intermediate and death domains of MyD88 (25). Clustered IRAK4 molecules undergo autophosphorylation and kinase activation, leading to IRAK4 → IRAK1/2 phosphorylation and induction of kinase activity (23, 26), recruitment, and Lys-63-linked ubiquitination of TNF receptor-associated factor (TRAF) 6 (4, 5, 27). IRAK1 also undergoes Lys-63-linked ubiquitination via recruitment of Pellinos and TRAF6, resulting in direct recruitment of IκB kinase (IKK)-γ to IRAK1 (28), whereas ubiquitinated TRAF6 recruits TGF-β-activated kinase (TAK) 1 by engaging ubiquitin recognition domains within inhibitor of NF-κB (IκB) kinase (IKK)-γ and TAK-interacting proteins (29). These processes activate TAK1 and the IKK complex and place them into close proximity, promoting TAK1-mediating activation of mitogen-activated protein kinases (MAPKs) and IKK-β, resulting in nuclear translocation of transcription factors that induce transcription of inflammatory genes (28, 30).
Mycobacterium tuberculosis is a causative agent of tuberculosis, one of the most ancient and devastating diseases (31). TLR2 detects M. tuberculosis and its structural components and plays an important role in host defense against M. tuberculosis infection (15, 32–35). Genome-wide association studies have identified several TIR domain-localized SNPs within TLR2, including R753Q and P631H, that have been linked to increased risk of M. tuberculosis infection and incidence of tuberculosis (36–39). Peripheral blood mononuclear cells isolated from patients expressing TLR2 SNPs had impaired NF-κB activation and cytokine secretion in response to M. tuberculosis-derived agonists (38, 39). However, the molecular basis of compromised functions of polymorphic TLR2 variants is unclear.
To determine mechanisms by which the R753Q polymorphism affects TLR2 signaling, we used transfection-based complementation of HEK293 cells with WT or R753Q YFP-TLR2 to study TLR2 signalosome assembly, induction of MAPKs, transcription factors, and cytokines. This study shows that compromised signaling capacity of R753Q TLR2 is not due to lower expression of the mutant receptor species but instead results from their deficient tyrosine phosphorylation, compromised TLR2-TLR6 assembly, and impaired recruitment of Mal and MyD88 to R753Q TLR2. These findings provide a novel mechanistic insight into how the R753Q polymorphism deregulates TLR2 signaling.
EXPERIMENTAL PROCEDURES
Reagents and Cell Culture
Recombinant human TNF-α, anti-GFP, and AU1 antibodies (Abs) were from Invitrogen, and Abs against TLR2, IκB-α, and tubulin were from Santa Cruz Biotechnology. Anti-phospho-p38 Ab was from Promega, and anti-phospho-tyrosine Ab PY20 was from BD Biosciences. S-[2,3-bis(Palmitoyloxy)-(2-RS)-propyl]-Cys-Ser-Lys(4)-OH (Pam2Cys), S-[2,3-bis(palmitoyloxy)-(2-RS)-propyl]-N-palmitoyl-(R)-Cys-Ser-Lys4-OH (Pam3Cys), and lipoarabinomannan (LAM) from Mycobacterium smegmatis were obtained from InvivoGen, irradiated H37V M. tuberculosis, M. tuberculosis cell wall (CW), culture filtrate preparations, and M. smegmatis LAM were obtained from the Biodefense and Emerging Infections Research Resources Repository. HEK293T and HEK293 cells were obtained from ATCC and maintained in DMEM medium supplemented with 10% FBS (HyClone), 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) (complete DMEM). WT and TLR2−/− J2 retrovirus-immortalized mouse bone marrow-derived macrophages (BMDMs) were kindly provided by Drs. Douglas Golenbock and Katherine Fitzgerald (University of Massachusetts Medical School, Worcester, MA) and maintained in complete DMEM.
Recombinant Plasmids, Site-directed Mutagenesis, and Transfection
pcDNA3-YFP-TLR2, pcDNA3-CD14, pELAM-luciferase (Luc), pTK-Renilla-Luc, pEFBOS-Flag-Mal, pcDNA3-AU1-MyD88, and pcDNA3-CFP-MyD88 were described previously (40, 41). pcDNA3-CFP-TLR6 was from Addgene (Addgene plasmid 13021), and pcDNA3-mCherry-Mal was constructed by restriction digestion of pEFBOS-Mal and ligation of the released Mal insert into pcDNA3-mCherry (kindly provided by Dr. Gregory Melikian (Emory University School of Medicine, Atlanta, GA)) by standard molecular biological techniques. The R753Q mutation was introduced into the YFP-TLR2 expression vector by site-directed mutagenesis, and its presence was confirmed by sequencing, as described previously (42). For transient transfection, HEK293T cells were cultured and plated in 100-mm dishes (for immunoprecipitation) or 6- or 24-well plates (for gene expression studies and reporter assays, respectively) and transfected for 3 h with the respective plasmids, using SuperFect transfection reagent (Qiagen). To obtain stable transfectants, HEK293 cells transfected with the plasmids encoding WT or R753Q YFP-TLR2 were selected in complete DMEM containing 1 mg/ml G418 (Sigma) and subcloned by limiting dilution techniques.
Nucleofection
pcDNA3-GFP and pcDNA3-YFP-TLR2 expression vectors encoding WT or R753Q variants were introduced into WT and TLR2−/− immortalized BMDMs (iBMDMs) by nucleofection, using the Nucleofector I device and the mouse macrophage nucleofection kit (Lonza), as recommended by the manufacturer. After recovery for 20 h, cells were treated for 3 h with medium or Pam3Cys (100 ng/ml), and RNA was isolated, reverse-transcribed, and analyzed by real-time quantitative PCR, using gene-specific primers.
Flow Cytometry
To examine TLR2 expression, untransfected HEK293 cells or HEK293 transfectants expressing WT or R753Q YFP-TLR2 variants were stained for 30 min on ice with phycoerythrin (PE)-conjugated anti-TLR2 (TL2.1, Santa Cruz Biotechnology), anti-HLA-A, -B, or -C-PE (positive control), or isotype control (BD Biosciences) Abs. Cells were washed with PBS, fixed for 15 min in 2% paraformaldehyde, and analyzed by FACS on an LSRII flow cytometer (BD Biosciences) to measure YFP (total YFP-TLR2 expression) and PE (cell surface TLR2 expression) fluorescence. The data were analyzed using the FlowJo software (Tree Star).
Confocal Microscopy
Confocal microscopy was performed as described previously (43). In brief, cells were seeded (4 × 105 cells) on poly-l-lysine-coated coverslips (Fischer Scientific) in phenol-free complete DMEM and cultured for 20 h. Transfection was performed using Lipofectamine 2000 (Invitrogen) transfection reagent followed by gentle washing and recovery for 48 h. Cells were fixed with 2% paraformaldehyde and mounted onto glass slides using a 1,4-diazabicyclo[2.2.2]octane-based antifade fluorescent mounting medium. Images were acquired using an Olympus FluoView 500 laser scanning confocal microscope (Olympus). Sequential scans were taken for CFP, YFP, and Cherry fluorophores, using excitation wavelengths at 458, 514, and 633 nm, respectively, as reported (44).
Isolation of RNA and Real-time Quantitative PCR Analysis
Total RNA was isolated using TRIzol (Invitrogen), residual genomic DNA was digested with DNase, and RNA was repurified as recommended by the manufacturer. cDNA was prepared from 1 μg of total RNA using the reverse transcription system (Promega) and examined by real-time quantitative PCR with primers for human IL-8, 5′-CACCGGAAGGAACCATCTCACT-3′ (forward) and 5′-TGCACCTTCACACAGAGCTGC-3′ (reverse); human hypoxanthine phosphoribosyltransferase (HPRT) 5′-ACCAGTCAACAGGGGACATAAAAG-3′ (forward) and 5′-GTCTGCATTGTTTTGCCAGTGTC-3′ (reverse); mouse hypoxanthine phosphoribosyltransferase 5′-GCTGACCTGCTGGATTACATT-3′ (forward) and 5′-GTTGAGAGATCATCTCCACCA-3′ (reverse); and mouse TNF-α, 5′-CCCAGGCAGTCAGATCATCTTC-3′ (forward) and 5′-GCTTGAGGGTTTGCTACAACATG-3′ (reverse) on a MyIQ real-time PCR machine (Bio-Rad). The data were analyzed by the 2−ΔΔCT method (45).
