

Abstract
Background
We examined the effect of the novel Alzheimer's disease (AD) risk variant rs11136000 single nucleotide polymorphism (SNP) in the clusterin gene (CLU) on longitudinal changes in resting state regional cerebral blood flow (rCBF) during normal aging and investigated its influence on cognitive decline in pre-symptomatic stages of disease progression.
Methods
Subjects were participants in the Baltimore Longitudinal Study of Aging. A subset of 88 cognitively normal older individuals had longitudinal 15O-water PET measurements of rCBF at baseline and up to 8 annual follow-up visits. We also analyzed trajectories of cognitive decline among CLU risk carriers and non-carriers both in individuals who remained cognitively normal (N=599) as well as in those who subsequently converted to mild cognitive impairment (MCI) or AD (N=95).
Results
Cognitively normal carriers of the CLU risk allele show significant and dose-dependent longitudinal increases in resting state rCBF in brain regions intrinsic to memory processes. There were no differences in trajectories of memory performance between CLU risk carriers and non-carriers who remained cognitively normal. However, in cognitively normal individuals who eventually convert to MCI or AD, CLU risk carriers show faster rates of decline in memory performance relative to non-carriers in the pre-symptomatic stages of disease progression.
Conclusions
The AD risk variant CLU influences longitudinal changes in brain function in asymptomatic individuals and is associated with faster cognitive decline in pre-symptomatic stages of disease progression. These results suggest mechanisms underlying the role of CLU in AD and may be important in monitoring disease progression in at-risk elderly.
Keywords: Clusterin, single nucleotide polymorphism, Alzheimer's disease, 15O-water PET, cerebral blood flow, memory
Introduction
Recent genome wide association studies (GWAS) identified polymorphisms in the clusterin (CLU) gene associated with risk for Alzheimer's disease (AD) (1, 2). Although this finding has subsequently been replicated (3, 4), the risk variant of CLU occurs commonly in the general population and exerts a small effect size in conferring risk for AD (5). It is therefore unlikely that this finding will be of clinical utility as a stand-alone predictor of disease risk in older individuals. Nevertheless, CLU and other novel genetic risk variants for AD may hold important clues to elucidating mechanisms relevant to AD pathogenesis, especially in at-risk older individuals.
Several lines of evidence suggest biological roles for clusterin in pathways relevant to AD pathogenesis including amyloid clearance, complement modulation and apoptosis (6–9). Recently, we showed that plasma concentration of clusterin protein was associated with brain atrophy, disease severity and clinical progression in AD patients as well as with brain fibrillar amyloid beta deposition in non-demented elderly (10). Our finding of an association between plasma clusterin concentration and disease severity in AD was recently replicated by Schrijvers and colleagues (11).
Our aim was to investigate the association between the principal single nucleotide polymorphism (SNP) in the CLU gene associated with AD risk and longitudinal changes in regional resting state cerebral blood flow (rCBF) evaluated by 15O-water positron emission tomography (PET) imaging. Together with regional cerebral glucose metabolism, rCBF is thought to be a reliable index of neuronal/synaptic function and both imaging modalities have been extensively used to study perturbations in neuronal function in asymptomatic individuals at increased risk for AD (12). In light of recent GWAS studies that reported a significantly reduced risk of AD in carriers of the T allele at SNP rs11136000 (1, 2), our first aim was to test the hypothesis that individuals with the alternative C-risk allele would show longitudinal changes in rCBF in brain regions vulnerable to AD pathology and/or important in memory processes.
We tested this hypothesis by examining longitudinal 15O-water PET data in older individuals who remained cognitively normal during the course of the study. While this hypothesis tested the effects of the AD risk variant CLU SNP on brain function in asymptomatic older individuals, it was also of interest to test whether this gene might influence the rate of progression in cognitive decline both during normal aging as well as in the presymptomatic stages of AD progression. Our second aim was therefore to test whether cognitively normal risk carriers of the AD risk variant CLU showed faster rates of decline in memory performance relative to non-carriers both in those who maintained cognitive health during aging as well among those who eventually converted to mild cognitive impairment or AD. We tested these hypotheses in non-demented older individuals in the Baltimore Longitudinal Study of Aging (BLSA) and in its neuroimaging substudy.