Co-immunoprecipitation and Immunoblotting
Cell lysates were prepared as described previously (46) and precleared with protein G-agarose beads (Roche Applied Science) for 4 h at 4 °C upon rotation. Precleared cell extracts were incubated overnight at 4 °C with the respective Abs in lysis buffer containing 20 mm HEPES (pH 7.4), 0.5% Triton X-100, 150 mm NaCl, 12.5 mm β-glycerophosphate, 50 mm NaF, 1 mm DTT, 1 mm sodium orthovanadate, 2 mm EDTA, 1 mm PMSF, and protease inhibitor mixture (Roche Applied Science). Protein G-agarose beads were added (45 μl/sample), and incubation continued for 4 h. Beads were washed five times with lysis buffer and resuspended in Laemmli sample loading buffer (50 mm Tris-Cl, pH 6.8, 10% glycerol, 2% SDS, 0.1% bromphenol blue, 5% 2-mercaptoethanol). Proteins were separated by SDS-PAGE on 4–20% mini-gels (Invitrogen), electrotransferred to Immobilon-P membranes, blocked, and probed with the respective Abs, as described (46, 47). Densitometric quantification was performed used the Quantity One program (Bio-Rad).
NF-κB Reporter Assays
Reporter assays were performed as reported previously (46–48). In brief, cells were transfected with plasmids, using SuperFect, recovered for 24 h, and treated for 5 h with medium or stimuli. Cells were lysed in a passive lysis buffer (Promega), and firefly luciferase versus Renilla luciferase activities were measured using the Dual-Luciferase reporter assay system (Promega) on a Berthold LB 9507 luminometer (Berthold Technologies).
Molecular Modeling Studies
Structural models of the WT and R753Q-containing TIR domains of TLR2 were built using the WT TLR2 structure (Protein Data Bank (PDB) accession code 1FYW) and the 3D-JIGSAW Protein Comparative Modeling Server (version 2.0) and visualized and analyzed using the Cn3D 4.3 macromolecular structure viewer (National Center for Biotechnology Information (NCBI), www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml) and PyMOL. Selenomethionine and dimethylarsenic cysteines modified residues from the TIR2 structure (1FYW) were stripped of modifications and replaced with Met and Cys, whereas retaining original conformations. Rossetta_backrub (49, 50) was used to evaluate 20 models for the impact of the R753Q mutation. Resulting models were visualized using PyMOL, and energy surface representations were calculated using the APBS (51, 52) plug-in in PyMOL.
Statistical Analysis
Statistical analysis was performed using the GraphPad Prism 5 program for Windows (GraphPad Software Inc.). Statistical differences were evaluated by the Student's t test with the level of significance set at p < 0.05. Data are expressed as mean ± S.D.
RESULTS
R753Q Polymorphism Does Not Significantly Diminish Expression Levels of TLR2 but Drastically Impairs TLR2-elicited NF-κB Activation
Because the magnitude of TLR signaling correlates with receptor expression (53, 54), we examined the impact of the R753Q polymorphism on TLR2 levels. To optimize detection, we transfected HEK293 cells with vectors encoding WT or R753Q TLR2 fused at the C terminus with YFP (42), which does not alter TLR cellular localization or functions (40, 44, 55). Fluorescent microscopy and FACS revealed similar YFP fluorescence in HEK293 cells transfected with WT or R753Q YFP-TLR2 (Fig. 1, A and B), and comparable levels of WT and R753Q YFP-TLR2 were immunoblotted with anti-GFP Ab (Fig. 1C) that cross-reacts with YFP (44, 55, 56). FACS analyses of 293/YFP-TLR2 stable transfectants expressing WT or R753Q variants and stained with PE-conjugated anti-TLR2 Ab showed minimal changes (<20%) in cell surface expression of WT versus R753Q TLR2 (Fig. 1B, right). To determine whether increasing levels of R753Q TLR2 overcomes its signaling deficiencies, HEK293T cells were transfected with different input amounts of plasmids encoding WT or mutant TLR2, and Pam3Cys-induced NF-κB transactivation was determined. Fig. 1D shows that ∼12-fold higher input amount of R753Q TLR2-expressing plasmid was required to achieve NF-κB reporter activation similar to that caused by vector encoding WT TLR2. Importantly, the plateau of NF-κB activation elicited by WT TLR2 was ∼3-fold higher when compared with the maximal response caused by the R753Q variant (Fig. 1D). Thus, drastic differences in signaling between WT and the R753Q TLR2 cannot be accounted for by minor differences in expression.
FIGURE 1.

Expression levels and signaling capacities of WT and R753Q YFP-TLR2 species. A–D, HEK293 cells were transiently (A, C, and D) or stably (B) transfected with expression vectors encoding WT or R753Q YFP-TLR2 variants. A, after recovery for 48 h, cells were analyzed by fluorescent and phase contrast microscopy to determine expression levels of the TLR2 variants. B, total expression of transfected YFP-TLR2 variants was determined by FACS analysis, using the YFP fluorescence intensity (FITC channel) (top panel). TLR2 cell surface expression was determined by staining of cells with TL2.1-PE (anti-TLR2 Ab) or isotype control IgG-PE followed by FACS analyses of PE fluorescence (bottom panel). % of Max, percentage of maximum. C, whole cell lysates were examined by immunoblotting using anti-GFP and anti-tubulin Abs. D, HEK293 cells were co-transfected with the indicated amount of plasmids expressing WT or R753Q YFP-TLR2, along with pELAM-Luc (0.2 μg/well). After recovery for 20 h, cells were treated for 5 h with medium or Pam3Cys (100 ng/ml), and firefly Luc activity was determined in cell lysates and normalized for protein levels. The data of a representative experiment (n = 12 (A); n = 5 (B); n = 3 (C and D)) are shown. *, p < 0.05.
Transfected R753Q TLR2 Elicits Diminished Phosphorylation of p38, Activation of NF-κB, and Expression of IL-8 Gene
To study how the R753Q polymorphism affects TLR2 signaling, we transiently transfected HEK293T cells with vectors encoding WT or mutant YFP-TLR2 and determined TLR2-driven activation of p38 and NF-κB and induction of IL-8 mRNA. Treatment of 293/WT YFP-TLR2 transfectants with Pam3Cys and LAM, TLR2-TLR1 and TLR2-TLR6 agonists, respectively (34, 57), led to marked phosphorylation of p38 (Fig. 2, A and C) and activation of the NF-κB luciferase reporter (Figs. 1D and 3B), whereas cells expressing R753Q YFP-TLR2 exhibited significantly blunted responses. In contrast to WT TLR2-expressing cells, 293/R753Q YFP-TLR2 transfectants failed to phosphorylate and degrade IκB-α (Fig. 2, B and C) and showed inhibited NF-κB reporter activation (Fig. 3A) when stimulated with irradiated M. tuberculosis, heat-killed Staphylococcus aureus, or M. smegmatis-derived LAM. Exposure of 293/R753Q YFP-TLR2 cells to M. tuberculosis-derived CW and culture filtrate fractions or LAM led to <2-fold NF-κB reporter induction when compared with robust (4.1–4.3-fold) activation elicited by WT TLR2 (Fig. 3B).
FIGURE 2.

The impact of R753Q polymorphism on TLR2-mediated phosphorylation of p38 and IκB-α and degradation of IκB-α. WT or R753Q YFP-TLR2 variants were overexpressed in HEK293T cells following transient transfection of the corresponding expression vectors. After recovery, cells were stimulated for the indicated times with 100 ng/ml Pam3Cys (A), irradiated M. tuberculosis (Mtb) (B, killed M. tuberculosis per HEK cell ratio 10:1), or 5 μg/ml LAM (C). YFP-TLR2 proteins were immunoprecipitated (IP) from whole cell lysates with anti-GFP Ab and analyzed by immunoblotting (IB) with anti-GFP Ab. Whole cell lysates were examined by Western blot analyses using Abs against GFP, phospho-p38 (p-p38), total p38, phospho-IκB-α (p-IκB-α), IκB-α, and tubulin. The results of a representative experiment (n = 3) are presented.
FIGURE 3.
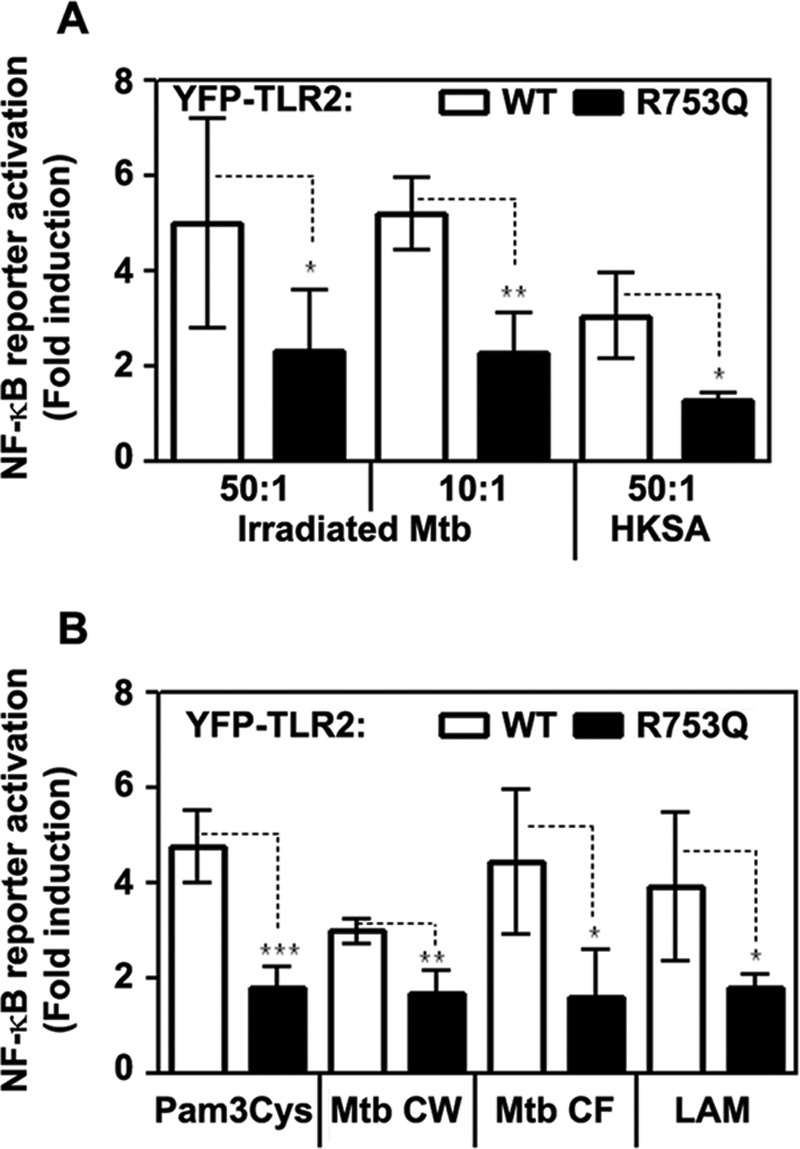
R753Q TLR2 mediates impaired activation of NF-κB luciferase reporter in response to inactivated M. tuberculosis, S. aureus, mycobacteria-derived cell wall, and culture filtrate fractions, LAM and Pam3Cys. HEK293T cells were transiently transfected with pcDNA3-YFP plasmids encoding WT or R753Q TLR2, along with pELAM-Luc (NF-κB reporter) and pTK-Renilla-Luc (to normalize for transfection efficiency). A, after recovery for 24 h, cells were stimulated with irradiated M. tuberculosis (Mtb) or heat-killed S. aureus (HKSA) using inactivated bacteria/HEK cell ratios 50:1 and 10:1. B, HEK transfectants were treated for 5 h with medium or stimulated with 100 ng/ml Pam3Cys, M. tuberculosis-derived CW and culture filtrate (CF) fractions, and LAM (5 μg/ml each). Firefly versus Renilla luciferase activities were determined in cell lysates, and the numbers in bacteria- or lipid-stimulated cells were normalized to medium control values and presented as -fold induction. The summary of three experiments (mean ± S.D.) is shown. *, p < 0.05, **, p < 0.01, ***, p < 0.005.
Because MAPKs and NF-κB control expression of pro-inflammatory mediators (5, 30), we next determined the impact of the R753Q mutation on TLR2-induced IL-8 gene expression. Although treatment of 293/WT YFP-TLR2 stable transfectants with Pam3Cys or M. tuberculosis-derived CW resulted in 42- and 15-fold induction of IL-8 mRNA, these stimuli elicited only 4- and 2.5-fold increase in IL-8 mRNA levels in cells expressing R753Q TLR2 (Fig. 4A). TNF-α induced comparable IL-8 gene expression in 293 cells expressing WT or R753Q TLR2 (Fig. 4A), indicating that the inhibitory impact of R753Q mutation is restricted to TLR signaling.
FIGURE 4.

The effect of R753Q polymorphism on TLR2-elicited cytokine gene expression. A, HEK293 cells stably transfected with plasmids encoding WT or R753Q YFP-TLR2 were treated for 3 h with medium, 1 μg/ml Pam3Cys, or 5 μg/ml M. tuberculosis (Mtb) CW preparation. B, mouse WT or TLR2−/− iBMDMs were nucleofected with pcDNA3-YFP or expression vectors encoding human WT or R753Q YFP-TLR2 variants. After recovery for 24 h, cells were treated for 3 h with medium or 1 μg/ml Pam3Cys. RNA was isolated, converted into cDNA, and subjected to real-time quantitative PCR analyses with the respective gene-specific primers. Data were processed according to the 2−ΔΔCT method. Shown is the summary (mean ± S.D.) of three independent experiments (A) and a representative experiment (mean ± S.E., n = 4). *, p < 0.05. ns, not significant.
We next sought to confirm our data in cells with the macrophage phenotype. To this end, we determined the effect of nucleofection-based complementation of TLR2−/− iBMDMs with human WT or R753Q YFP-TLR2 on Pam3Cys-mediated TNF-α gene expression. iBMDM cell lines replicate TLR responses of primary macrophages (58) and have been extensively used by several groups, including ours (43, 58, 59). As shown in Fig. 4B, Pam3Cys induced 9.2-fold induction of TNF-α mRNA in WT iBMDMs, whereas TLR2−/− iBMDMs showed no responsiveness. Nucleofection of expression vector encoding human WT YFP-TLR2 endowed Pam3Cys sensitivity, leading to ∼4-fold induction of TNF-α mRNA levels, whereas only 1.9-fold increase was seen in cells nucleofected with R753Q YFP-TLR2 (Fig. 4B). Quantitative real-time PCR analyses revealed similar relative levels of human TLR2 mRNA in TLR2−/− iBMDMs complemented with WT versus D299G TLR2 (supplemental Fig. S1), indicating comparable expression of transfected YFP-TLR2 species. Thus, the R753Q polymorphism reduces the capacity of TLR2 to activate p38 and NF-κB and induce IL-8 gene expression in response to Pam3Cys, inactivated M. tuberculosis, and mycobacteria-derived CW, culture filtrate, or LAM, whereas not affecting TNF-α-driven expression of IL-8 mRNA.
Molecular Modeling of the Impact of the R753Q Mutation on the Structure of the TIR Domain of TLR2
The TIR2 structure (PDB code 1FYW) was used as a template structure to model the point mutation R753Q using Rosetta backrub (49). Analysis of 20 modeled Gln-753 TIR2 structures showed no gross conformational changes in nearby or distant main chain atoms with respect to the WT TIR2 (Fig. 5, A–C). However, we noted discrete main and side chain differences, affecting residues Pro-746, Arg-748, and Lys-751 and the R753Q mutation itself (Fig. 5D), thought to be involved in TIR2 interactions with TIR1 (Arg-748 and Arg-753) and TIR6 (Arg-748 and Lys-751) (61). We also observed a notable change in the electrostatic potential within the DD loop and αD region resulting from the R753Q mutation, with a relative decrease in the net positive charge (Fig. 5, E and F). Thus, the R753Q mutation is likely to change the interaction surface within the TIR domain via altered electrostatic potential of or conformational effects on the DD loop, possibly affecting TLR2 dimerization with TLR1 and/or TLR6 and recruitment of adapter proteins Mal and MyD88.