Subjects and Methods
This study analyzed two complementary datasets from participants in the Baltimore Longitudinal Study of Aging (BLSA) (Figure 1). The first was the neuroimaging substudy of the main BLSA study where longitudinal 15O-water PET data were collected annually between 1994 and 2004. PET data analyzed in this report were acquired from 88 (mean age 69 years; range 56–86 years) non-demented participants followed over a mean interval of 7.5 years (range 4–8 years (13). These participants represent all neuroimaging substudy participants with a minimum of three resting state 15O-water PET scans in whom genome-wide genotyping data were available, with the exception of the following exclusions. We excluded individuals with clinical strokes, severe head trauma and CNS infection. We also excluded participants meeting criteria for AD (NINCDS-ADRDA) and mild cognitive impairment (MCI) as determined by consensus case conference (14, 15) from the PET analysis because the numbers of neuroimaging study participants who developed cognitive impairment were too few to stratify by the CLU risk allele.
Figure-1. Participants in the Baltimore Longitudinal Study of Aging (BLSA) and its neuroimaging substudy.

Schematic illustration of the main BLSA and neuroimaging sub-studies showing the selection of participants whose longitudinal 15O-water PET and cognitive data were analyzed in this study.
The second dataset that was analyzed was the main BLSA study and also included participants from the neuroimaging substudy. The objective of this analysis was to examine the effect of the rs11136000 SNP on rates of cognitive decline in individuals who remained cognitively normal (NC) as well as in those converting to MCI/AD (converters). In this analysis, we used all available data from the main BLSA and neuroimaging substudies. We selected BLSA participants who were cognitively normal at the time of entry into the study, were assessed annually or every 2 years by neuropsychological testing and had genome-wide genotyping data available. The first time point selected for analysis of rates of cognitive decline was the earliest visit where two tests of memory performance were administered, i.e. Benton Visual Retention Test (BVRT) and California Verbal Learning Test (CVLT). We excluded assessments at and after the onset of cognitive impairment or AD for the converters group. Assessments before the age 60 years were also excluded in the NC group to ensure that the range of participant ages in the two groups were similar. In the `NC' group, longitudinal cognitive data were available in 599 participants (mean age at first assessment; 67.5 years; range 60–93 years) who remained cognitively normal during the course of this study (mean follow-up interval of 6.6 years; range 0–15 years). In the `converters' group, 95 participants who were initially cognitively normal (mean age at first assessment; 75.9 years; range 60–91 years) eventually declined to either MCI (N=45) or AD (N=50). These analyses were performed on longitudinal cognitive data containing 435 serial assessments (45 data points in risk non-carriers and 390 data points in risk allele carriers) in the `converter' group and 2859 serial cognitive assessments (530 data points in risk non-carriers and 2329 data points in risk allele carriers) in the `NC' group over 15 years from 1993 to 2008.
The 599 individuals who remained cognitively normal through the duration of follow up also included the 88 individuals who participated in the 15O-water PET experiments. All 954 BLSA participants (i.e. decliners to MCI/AD; N=106 and cognitively normal through the course of the study; N=848) (mean age 64 years) were cognitively normal at the time of entry into the study, were assessed annually or every 2 years by neuropsychological testing and had genome-wide genotyping data available. The first time point selected for analysis of rates of cognitive decline was the earliest visit where two tests of memory performance were administered, i.e. BVRT and CVLT. Tables 1 and 2 show the sample characteristics of participants included in this study. This study was approved by the local institutional review board. All participants provided written informed consent prior to each assessment.
Table-1.