FIGURE 5.

Molecular modeling of the structures of the TIR domains of WT and R753Q TIR2 species. A–C, ribbon models of WT Arg-753 (A) and mutant Gln-753 (MUT Q753) (B) TIR domains of TLR2 and their overlay (C) are shown. D, a close-up of the DD-loop-αD region encircled in C. E and F, the electrostatic surfaces of the WT (Arg-753) (E) versus mutant Gln-753 (F). The electrostatic potential was calculated by solving the Poisson-Boltzmann equation using the APBS PyMOL plug-in and depict a range from − (red)/+ (blue) 1kbT/ec, where kb is the Boltzmann constant, T is the temperature, and ec is the charge of the electron. Surfaces of the TIR2 domains are depicted.
Impact of R753Q Polymorphism on TLR2 Tyrosine Phosphorylation upon Stimulation with Pam3Cys and Irradiated M. tuberculosis
Previous studies from our group and other laboratories demonstrated a critical role of tyrosine phosphorylation of TLR2 for initiating signal transduction (20, 40). Because the R753Q polymorphism may alter conformation and the electrostatic potential of the TIR2 domain (Fig. 5), it is possible that such changes could affect recruitment of protein-tyrosine kinases (PTKs) or accessibility of tyrosine residues for PTKs. Therefore, we studied agonist-inducible tyrosine phosphorylation of WT versus R753Q YFP-TLR2 immunoprecipitated with anti-GFP (Fig. 6A) or anti-TLR2 (Fig. 6B) Abs from 293/TLR2 stable cell lines. Immunoblotting of YFP-TLR2 immune complexes with anti-phospho-tyrosine Ab revealed marked induction of tyrosine phosphorylation of WT, but not R753Q, YFP-TLR2 in HEK293 transfectants stimulated with Pam3Cys or irradiated M. tuberculosis (4.8- and 7.5-fold, respectively, Fig. 6, A–D). Immunoprecipitation and immunoblotting with anti-GFP Ab revealed comparable total levels of WT and R753Q YFP-TLR2 (Fig. 6, A and B), indicating that different tyrosine phosphorylation was not due to lower expression of the mutant TLR2. Our data indicate that the R753Q polymorphism blocks the ability of TLR2 to undergo tyrosine phosphorylation upon stimulation with Pam3Cys or irradiated M. tuberculosis.
FIGURE 6.

The impact of R753Q polymorphism on agonist-induced tyrosine phosphorylation of TLR2. A and B, HEK293 stable transfectants expressing WT or R753Q TLR2 were stimulated for the indicated time points with 100 ng/ml Pam3Cys (A) or treated for 15 min with medium or irradiated M. tuberculosis (Mtb) (inactivated M. tuberculosis/HEK cell ratios 50:1) (B). Cell lysates were immunoprecipitated (IP) with anti-GFP (A) or anti-TLR2 (B) Abs and subjected to immunoblot (IB) analyses with anti-GFP and anti-phospho-tyrosine Abs (α-p-Tyr). Total YFP-TLR2 levels were examined by immunoblotting of cell lysates with anti-GFP Ab, and protein loading was controlled by immunoblotting with anti-tubulin Ab (B). p-YFP-TLR2, phospho-YFP-TLR2. C and D represent quantification of Western blot data shown in A and B, respectively, with the numbers in the parentheses indicating -fold increases in agonist-mediated tyrosine phosphorylation of TLR2 normalized to medium-treated values. Shown are the data of a representative (n = 3) experiment.
R753Q Polymorphism Interferes with Agonist-inducible TLR2 Hetero-dimerization with TLR6
Given that TLR2 senses M. tuberculosis and M. tuberculosis-associated lipids in cooperation with TLR6 (15, 16, 33), we studied whether the R753Q polymorphism affects TLR2-TLR6 assembly. To this end, 293/YFP-TLR2 stable cell lines expressing WT or R753Q species were transfected with plasmids encoding CFP-TLR6, and Pam2Cys-inducible TLR2-TLR6 hetero-dimerization was assessed by co-immunoprecipitation and Western blot analyses. Pam2Cys stimulation resulted in 5.9–8-fold increases in the accumulation of TLR2-associated TLR6 proteins in 293/WT YFP-TLR2 transfectants, whereas significantly impaired TLR2-TLR6 hetero-dimerization was observed in cells expressing the R753Q species (Fig. 7, A and B). Comparable total levels of CFP-TLR6, WT, and R753Q YFP-TLR2 were observed (Fig. 7A), indicating that differences in dimerization with TLR6 exhibited by WT versus R753Q TLR2 variants were not due to variations in the total levels of the interacting proteins. These results suggest that changes in conformation and/or electrostatic potential of the TIR domain imposed by the R753Q polymorphism (Fig. 5) contribute to deficient agonist-inducible TLR2-TLR6 association.
FIGURE 7.

The R753Q polymorphism impairs the ability of TLR2 to hetero-dimerize with TLR6 upon Pam2Cys stimulation. A, 293/YFP-TLR2 stable cell lines expressing WT or R753Q YFP-TLR2 variants were co-transfected with pcDNA3-CFP-TLR6. After recovery for 20 h, cells were treated for the indicated time points with medium or 100 ng/ml Pam2Cys. Whole cell lysates were prepared, and TLR2 species were immunoprecipitated (IP) with anti-TLR2 Ab (TL2.1) and analyzed by immunoblotting (IB) with anti-TLR6 Ab to detect TLR2-associated TLR6. Total levels of transfected YFP-TLR2 and CFP-TLR6 proteins were assessed by Western blot analyses of whole cell lysates with anti-TLR2 and anti-TLR6 Abs, respectively. Immunoblotting of cell lysates with anti-tubulin Ab was used to control for protein loading. The results of a representative (n = 3) experiment are shown. B, quantification of the results shown in A. TLR2-TLR6 arbitrary units were calculated by measuring densitometric values for TLR2-TLR6, TLR2, TLR6, and tubulin at each time point, subtracting background values, normalizing for tubulin, and then dividing TLR2-TLR6 values by those calculated for TLR2 and by TLR6.
R753Q TLR2s Show Impaired Recruitment of Mal and MyD88
Upon ligand recognition, TLR2 hetero-dimerizes with TLR1 or TLR6, resulting in docking platform assembly within their TIR domains to enable recruitment of adapter proteins Mal and MyD88 (4, 5, 22, 30, 62, 63). In our next series of experiments, we examined whether compromised ability of R753Q TLR2 to hetero-dimerize with TLR6 affects the ability of TLR2 to associate with Mal and to recruit MyD88. 293/YFP-TLR2 stable cell lines expressing WT or R753Q TLR2 were co-transfected with plasmids encoding Flag-Mal or AU1-MyD88, and agonist-mediated recruitment of epitope-tagged adapters to YFP-TLR2 was determined by co-immunoprecipitation. Pam3Cys stimulation for 10 and 30 min up-regulated the amounts of Flag-Mal associated with WT YFP-TLR2, whereas it failed to increase Flag-Mal interactions with the R753Q YFP-TLR2 species over basal levels seen in medium-treated cells (Fig. 8A). Exposure to irradiated M. tuberculosis induced robust recruitment of AU1-MyD88 to WT YFP-TLR2 evident within 15 min of stimulation followed by its rapid dissociation (Fig. 8B). In contrast, irradiated M. tuberculosis failed to induce AU1-MyD88 recruitment to the R753Q TLR2 within the time course analyzed (Fig. 8B). Similar amounts of total transfected WT and R753Q YFP-TLR2 proteins Flag-Mal and AU1-MyD88 were seen in all samples (Fig. 8), indicating that differences in MyD88 recruitment to WT versus R753Q TLR2 cannot be attributed to variations in total expression of the interacting transfected proteins. To confirm our results obtained by immunoprecipitation, we used confocal microscopy as an alternative approach to study co-localization of transfected YFP-TLR2, mCherry-Mal, and CFP-MyD88. Pam3Cys induced increased co-localization of mCherry-Mal and CFP-MyD88 with WT YFP-TLR2, whereas the R753Q YFP-TLR2 species showed deficient co-localization (supplemental Figs. S2 and S3). Taken collectively, these results demonstrate impaired capacities of R753Q TLR2 to associate with Mal and to recruit MyD88 after stimulation with Pam3Cys or irradiated M. tuberculosis.