Sample characteristics of participants included in the 15O-water PET studies (BLSA neuroimaging substudy)
| rs11136000 Genotype | N | APOE ε4 status | Sex | Race | Education (years) (±SD) | Age at baseline (years) (±SD) | Follow-up duration (years) (±SD) | Framingham Risk Score (±SD) |
|---|---|---|---|---|---|---|---|---|
| Whole Sample | 88 | 59 ε4− 29 ε4+ |
36 F 52 M |
78 C 10 AA |
16.4 (2.8) | 69.3 (7.3) | 7.5 (0.9) | 13.4 (7.5) |
| CC | 29 | 21 ε4− 8 ε4+ |
9 F 20 M |
28 C 1 AA |
16.7 (2.8) | 69.8 (6.5) | 7.8(0.6) | 13.9 (7.5) |
| TC | 45 | 31 ε4− 14 ε4+ |
24 F 21 M |
39 C 1 AA |
16.1 (2.5) | 69.0 (7.4) | 7.3 (1.0) | 12.1 (7.2) |
| TT | 14 | 7 ε4− 7 ε4+ |
3 F 11 M |
11 C 3 AA |
16.8 (3.5) | 69.3 (8.8) | 7.4 (0.9) | 16.8 (8.0) |
| p-value for difference | 0.32 | 0.044 | 0.13 | 0.61 | 0.88 | 0.11 | 0.13 |
F= female; M= male, C= Caucasian; AA= African-American
Genotyping
Genome-wide genotyping was performed using the Illumina Infinium HumanHap550 genotyping chip, assaying >555,000 unique SNPs per sample. Though many polymorphisms were genotyped, this study deals only with the AD risk variant rs11136000 polymorphism found within the CLU gene. Standard quality control of genotyping data was conducted as described previously (16). In brief, individuals were excluded due to call rate < 95% genome-wide, cryptic relatedness due to proportional sharing (pi_hat) > 0.125 with another participant in the BLSA (effectively excluding first degree relatives), and non-European ancestry ascertained from multi-dimensional scaling analyses using HapMap reference populations. SNPs were excluded due to minor allele frequencies < 1%, a missingness rate > 5%, Hardy-Weinberg equilibrium p-values < 1E-5, and non-random missingness by haplotype p-values < 1E-5. All quality control of genotype data was undertaken using PLINKv1.05 [PMID: 17701901]. Among individuals included in the 15O-water PET studies, frequencies of genotypes in the rs11136000 polymorphism were T/T in 14 subjects (15.9%), C/T in 45 subjects (51.1%), and C/C in 29 subjects (32.9%). Thus 84% of our participants in the 15O-water PET studies carried the risk C-allele. The frequency for the minor (T) allele (MAF) in our sample was 41.48%. Among individuals included in the analysis of cognitive decline in those converting to MCI/AD, the frequencies of genotypes in the rs11136000 polymorphism were T/T in 11 subjects (10.3%), C/T in 57 subjects (53.7%), and C/C in 38 subjects (35.8%). APOE genotype analysis was performed separately on DNA extracted from fresh blood by restriction enzyme isoform genotyping in all participants.
Neuropsychological testing
During each annual neuroimaging visit, participants completed a battery of neuropsychological tests evaluating six cognitive domains. Mental status was assessed with the Mini-Mental State Examination (MMSE), memory was assessed using the California Verbal Learning Test (CVLT) and Benton Visual Retention Test (BVRT). Word knowledge and verbal ability were measured using Primary Mental Abilities Vocabulary (PMA). Verbal fluency was assessed by Letter (i.e. FAS) and Category fluency tests. Attention and working memory were measured by the Digit Span Test of the Wechsler Adult Intelligence Scale-Revised, and the Trail Making Test. Digits Backward, Trails B, and Verbal Fluency (categories and letters) assessed executive function. The Card Rotations Test assessed visuospatial function. Data from evaluations at each time point were used to examine differences in change in cognitive performance over time between CLU risk carriers and non-carriers in participants undergoing 15O-water PET studies.
In the analyses comparing trajectories of cognitive decline between CLU risk carriers and non-carriers in individuals remaining cognitively normal and those eventually converting to MCI and AD, we formulated an a priori hypothesis, based on the regional distribution of rCBF changes in our 15O-water PET studies and examined differences in memory performance over the follow-up interval. In this analysis, we used all data points from the first assessment (i.e. earliest visit where both CVLT and BVRT tests were administered) until the onset of MCI or AD, thereby capturing changes in rates of decline in memory performance in the pre-symptomatic stages of disease progression. Similar to the 15O-water PET studies, the main aim of this analysis was to examine the effects of the CLU risk allele on the pre-symptomatic stages of disease progression. Hence data from time points at and after the onset of MCI/AD were not included.
Linear mixed effects models were used in these longitudinal analyses to investigate the effects of the CLU rs11136000 SNP on both baseline memory scores as well as rates of change in memory performance (17). The models were fit using PROC MIXED in SAS 9.1 (SAS Institute, Cary, NC) software. Performance scores on the CVLT and BVRT tests were entered as dependent variables. The fixed effects part of the model included the following predictors; baseline age (age0), sex, race, APOE ε4 status (carriers coded 1 and non-carriers coded 0), CLU risk carrier status (CC/CT coded 1 and TT coded 0), group (NC coded as 0, converters coded as 1) time (i.e. follow up time from baseline), age0*time, sex* time, race* time, APOE ε4 status* time, CLU risk carrier status* time, group*time and group* CLU risk carrier status* time. Random effects included intercept and time. These models allowed us to test firstly if the effects of the clusterin risk allele on rates of change in memory performance are the same between NC group and Converter group after adjusting for baseline age, sex, race and APOE ε4 status, secondly the effect of clusterin risk allele on rates of change in memory performance separately in NC group and converter group. In exploratory analyses, CLU risk carrier status was also coded additively for the number of risk alleles (C) as 0, 1 or 2.
PET scanning parameters
Participants underwent PET scans at baseline (year-1) and up to eight annual follow-ups. Each imaging session included a resting scan in which participants were instructed to keep their eyes open and focused on a computer screen covered by a black cloth.
PET measures of rCBF were obtained using [15O] water. For each scan, 75 mCi of [15O] water were injected as a bolus. Scans were performed on a GE 4096+ scanner, which provides 15 slices of 6.5 mm thickness. Images were acquired for 60 seconds from the time total radioactivity counts in the brain reached threshold level. Attenuation correction was performed using a transmission scan acquired prior to the emission scans.
PET data analysis
Data from PET scans obtained annually from baseline to the last available follow-up time points were used in the analyses. The mean interval between baseline and last follow-up PET scans was 7.5 (±0.9 SD) years. The PET scans were realigned and spatially normalized into standard stereotactic space and smoothed to full width at half maximum of 12×12×12 mm in the x, y, and z planes using a Gaussian filter. Next, to control for variability in global flow, rCBF values at each voxel were ratio adjusted to the mean global flow estimated from gray matter intensity values and scaled to 50 ml/100g/min for each scan, then thresholded at 0.80 of the mean image intensity value of each scan to exclude peripheral signal scatter in the images. For each participant, change in rCBF was calculated across all preprocessed scans using linear modeling to estimate the rates of change over time and extract the estimated fit parameter for each voxel. An image of the longitudinal rates of change at each voxel (i.e. slope or linear temporal trends image) was then created for each subject (Statistical Parametric Mapping software, SPM2, Wellcome Trust Centre for Neuroimaging, UCL, London).
We used the slope images from all subjects in a voxel-based multiple regression analysis (SPM5) with the number of C-alleles of the rs11136000 SNP as an independent predictor of longitudinal changes in rCBF. Alleles were coded for association using an additive model (i.e. 0, 1 and 2 representing individuals with 0, 1 and 2 copies of the C allele respectively). The associations were adjusted for baseline age, sex and the interval between baseline and last scan. In a secondary analysis, we also included APOE ε4 status (ε4 carriers coded as 1; N=29 and ε4 non-carriers coded as 0; N=59) as a covariate in the analysis.
In order to reduce the risk of type-I error due to multiple comparisons, we applied the following procedures. First, we adopted a statistical magnitude threshold recommended by the PET Working Group of theNIH/NIA Neuroimaging Initiative (http://www.nia.nih.gov/ResearchInformation/ExtramuralPrograms/NeuroscienceOfAging/Summary+%E2%80%93+PET+Working+Group.htm). Secondly, we applied a spatial extent threshold of at least 50 voxels within the regions meeting the statistical threshold of p<0.005. Finally, in those regions that met both the statistical (p<0.005) and spatial extent criteria (50 voxels), we applied a small volume correction with false discovery rate (FDR) adjusted p-value of <0.05. In those regions showing significant associations between the number of C-alleles of rs11136000 and longitudinal changes in rCBF, baseline rCBF as well as the magnitude of change in rCBF were extracted at the local maxima for each region using a 4-mm spherical search area.
Results
Sample characteristics (15O-water-PET study)
Group differences in continuous variables were examined using t-tests, and differences in categorical variables by the Fisher-exact test. The three groups (CC, CT and TT) did not differ significantly in age at baseline, number of years of education or APOE ε4 status. Their Mini Mental State Examination (MMSE) scores did not differ significantly either at baseline or at the last follow-up. The Framingham cardiovascular risk score at the baseline imaging visit was also calculated and provided an assessment of the 10-year risk profile for coronary heart disease (CHD) (18). This composite score of cardiovascular risk was based on the presence of the following specific risk factors: age, total serum cholesterol concentration, blood pressure, diabetes mellitus and smoking. There were no significant differences in the cardiovascular risk profiles as determined by the composite Framingham risk score for each group (table-1). The annual rates of change in performance on each of the cognitive tests also did not differ significantly between these groups (table S1 in the Supplement).
Effect of CLU rs11136000 SNP on longitudinal changes in rCBF
We observed significant longitudinal increases in rCBF in several brain regions in carriers of the C-allele of the rs11136000 SNP (Figure 2 and Table 3). The direction of the effect indicated greater longitudinal increases in rCBF in those with two copies of the C-allele (CC) relative to CT and TT genotypes. The increments in rCBF were thus greatest in individuals with CC, intermediate in CT and lowest in the TT genotypes. The brain regions showing these longitudinal increments in rCBF included the right and left hippocampus, and the right anterior cingulate cortex. The right orbitofrontal cortex approached statistical significance (FDR-adjusted p=0.06). These associations remained significant even after co-varying for APOE ε4 status. We also calculated baseline rCBF and magnitude of change in rCBF over time within these regions and confirmed that carriers of the C-allele of rs11136000 SNP showed greater magnitude of rCBF increase over time (table-4).