FIGURE 8.

R753Q TLR2 shows deficient capacity to recruit Mal and MyD88 in response to agonist stimulation. HEK293T cells were transiently transfected with pEFBOS-Flag-Mal (A) or pcDNA3-AU1-MyD88 (B) in combination with pcDNA3-WT YFP-TLR2 or pcDNA3-R753Q YFP-TLR2, and control cells were transfected with pcDNA3. IP, immunoprecipitation; IB, immunoblot. After recovery for 24 h, cells were treated with medium or stimulated with 100 ng/ml Pam3Cys (A) or irradiated M. tuberculosis (Mtb) (inactivated M. tuberculosis/HEK cell ratio 50:1) (B) for the indicated time points, and cell lysates were prepared and subjected to immunoprecipitation with anti-TLR2 Ab. Flag-Mal and TLR2 immune complexes were examined by Western blotting with anti-GFP and anti-AU1 Abs. Whole cell lysates were subjected to immunoblotting with anti-AU1 and anti-tubulin Abs to analyze total AU1-MyD88 expression and to control for protein loading, respectively. The results of a representative (n = 2) experiment are shown.
DISCUSSION
The R753Q TLR2 polymorphism has been associated with increased prevalence of leprosy and tuberculosis (36–38, 64). More than 10% of patients with atopic dermatitis express the TLR2 R753Q SNP and exhibit severe eczema (65, 66). Furthermore, patients with liver transplants expressing this mutation exhibited a trend toward a higher rate of CMV infection (67), recurrence of Gram-positive infections, and septic shock (68). Studies with peripheral blood mononuclear cells obtained from patients with leprosy or tuberculosis expressing the R753Q polymorphism and experiments with HEK293 cells transfected with the corresponding mutant TLR2 revealed attenuated NF-κB activation and cytokine release in response to stimulation with M. tuberculosis and defined TLR2 agonists (38, 39, 42, 69). However, the molecular basis by which the R753Q polymorphism compromises TLR2-mediated host innate defense, promoting bacterial infections, remains poorly understood.
In this study, we employed overexpression of WT and mutant YFP-TLR2 in HEK293 cells to determine the impact of the R753Q polymorphism on TLR2 expression, tyrosine phosphorylation, and signalosome assembly, information that has not been reported previously. Because the magnitude of TLR signaling depends on relative TLR protein levels (53), we first determined the impact of the R753Q polymorphism on TLR2 expression. Using fluorescent microscopy, immunoprecipitation, and immunoblot analyses, we found comparable total levels of WT and R753Q TLR2 proteins and only slightly (<20%) reduced cell surface expression of the mutant species. Our dose-response experiments showed that even transfection of ∼100-times higher amount of the R753Q TLR2-encoding vector failed to elicit the magnitude of NF-κB reporter activation comparable with the plateau of WT TLR2-inducible response. These results indicate that impaired signaling via R753Q TLR2 cannot be attributed to its slightly diminished cell surface expression. Under conditions of comparable expression of WT and mutant TLR2s, we observed significant deficiencies of the R753Q TLR2 species to activate p38 and NF-κB and to induce IL-8 gene expression upon stimulation with inactivated M. tuberculosis and M. tuberculosis-derived cell wall and culture filtrate fractions, LAM and Pam3Cys. Nucleofection-based complementation of TLR2−/− iBMDMs with human WT TLR2 partially restored their responsiveness to Pam3Cys, whereas the R753Q TLR2 version failed to impart Pam3Cys sensitivity, confirming our results in cells with the macrophage background. In contrast, TNF-α induced comparable levels of IL-8 mRNA in 293/TLR2 transfectants that express WT or R753Q TLR2, demonstrating the specific impact of the R753Q mutation on TLR2 signaling.
Very little and controversial information has been reported in the literature regarding the impact of the R753Q polymorphism on TLR2 expression. One study showed no significant changes in expression levels of TLR2 in healthy volunteers and patients with familial Mediterranean fever expressing WT and R753Q TLR2 (70). In contrast, another group found lower α-CD3 Ab-inducible expression of R753Q versus WT TLR2 in T cells obtained from healthy volunteers or patients with atopic dermatitis, with distinct modulation of WT and R753Q TLR2 levels upon stimulation with lipoteichoic acid (69). Of note, two other loss-of-function TIR2 domain mutations, P681H and P631H, do not affect TLR2 expression levels (50, 71). Our data support findings showing the lack of the impact of the TIR2 mutations on TLR2 expression levels. However, we cannot exclude that the R753Q polymorphism could affect TLR2 trafficking and localization, an important regulatory mechanism of TLR2 signaling (72), analogous to the reported effect of the P6531H mutation (71).
To gain initial insights into how the R753Q mutation could interfere with TLR2 signaling, we undertook molecular modeling of the secondary structures of WT- and R753Q-containing TIR domains. These studies suggested significant changes in the electrostatic potential of the DD loop and α-D region with accumulation of the net positive charge in conjunction with discrete main and side chain differences, affecting Pro-746, Arg-748, and Lys-751, and the R753Q mutation itself. Notably, one model of TLR2 hetero-dimerization postulates that the dimerization interface involves the interaction of the DD loop region of the TIR2 and the BB loops of TIR1 or TIR6 (61). Interestingly, according to our molecular modeling, changes in electrostatic potential and discrete main and side chain differences within the DD loop and α-D region imposed by the R753Q mutation affect residues involved in TIR2 interactions with TIR1 (Arg-748 and Arg-753) and TIR6 (Arg-748 and Lys-751) (61). Because TLR2 hetero-dimerization with TLR6 is the initial step required for cell activation in response to M. tuberculosis and M. tuberculosis-derived lipids (15, 32–35), we sought to obtain direct biochemical evidence on whether the R753Q polymorphism affects agonist-inducible TLR2-TLR6 assembly. Using co-immunoprecipitation and Western blot analyses, we found a significantly lower ability of the R753Q TLR2 to associate with TLR6 upon stimulation with Pam2Cys, a defined TLR2-TLR6 agonist (73), confirming the direct impact of the mutation of TLR2-TLR6 hetero-dimerization predicted by molecular modeling.
The molecular basis by which the R753Q mutation interferes with TLR2-TLR6 dimerization is unclear. TLR2 hetero-dimerization with TLR6 is initiated via ligand recognition by leucine-rich repeat-containing receptor ectodomains (17, 74, 75) and is thought to involve subsequent cooperative engagement of their respective TIR domains (17). Because the R753Q polymorphism is localized to the TIR domain and spaced at a significant distance from the ligand-recognition interface in the ectodomain, direct impacts of the mutation on ligand recognition are unlikely. It is tempting to speculate that the R753Q mutation affects cooperative TIR2-TIR6 interactions due to changes in the electrostatic potential and conformation within the DD loop region imposed by the mutation. Further structural and biochemical studies are required to determine the precise molecular mechanism of R753Q-mediated interference.
Agonist-mediated dimerization of the TLR2 TIR domains is thought to form a scaffold to which TIR domain containing adaptor proteins (TIRAP/Mal and MyD88 adapters) and different kinases, including PTKs and IRAKs, are recruited (4, 5, 21, 22, 30, 63, 76). Previous studies by us and others demonstrated the importance of agonist-inducible tyrosine phosphorylation of TLR2, TLR3, TLR4, and TLR9 for endowing signaling competence (20, 40, 60, 76–78). They revealed c-Src as a likely PTK candidate phosphorylating TLR3 (60) and members of the Src kinase and Bruton tyrosine kinase families as putative PTKs that phosphorylate TLR4 (40). However, the identity of PTKs involved in tyrosine phosphorylation of TLR2 is unknown. Because the R753Q polymorphism inhibits TLR2-TLR6 hetero-dimerization that assembles a scaffold TIR interface to which PTKs are recruited, this could translate into the failure of the mutant TLR2 to become tyrosine-phosphorylated. Indeed, this study demonstrates for the first time that in contrast to WT TLR2, the R753Q TLR2 fails to undergo tyrosine phosphorylation upon stimulation with Pam2Cys or irradiated M. tuberculosis, known TLR2-TLR6 agonists. Experiments are ongoing to determine the mechanisms of impaired agonist-inducible tyrosine phosphorylation of R753Q TLR2 and to determine the PTKs involved in TLR2 tyrosine phosphorylation.
TLR2-TLR6 hetero-dimerization and tyrosine phosphorylation of TLR2 are important for assembly of TIR docking platforms to enable recruitment of downstream adapter proteins and kinases. Significant and novel findings reported herein are that the R753Q polymorphism alters agonist-inducible association of the mutant TLR2 with a sorting adapter protein Mal and blocks recruitment of the signaling adapter MyD88. Consistent with dysregulated R753Q TLR2 signalosome assembly, the mutant receptor exhibited deficient phosphorylation of p38, activation of NF-κB reporter, and expression of IL-8 mRNA in 293/TLR2 transfectants stimulated with irradiated M. tuberculosis, M. tuberculosis-derived components, and Pam3Cys. It is plausible that deficient signal transduction of R753Q TLR2 could promote M. tuberculosis replication in infected macrophages, underlying a decreased resistance of R753Q carriers to M. tuberculosis infection and increased incidence of tuberculosis.
In summary, this study shows for the first time that the R753Q mutation renders TLR2 signaling-deficient by impairing TLR2-TLR6 hetero-dimerization, tyrosine phosphorylation, and recruitment of Mal and MyD88, whereas not affecting TLR2 expression. We also provide important and novel mechanistic insights into R753Q-mediated disruption of TLR2 signaling. Furthermore, our data suggest that peptide mimics or small molecule components promoting TLR2-TLR6 hetero-dimerization may overcome the signaling deficiency of the R753Q TLR2 and could serve as an important therapeutic modality for treatment of TLR2-deficient patients.
Supplementary Material
Acknowledgments
We are grateful to Drs. Katherine Fitzgerald and Douglas T. Golenbock (University of Massachusetts Medical School, Worcester, MA) for providing us with cell lines and expression vectors, to Dr. Gregory Melikian (Emory University School of Medicine, Atlanta, GA) for providing pcDNA3-mCherry, and to Dr. Adam Puche (University of Maryland School of Medicine) for guidance and advice with confocal microscopy analyses.
This work was supported, in whole or in part, by National Institutes of Health Grants R21 AI067468 and RO1 AI059524 and the University of Maryland Baltimore-University of Maryland College Park National Institutes of Health Seed Grant (to A. E. M.).

This article contains supplemental Figs. S1–S3.
- TLR
- Toll-like receptor
- TIR
- Toll-IL-1-receptor
- MyD
- myeloid differentiation primary response protein
- Mal
- MyD88 adapter-like
- IRAK
- IL-1 receptor-associated kinase
- TRAF
- TNF receptor-associated factor
- IKK
- IκB kinase
- TAK
- TGF-β-activated kinase
- Ab
- antibody
- LAM
- lipoarabinomannan
- PE
- phycoerythrin
- RT
- real-time
- q
- quantitative
- PTK
- protein-tyrosine kinase
- CW
- cell wall
- BMDM
- bone marrow-derived macrophage
- iBMDM
- immortalized BMDM
- Pam2Cys
- S-[2,3-bis(palmitoyloxy)-(2-RS)-propyl]-Cys-Ser-Lys(4)-OH
- Pam3Cys
- S-[2,3-bis(palmitoyloxy)-(2-RS)-propyl]-N-palmitoyl-(R)-Cys-Ser-Lys4-OH
- Luc
- luciferase.
REFERENCES
- 1. Janeway C. A., Jr., Medzhitov R. (2002) Innate immune recognition. Annu. Rev. Immunol. 20, 197–216 [DOI] [PubMed] [Google Scholar]
- 2. Poltorak A., He X., Smirnova I., Liu M. Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 [DOI] [PubMed] [Google Scholar]
- 3. Medzhitov R., Preston-Hurlburt P., Janeway C. A., Jr. (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397 [DOI] [PubMed] [Google Scholar]
- 4. Beutler B. (2009) Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol. Rev. 227, 248–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doyle S. L., O'Neill L. A. (2006) Toll-like receptors: from the discovery of NFκB to new insights into transcriptional regulations in innate immunity. Biochem. Pharmacol. 72, 1102–1113 [DOI] [PubMed] [Google Scholar]
- 6. Gewirtz A. T., Navas T. A., Lyons S., Godowski P. J., Madara J. L. (2001) Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J. Immunol. 167, 1882–1885 [DOI] [PubMed] [Google Scholar]
- 7. Alexopoulou L., Holt A. C., Medzhitov R., Flavell R. A. (2001) Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413, 732–738 [DOI] [PubMed] [Google Scholar]
- 8. Diebold S. S., Kaisho T., Hemmi H., Akira S., Reis e Sousa C. (2004) Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531 [DOI] [PubMed] [Google Scholar]
- 9. Heil F., Hemmi H., Hochrein H., Ampenberger F., Kirschning C., Akira S., Lipford G., Wagner H., Bauer S. (2004) Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science 303, 1526–1529 [DOI] [PubMed] [Google Scholar]
- 10. Hemmi H., Takeuchi O., Kawai T., Kaisho T., Sato S., Sanjo H., Matsumoto M., Hoshino K., Wagner H., Takeda K., Akira S. (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 [DOI] [PubMed] [Google Scholar]
- 11. Nakamura K., Miyazato A., Xiao G., Hatta M., Inden K., Aoyagi T., Shiratori K., Takeda K., Akira S., Saijo S., Iwakura Y., Adachi Y., Ohno N., Suzuki K., Fujita J., Kaku M., Kawakami K. (2008) Deoxynucleic acids from Cryptococcus neoformans activate myeloid dendritic cells via a TLR9-dependent pathway. J. Immunol. 180, 4067–4074 [DOI] [PubMed] [Google Scholar]
- 12. Krug A., French A. R., Barchet W., Fischer J. A., Dzionek A., Pingel J. T., Orihuela M. M., Akira S., Yokoyama W. M., Colonna M. (2004) TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 21, 107–119 [DOI] [PubMed] [Google Scholar]
- 13. Abe T., Hemmi H., Miyamoto H., Moriishi K., Tamura S., Takaku H., Akira S., Matsuura Y. (2005) Involvement of the Toll-like receptor 9 signaling pathway in the induction of innate immunity by baculovirus. J. Virol. 79, 2847–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guggemoos S., Hangel D., Hamm S., Heit A., Bauer S., Adler H. (2008) TLR9 contributes to antiviral immunity during gammaherpesvirus infection. J. Immunol. 180, 438–443 [DOI] [PubMed] [Google Scholar]
- 15. Bulut Y., Faure E., Thomas L., Equils O., Arditi M. (2001) Cooperation of Toll-like receptor 2 and 6 for cellular activation by soluble tuberculosis factor and Borrelia burgdorferi outer surface protein A lipoprotein: role of Toll-interacting protein and IL-1 receptor signaling molecules in Toll-like receptor 2 signaling. J. Immunol. 167, 987–994 [DOI] [PubMed] [Google Scholar]
- 16. Lien E., Sellati T. J., Yoshimura A., Flo T. H., Rawadi G., Finberg R. W., Carroll J. D., Espevik T., Ingalls R. R., Radolf J. D., Golenbock D. T. (1999) Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J. Biol. Chem. 274, 33419–33425 [DOI] [PubMed] [Google Scholar]
- 17. Ozinsky A., Underhill D. M., Fontenot J. D., Hajjar A. M., Smith K. D., Wilson C. B., Schroeder L., Aderem A. (2000) The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc. Natl. Acad. Sci. U.S.A. 97, 13766–13771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takeuchi O., Sato S., Horiuchi T., Hoshino K., Takeda K., Dong Z., Modlin R. L., Akira S. (2002) Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 169, 10–14 [DOI] [PubMed] [Google Scholar]
- 19. Jin M. S., Kim S. E., Heo J. Y., Lee M. E., Kim H. M., Paik S. G., Lee H., Lee J. O. (2007) Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130, 1071–1082 [DOI] [PubMed] [Google Scholar]
- 20. Arbibe L., Mira J. P., Teusch N., Kline L., Guha M., Mackman N., Godowski P. J., Ulevitch R. J., Knaus U. G. (2000) Toll-like receptor 2-mediated NF-κB activation requires a Rac1-dependent pathway. Nat. Immunol. 1, 533–540 [DOI] [PubMed] [Google Scholar]