Figure-2. Associations between longitudinal changes in resting regional cerebral blood flow (rCBF) and the AD risk variant rs11136000 SNP in the clusterin gene (CLU) in cognitively normal older individuals.
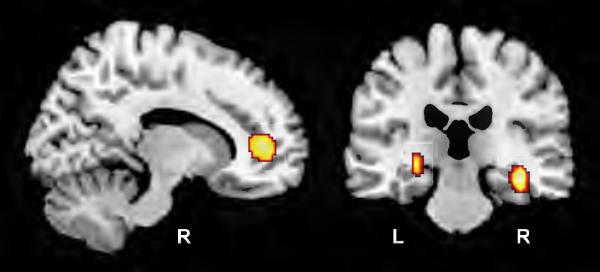
Carriers of the C-allele of the rs11136000 SNP were coded additively (CC=2, CT=1 and TT=0) in a voxel-based multiple regression analysis (SPM5). Highlighted regions show significantly greater longitudinal increases in rCBF in carriers of the risk variant C-allele within the right anterior cingulate cortex (left panel) andbilateral hippocampi (right panel) (R and L; right and left cerebral hemispheres respectively).
Table-3. Local maxima within areas of significant longitudinal increases in rCBF in carriers of the C-allele of the rs11136000 snp in the clusterin gene. Coordinates are in stereotactic space and Brodmann areas are in parentheses.
Carriers of the C-allele of the rs11136000 SNP were coded additively (CC=2, CT=1 and TT=0) in a voxel-based multiple regression analysis (SPM5).
| Coordinates | ||||||||
|---|---|---|---|---|---|---|---|---|
| Region | side | x | y | z | t value | p value | FDR-adjusted p value | # voxels |
| Orbitofrontal Cortex (47) | R | 34 | 30 | −22 | 3.73 | <0.001 | 0.06 | 130 |
| Ant Cingulate Cortex (32) | R | 14 | 46 | 4 | 4.13 | <0.001 | 0.02 | 245 |
| Hippocampus | R | 36 | −24 | −14 | 4.10 | <0.001 | 0.02 | 137 |
| Hippocampus | L | −26 | −22 | −6 | 3.35 | 0.001 | 0.03 | 90 |
Table-4. Mean annual rates of change (ml/100g/minute) and the corresponding standard errors for resting state regional cerebral blood flow (rCBF) in TT, CT and CC groups of the rs11136000 CLU SNP.
The magnitude of change in rCBF over time was calculated within those regions showing significant associations between the number of C-alleles of rs11136000 and longitudinal changes in rCBF in the primary SPM analysis (table-3). This was performed by extracting the adjusted rCBF values at the local maxima for each region shown in table-3 using a 4-mm spherical search area. The brain regions shown above are components of the default mode network, an interacting set of brain regions involved in intrinsic memory processes.
| CC | CT | TT | |
|---|---|---|---|
| Lt. Hippocampus | 0.65 (0.11) | 0.37 (0.09) | 0.12 (0.13) |
| Rt. Hippocampus | 0.30 (0.09) | 0.10 (0.07) | −0.35 (0.16) |
| Rt. Anterior cingulate cortex | 0.31 (0.09) | 0.07 (0.08) | −0.37 (0.11) |
Effect of CLU rs11136000 SNP on rates of cognitive decline
The effects of the CLU rs11136000 risk allele on rates of change in memory performance as measured by the California Verbal Learning Test (CVLT) were significantly different between the `NC' group and `converter' groups. These effects were observed both for verbal immediate (sum of 5 CVLT List A trials) (p=0.027) and delayed free recall scores (p=0.0098). Among individuals who remained cognitively normal during the course of the study (N=599) there were no significant differences in rates of change in memory performance between CLU risk carriers and non-carriers as observed in both verbal immediate (sum of 5 CVLT List A trials) (p=0.99) and delayed free recall scores (p=0.85). Among those participants who were cognitively normal at the first assessment, but subsequently declined to MCI/AD (N=95), CLU risk carriers showed significantly faster rates of decline than CLU risk non-carriers in both verbal immediate (sum of 5 CVLT List A trials) (p=0.0032) and delayed free recall scores (p=0.032).