- 21. Fitzgerald K. A., Palsson-McDermott E. M., Bowie A. G., Jefferies C. A., Mansell A. S., Brady G., Brint E., Dunne A., Gray P., Harte M. T., McMurray D., Smith D. E., Sims J. E., Bird T. A., O'Neill L. A. (2001) Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 413, 78–83 [DOI] [PubMed] [Google Scholar]
- 22. Horng T., Barton G. M., Medzhitov R. (2001) TIRAP: an adapter molecule in the Toll signaling pathway. Nat. Immunol. 2, 835–841 [DOI] [PubMed] [Google Scholar]
- 23. Kawagoe T., Sato S., Matsushita K., Kato H., Matsui K., Kumagai Y., Saitoh T., Kawai T., Takeuchi O., Akira S. (2008) Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat. Immunol. 9, 684–691 [DOI] [PubMed] [Google Scholar]
- 24. Li S., Strelow A., Fontana E. J., Wesche H. (2002) IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc. Natl. Acad. Sci. U.S.A. 99, 5567–5572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burns K., Janssens S., Brissoni B., Olivos N., Beyaert R., Tschopp J. (2003) Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J. Exp. Med. 197, 263–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheng H., Addona T., Keshishian H., Dahlstrand E., Lu C., Dorsch M., Li Z., Wang A., Ocain T. D., Li P., Parsons T. F., Jaffee B., Xu Y. (2007) Regulation of IRAK-4 kinase activity via autophosphorylation within its activation loop. Biochem. Biophys. Res. Commun. 352, 609–616 [DOI] [PubMed] [Google Scholar]
- 27. Li X. (2008) IRAK4 in TLR/IL-1R signaling: possible clinical applications. Eur. J. Immunol. 38, 614–618 [DOI] [PubMed] [Google Scholar]
- 28. Conze D. B., Wu C. J., Thomas J. A., Landstrom A., Ashwell J. D. (2008) Lys-63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and Toll-like receptor-mediated NF-κB activation. Mol. Cell. Biol. 28, 3538–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lamothe B., Campos A. D., Webster W. K., Gopinathan A., Hur L., Darnay B. G. (2008) The RING domain and first zinc finger of TRAF6 coordinate signaling by interleukin-1, lipopolysaccharide, and RANKL. J. Biol. Chem. 283, 24871–24880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Neill L. A. (2008) When signaling pathways collide: positive and negative regulation of Toll-like receptor signal transduction. Immunity 29, 12–20 [DOI] [PubMed] [Google Scholar]
- 31. Kaufmann S. H. (2006) Tuberculosis: back on the immunologists' agenda. Immunity 24, 351–357 [DOI] [PubMed] [Google Scholar]
- 32. Bowdish D. M., Sakamoto K., Kim M. J., Kroos M., Mukhopadhyay S., Leifer C. A., Tryggvason K., Gordon S., Russell D. G. (2009) MARCO, TLR2, and CD14 are required for macrophage cytokine responses to mycobacterial trehalose dimycolate and Mycobacterium tuberculosis. PLoS Pathog. 5, e1000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Drage M. G., Pecora N. D., Hise A. G., Febbraio M., Silverstein R. L., Golenbock D. T., Boom W. H., Harding C. V. (2009) TLR2 and its co-receptors determine responses of macrophages and dendritic cells to lipoproteins of Mycobacterium tuberculosis. Cell. Immunol. 258, 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Means T. K., Wang S., Lien E., Yoshimura A., Golenbock D. T., Fenton M. J. (1999) Human Toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J. Immunol. 163, 3920–3927 [PubMed] [Google Scholar]
- 35. Drennan M. B., Nicolle D., Quesniaux V. J., Jacobs M., Allie N., Mpagi J., Frémond C., Wagner H., Kirschning C., Ryffel B. (2004) Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am. J. Pathol. 164, 49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ogus A. C., Yoldas B., Ozdemir T., Uguz A., Olcen S., Keser I., Coskun M., Cilli A., Yegin O. (2004) The Arg753Gln polymorphism of the human Toll-like receptor 2 gene in tuberculosis disease. Eur. Respir. J. 23, 219–223 [DOI] [PubMed] [Google Scholar]
- 37. Ben-Ali M., Barbouche M. R., Bousnina S., Chabbou A., Dellagi K. (2004) Toll-like receptor 2 Arg677Trp polymorphism is associated with susceptibility to tuberculosis in Tunisian patients. Clin. Diagn. Lab. Immunol. 11, 625–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kang T. J., Chae G. T. (2001) Detection of Toll-like receptor 2 (TLR2) mutation in the lepromatous leprosy patients. FEMS Immunol. Med. Microbiol. 31, 53–58 [DOI] [PubMed] [Google Scholar]
- 39. Kang T. J., Lee S. B., Chae G. T. (2002) A polymorphism in the Toll-like receptor 2 is associated with IL-12 production from monocyte in lepromatous leprosy. Cytokine 20, 56–62 [DOI] [PubMed] [Google Scholar]
- 40. Medvedev A. E., Piao W., Shoenfelt J., Rhee S. H., Chen H., Basu S., Wahl L. M., Fenton M. J., Vogel S. N. (2007) Role of TLR4 tyrosine phosphorylation in signal transduction and endotoxin tolerance. J. Biol. Chem. 282, 16042–16053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Medvedev A. E., Thomas K., Awomoyi A., Kuhns D. B., Gallin J. I., Li X., Vogel S. N. (2005) Cutting edge: expression of IL-1 receptor-associated kinase-4 (IRAK-4) proteins with mutations identified in a patient with recurrent bacterial infections alters normal IRAK-4 interaction with components of the IL-1 receptor complex. J. Immunol. 174, 6587–6591 [DOI] [PubMed] [Google Scholar]
- 42. Quevedo-Diaz M. A., Song C., Xiong Y., Chen H., Wahl L. M., Radulovic S., Medvedev A. E. (2010) Involvement of TLR2 and TLR4 in cell responses to Rickettsia akari. J. Leukoc. Biol. 88, 675–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Figueroa L., Xiong Y., Song C., Piao W., Vogel S. N., Medvedev A. E. (2012) The Asp299Gly polymorphism alters TLR4 signaling by interfering with recruitment of MyD88 and TRIF. J. Immunol. 188, 4506–4515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Visintin A., Latz E., Monks B. G., Espevik T., Golenbock D. T. (2003) Lysines 128 and 132 enable lipopolysaccharide binding to MD-2, leading to Toll-like receptor-4 aggregation and signal transduction. J. Biol. Chem. 278, 48313–48320 [DOI] [PubMed] [Google Scholar]
- 45. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 46. Piao W., Song C., Chen H., Wahl L. M., Fitzgerald K. A., O'Neill L. A., Medvedev A. E. (2008) Tyrosine phosphorylation of MyD88 adapter-like (Mal) is critical for signal transduction and blocked in endotoxin tolerance. J. Biol. Chem. 283, 3109–3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xiong Y., Qiu F., Piao W., Song C., Wahl L. M., Medvedev A. E. (2011) Endotoxin tolerance impairs IL-1 receptor-associated kinase (IRAK) 4 and TGF-β-activated kinase 1 activation, K63-linked polyubiquitination and assembly of IRAK1, TNF receptor-associated factor 6, and IκB kinase γ and increases A20 expression. J. Biol. Chem. 286, 7905–7916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Piao W., Song C., Chen H., Diaz M. A., Wahl L. M., Fitzgerald K. A., Li L., Medvedev A. E. (2009) Endotoxin tolerance dysregulates MyD88- and Toll/IL-1R domain-containing adapter inducing IFN-β-dependent pathways and increases expression of negative regulators of TLR signaling. J. Leukoc. Biol. 86, 863–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith C. A., Kortemme T. (2008) Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J. Mol. Biol. 380, 742–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu Y., Tao X., Shen B., Horng T., Medzhitov R., Manley J. L., Tong L. (2000) Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature 408, 111–115 [DOI] [PubMed] [Google Scholar]