We did not find significant differential effects of the CLU risk allele on rates of change in visual memory performance as measured by the Benton Visual Retention Test (BVRT) between the `NC' group and `converter' groups (p=0.52). Moreover, the effects of the CLU risk allele on rates of change in BVRT performance in both groups was not significant (NC group p = 0.80, converter group, p = 0.90). We did not observe any significant effects of CLU on rates of decline in memory performance when the risk alleles were entered additively (i.e. 0, 1 or 2 copies of the C allele).
Discussion
Our main aim was to study the effect of the AD risk variant CLU SNP on brain function during normal aging using 15O-water PET. We observe robust changes in rCBF over time in cognitively normal older individuals carrying the C-allele of the rs11136000 SNP in the CLU gene which confers increased risk of AD. These changes reflect significant longitudinal increases in rCBF in the hippocampus and anterior cingulate cortex; brain regions especially important to memory function and key components of the default mode network (DMN), an interacting set of brain regions involved in intrinsic memory processes (19). We observed the largest incremental change in rCBF in individuals with two copies of the C-allele of the rs11136000 SNP. These findings remained significant after adjusting for age, sex and APOE ε4 status.
Previous studies have reported changes in brain function in asymptomatic carriers of the APOE ε4 allele, the most robust genetic risk factor for late-onset AD (12, 20). Most of these studies have been cross sectional, and the most consistent observations were that of reductions in cerebral metabolic rate of glucose consumption in the temporal and parietal cortices (21, 22). In a recent longitudinal study, we reported decreases in resting rCBF in the frontal, parietal and temporal cortices as well as increases in the insular cortex in non-demented older APOE ε4 carriers (23). In functional magnetic resonance imaging (fMRI) studies, asymptomatic APOE ε4 carriers show greater magnitude and extent of brain activation while performing a memory task in comparison to non-carriers in several regions including hippocampus, parietal and prefrontal regions (24, 25). This pattern of greater brain activation in asymptomatic APOE ε4 carriers must be interpreted in the light of studies that have shown a significant age × APOE interaction in the pattern of increased neuronal activity observed in fMRI paradigms. Fillipini et al. have demonstrated recently that overactivity of brain function in young ε4-carriers is disproportionately reduced with advancing age even before the onset of measurable memory impairment (26). More recently, Linden and colleagues demonstrated neural hyperactivation in the frontal cortex, posterior cingulate cortex and hippocampus in healthy young adult carriers (median age; 29.1 years) of the AD risk variant of CLU during performance of a working memory task (27). Increased neural activation has been hypothesized to represent compensatory mechanisms wherein greater cognitive effort is required in at-risk individuals to achieve equivalent levels of performance to risk non-carriers (25). Similarly, increased hippocampal activity during memory processes in individuals with mild cognitive impairment (MCI) has also been observed (28).
Our current results extend these findings substantially by demonstrating that in carriers of the recently discovered AD risk variant of CLU, longitudinal increments in neural activity also occur in the resting state within brain regions critical to memory processes, perhaps to maintain their normal physiological function in cognitively normal at-risk individuals. The regions implicated in this study are known to place a high demand on the brain's energy resources and are also include some areas that are vulnerable to disruption by deposition of beta amyloid even in the non-demented elderly (29, 30). It is plausible that sustaining the observed increments in resting state rCBF over several years places an especially high burden on the brain's energy resources. The eventual failure of these presumed compensatory increments in neural activity in some individuals may be the threshold beyond which early cognitive impairment begins to manifest, marking their transition from normal aging to Alzheimer's disease.
Given the above observations in resting state rCBF in asymptomatic carriers of the AD risk variant of CLU, we asked whether risk carriers of this allele also show changes in cognitive performance over time in pre-symptomatic stages of disease progression. Given the observed pattern of changes within regions critical for memory processes in asymptomatic CLU risk carriers, we hypothesized that CLU risk carriers eventually converting to MCI/AD would show faster rates of decline in memory performance relative to non-carriers in the pre-symptomatic stages of disease progression. Moreover, we previously showed that decline in episodic memory performance is the earliest change in cognition in the presymptomatic stages of AD (31).