- 51. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Grell L., Parkin C., Slatest L., Craig P. A. (2006) EZ-Viz, a tool for simplifying molecular viewing in PyMOL. Biochem. Mol. Biol. Educ. 34, 402–407 [DOI] [PubMed] [Google Scholar]
- 53. Du X., Poltorak A., Silva M., Beutler B. (1999) Analysis of Tlr4-mediated LPS signal transduction in macrophages by mutational modification of the receptor. Blood Cells Mol. Dis. 25, 328–338 [DOI] [PubMed] [Google Scholar]
- 54. Heggelund L., Müller F., Lien E., Yndestad A., Ueland T., Kristiansen K. I., Espevik T., Aukrust P., Frøland S. S. (2004) Increased expression of Toll-like receptor 2 on monocytes in HIV infection: possible roles in inflammation and viral replication. Clin. Infect. Dis. 39, 264–269 [DOI] [PubMed] [Google Scholar]
- 55. Latz E., Visintin A., Lien E., Fitzgerald K. A., Monks B. G., Kurt-Jones E. A., Golenbock D. T., Espevik T. (2002) Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the Toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J. Biol. Chem. 277, 47834–47843 [DOI] [PubMed] [Google Scholar]
- 56. Chow J. C., Young D. W., Golenbock D. T., Christ W. J., Gusovsky F. (1999) Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J. Biol. Chem. 274, 10689–10692 [DOI] [PubMed] [Google Scholar]
- 57. Aliprantis A. O., Yang R. B., Mark M. R., Suggett S., Devaux B., Radolf J. D., Klimpel G. R., Godowski P., Zychlinsky A. (1999) Cell activation and apoptosis by bacterial lipoproteins through Toll-like receptor-2. Science 285, 736–739 [DOI] [PubMed] [Google Scholar]
- 58. Nagpal K., Plantinga T. S., Wong J., Monks B. G., Gay N. J., Netea M. G., Fitzgerald K. A., Golenbock D. T. (2009) A TIR domain variant of MyD88 adapter-like (Mal)/TIRAP results in loss of MyD88 binding and reduced TLR2/TLR4 signaling. J. Biol. Chem. 284, 25742–25748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hayes M. W., Carrion R., Jr., Nunneley J., Medvedev A. E., Salvato M. S., Lukashevich I. S. (2012) Pathogenic Old World arenaviruses inhibit TLR2/Mal-dependent proinflammatory cytokines in vitro. J. Virol. 86, 7216–7226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Johnsen I. B., Nguyen T. T., Ringdal M., Tryggestad A. M., Bakke O., Lien E., Espevik T., Anthonsen M. W. (2006) Toll-like receptor 3 associates with c-Src tyrosine kinase on endosomes to initiate antiviral signaling. EMBO J. 25, 3335–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Basith S., Manavalan B., Govindaraj R. G., Choi S. (2011) In silico approach to inhibition of signaling pathways of Toll-like receptors 2 and 4 by ST2L. PLoS One 6, e23989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yamamoto M., Sato S., Hemmi H., Sanjo H., Uematsu S., Kaisho T., Hoshino K., Takeuchi O., Kobayashi M., Fujita T., Takeda K., Akira S. (2002) Essential role for TIRAP in activation of the signaling cascade shared by TLR2 and TLR4. Nature 420, 324–329 [DOI] [PubMed] [Google Scholar]
- 63. Kagan J. C., Medzhitov R. (2006) Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 125, 943–955 [DOI] [PubMed] [Google Scholar]
- 64. Ferwerda B., Kibiki G. S., Netea M. G., Dolmans W. M., van der Ven A. J. (2007) The Toll-like receptor 4 Asp299Gly variant and tuberculosis susceptibility in HIV-infected patients in Tanzania. AIDS 21, 1375–1377 [DOI] [PubMed] [Google Scholar]
- 65. Salpietro C., Rigoli L., Miraglia Del Giudice M., Cuppari C., Di Bella C., Salpietro A., Maiello N., La Rosa M., Marseglia G. L., Leonardi S., Briuglia S., Ciprandi G. (2011) TLR2 and TLR4 gene polymorphisms and atopic dermatitis in Italian children: a multicenter study. Int. J. Immunopathol. Pharmacol. 24, Supp. 4, 33–40 [DOI] [PubMed] [Google Scholar]
- 66. Ahmad-Nejad P., Mrabet-Dahbi S., Breuer K., Klotz M., Werfel T., Herz U., Heeg K., Neumaier M., Renz H. (2004) The Toll-like receptor 2 R753Q polymorphism defines a subgroup of patients with atopic dermatitis having severe phenotype. J. Allergy Clin. Immunol. 113, 565–567 [DOI] [PubMed] [Google Scholar]
- 67. Kang S. H., Abdel-Massih R. C., Brown R. A., Dierkhising R. A., Kremers W. K., Razonable R. R. (2012) Homozygosity for the Toll-like receptor 2 R753Q single-nucleotide polymorphism is a risk factor for cytomegalovirus disease after liver transplantation. J. Infect. Dis. 205, 639–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee S. O., Brown R. A., Kang S. H., Abdel-Massih R. C., Razonable R. R. (2011) Toll-like receptor 2 polymorphism and Gram-positive bacterial infections after liver transplantation. Liver Transpl. 17, 1081–1088 [DOI] [PubMed] [Google Scholar]
- 69. Mrabet-Dahbi S., Dalpke A. H., Niebuhr M., Frey M., Draing C., Brand S., Heeg K., Werfel T., Renz H. (2008) The Toll-like receptor 2 R753Q mutation modifies cytokine production and Toll-like receptor expression in atopic dermatitis. J. Allergy Clin. Immunol. 121, 1013–1019 [DOI] [PubMed] [Google Scholar]
- 70. Soylu A., Ateş H., Cingöz S., Türkmen M., Demir B. K., Tunca M., Sakızlı M., Cirit M., Ersoy R., Ulgenalp A., Kavukçu S. (2011) TLR polymorphisms in FMF: association of TLR-2 (Arg753Gln) and TLR-4 (Asp299Gly, Thre399Ile) polymorphisms and myeloid cell TLR-2 and TLR-4 expression with the development of secondary amyloidosis in FMF. Inflammation 34, 379–387 [DOI] [PubMed] [Google Scholar]
- 71. Etokebe G. E., Skjeldal F., Nilsen N., Rodionov D., Knezevic J., Bulat-Kardum L., Espevik T., Bakke O., Dembic Z. (2010) Toll-like receptor 2 (P631H) mutant impairs membrane internalization and is a dominant negative allele. Scand. J. Immunol. 71, 369–381 [DOI] [PubMed] [Google Scholar]
- 72. McGettrick A. F., O'Neill L. A. (2010) Localization and trafficking of Toll-like receptors: an important mode of regulation. Curr. Opin. Immunol. 22, 20–27 [DOI] [PubMed] [Google Scholar]
- 73. Buwitt-Beckmann U., Heine H., Wiesmüller K. H., Jung G., Brock R., Akira S., Ulmer A. J. (2005) Toll-like receptor 6-independent signaling by diacylated lipopeptides. Eur. J. Immunol. 35, 282–289 [DOI] [PubMed] [Google Scholar]
- 74. Kajava A. V., Vasselon T. (2010) A network of hydrogen bonds on the surface of TLR2 controls ligand positioning and cell signaling. J. Biol. Chem. 285, 6227–6234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kang J. Y., Nan X., Jin M. S., Youn S. J., Ryu Y. H., Mah S., Han S. H., Lee H., Paik S. G., Lee J. O. (2009) Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31, 873–884 [DOI] [PubMed] [Google Scholar]
- 76. Chockalingam A., Rose W. A., 2nd., Hasan M., Ju C. H., Leifer C. A. (2012) Cutting edge: a TLR9 cytoplasmic tyrosine motif is selectively required for proinflammatory cytokine production. J. Immunol. 188, 527–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sarkar S. N., Elco C. P., Peters K. L., Chattopadhyay S., Sen G. C. (2007) Two tyrosine residues of Toll-like receptor 3 trigger different steps of NF-κB activation. J. Biol. Chem. 282, 3423–3427 [DOI] [PubMed] [Google Scholar]
- 78. Sarkar S. N., Peters K. L., Elco C. P., Sakamoto S., Pal S., Sen G. C. (2004) Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat. Struct. Mol. Biol. 11, 1060–1067 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.