We confirmed our hypothesis by demonstrating significantly faster rates of decline in verbal memory performance scores in CLU risk carriers. By focusing on individuals eventually converting to MCI/AD and only using longitudinal cognitive data from baseline until the onset of MCI/AD, our analysis was targeted specifically towards delineating effects of CLU on pre-symptomatic stages of disease progression. We combined the MCI and AD groups to increase the numbers of participants and thereby acquire more power to detect differences between the risk and non-risk groups. While the underlying assumption in doing so is that MCI and AD are part of a spectrum of disease severity, it must be acknowledged that this is a possible limitation of the study. It must be noted in this context that the effects of the CLU risk variant on AD risk are small, and not surprisingly, a recent study did not observe any significant effects of CLU on memory performance over time in individuals who remained cognitively normal (32), a finding consistent with our own observations in the present analysis. However, it is also plausible that with a longer follow up interval, greater numbers of individuals in the cognitively normal group may show significant differences in changes in memory performance over time between the CLU risk and non-risk groups. It is also likely that the small effect of the CLU risk variant on cognition did not allow us to detect a dose effect when we coded the risk additively for the number of C alleles.
Conclusions
Our results suggest that the novel AD risk variant CLU influences neuronal activity within memory circuits in asymptomatic risk carriers who remain cognitively normal. Moreover, CLU also exerts effects on early, pre-symptomatic stages of disease progression by accelerating decline in memory performance in risk carriers who eventually convert to MCI/AD. These findings provide evidence implicating CLU in early events in AD pathogenesis and may be important in monitoring disease progression in carriers of this common risk variant.
Supplementary Material
Table-2a.
Sample characteristics of participants included in the analysis of rates of decline in memory performance in individuals remaining cognitively normal (NC)
| rs11136000 Genotype | N | APOE ε4 status | Sex | Race | Education (years) (±SD) | Age at baseline (years) (±SD) | Follow-up duration (years) (±SD) | Total number of Data points |
|---|---|---|---|---|---|---|---|---|
| Whole Sample | 599 119 TT 288 TC 192 CC |
426 ε4− 153 ε4+ |
257 F 342 M |
134 AA 465 C |
16.5 (2.5) | 67.5 (7.5) | 6.6 (4.6) | 2859 |
| TT | 119 | 77 ε4− 39 ε4+ |
52 F 67 M |
49 AA 79 C |
16.7 (2.4) | 67.7 (7.2) | 6.3 (4.3) | 530 |
| TC,CC | 480 | 349 ε4− 114 ε4+ |
205 F 275 M |
94 AA 386 C |
16.4 (2.5) | 67.5 (7.6) | 6.7 (4.7) | 2329 |
| p-value for difference | 0.049 | 0.85 | 0.001 | 0.22 | 0.83 | 0.35 |
F= female; M= male, C= Caucasian; AA= African-American
Table-2b.
Sample characteristics of participants included in the analysis of rates of decline in memory performance in cognitively normal individuals who eventually converted to MCI/AD (converters) rs11136000 Genotype
| N | Diagnosis | APOE ε4 status | Sex | Race | Education (years) (±SD) | Age at baseline (years) (±SD) | Follow-up duration (years) (±SD) | Total number of Data points | |
|---|---|---|---|---|---|---|---|---|---|
| Whole Sample | 95 9 TT 52 TC 34 CC |
45 MCI 50 AD |
70 ε4− 25 ε4+ |
41 F 54 M |
7 AA 88 C |
16.2 (3.1) | 75.9 (7.1) | 5.4 (4.2) | 435 |
| TT | 9 | 4 MCI 5 AD |
8 ε4− 1 ε4+ |
6 F 3 M |
1 AA 8 C |
17.1 (1.7) | 78.6 (8.1) | 5.3 (3.8) | 45 |
| TC,CC | 86 | 41 MCI 45 AD |
62 ε4− 24 ε4+ |
35 F 51 M |
6 AA 80 C |
16.1 (3.2) | 75.6 (6.9) | 5.4 (4.2) | 390 |
| p-value for difference | 1.00 | 0.44 | 0.17 | 0.51 | 0.37 | 0.22 | 0.98 |
Acknowledgments
This work was supported in part by research and development contract N01-AG-3-2124 from the Intramural Research Program, National Institute onAging, National Institutes of Health and by a research and development contract with MedStar Research Institute. We are grateful to the Baltimore Longitudinal Study of Aging participants and neuroimaging staff for their dedication to these studies and the staff of the Johns Hopkins University PET facility for their assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Potential conflicts of interest: Dr. Thambisetty is named as an inventor on a patent application filed by his previous employer, Kings College London (KCL) on biomarkers for Alzheimer's disease. Dr. Lovestone is an inventor on patents held by KCL for biomarkers of Alzheimer's disease. He is a research collaborator on studies funded by J and J, GSK, Proteome Sciences and Merck Millipore. He is a member of the Lundbeck Neurocience speaker panel. He does not report any personal financial rewards from the above entities. None of the other authors report any biomedical financial interests or potential conflicts of interest.
REFERENCES
- 1.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 3.Carrasquillo MM, Belbin O, Hunter TA, Ma L, Bisceglio GD, Zou F, et al. Replication of CLU, CR1, and PICALM associations with alzheimer disease. Arch Neurol. 2010;67:961–964. doi: 10.1001/archneurol.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corneveaux JJ, Myers AJ, Allen AN, Pruzin JJ, Ramirez M, Engel A, et al. Association of CR1, CLU and PICALM with Alzheimer's disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum Mol Genet. 2010;19:3295–3301. doi: 10.1093/hmg/ddq221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW, et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004;41:193–202. doi: 10.1016/s0896-6273(03)00850-x. [DOI] [PubMed] [Google Scholar]
- 7.DeMattos RB, O'Dell MA, Parsadanian M, Taylor JW, Harmony JA, Bales KR, et al. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2002;99:10843–10848. doi: 10.1073/pnas.162228299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.French LE, Wohlwend A, Sappino AP, Tschopp J, Schifferli JA. Human clusterin gene expression is confined to surviving cells during in vitro programmed cell death. J Clin Invest. 1994;93:877–884. doi: 10.1172/JCI117043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nuutinen T, Suuronen T, Kauppinen A, Salminen A. Clusterin: a forgotten player in Alzheimer's disease. Brain Res Rev. 2009;61:89–104. doi: 10.1016/j.brainresrev.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 10.Thambisetty M, Simmons A, Velayudhan L, Hye A, Campbell J, Zhang Y, et al. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry. 2010;67:739–748. doi: 10.1001/archgenpsychiatry.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schrijvers EM, Koudstaal PJ, Hofman A, Breteler MM. Plasma clusterin and the risk of Alzheimer disease. JAMA. 2011;305:1322–1326. doi: 10.1001/jama.2011.381. [DOI] [PubMed] [Google Scholar]
- 12.Scarmeas N, Stern Y. Imaging studies and APOE genotype in persons at risk for Alzheimer's disease. Curr Psychiatry Rep. 2006;8:11–17. doi: 10.1007/s11920-006-0076-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Resnick SM, Goldszal AF, Davatzikos C, Golski S, Kraut MA, Metter EJ, et al. One-year age changes in MRI brain volumes in older adults. Cereb Cortex. 2000;10:464–472. doi: 10.1093/cercor/10.5.464. [DOI] [PubMed] [Google Scholar]
- 14.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 15.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 16.Terracciano A, Balaci L, Thayer J, Scally M, Kokinos S, Ferrucci L, et al. Variants of the serotonin transporter gene and NEO-PI-R Neuroticism: No association in the BLSA and SardiNIA samples. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:1070–1077. doi: 10.1002/ajmg.b.30932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38:963–974. [PubMed] [Google Scholar]
- 18.Wilson PW, D'Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 19.Greicius MD, Menon V. Default-mode activity during a passive sensory task: uncoupled from deactivation but impacting activation. J Cogn Neurosci. 2004;16:1484–1492. doi: 10.1162/0898929042568532. [DOI] [PubMed] [Google Scholar]
- 20.Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer's disease. J Clin Psychiatry. 2007;68:613–618. doi: 10.4088/jcp.v68n0419. [DOI] [PubMed] [Google Scholar]
- 21.Reiman EM, Caselli RJ, Chen K, Alexander GE, Bandy D, Frost J. Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:3334–3339. doi: 10.1073/pnas.061509598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Small GW, Ercoli LM, Silverman DH, Huang SC, Komo S, Bookheimer SY, et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer's disease. Proc Natl Acad Sci U S A. 2000;97:6037–6042. doi: 10.1073/pnas.090106797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thambisetty M, Beason-Held L, An Y, Kraut MA, Resnick SM. APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch Neurol. 2010;67:93–98. doi: 10.1001/archneurol.2009.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bondi MW, Houston WS, Eyler LT, Brown GG. fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease. Neurology. 2005;64:501–508. doi: 10.1212/01.WNL.0000150885.00929.7E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bookheimer S, Burggren A. APOE-4 genotype and neurophysiological vulnerability to Alzheimer's and cognitive aging. Annu Rev Clin Psychol. 2009;5:343–362. doi: 10.1146/annurev.clinpsy.032408.153625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Filippini N, Ebmeier KP, MacIntosh BJ, Trachtenberg AJ, Frisoni GB, Wilcock GK, et al. Differential effects of the APOE genotype on brain function across the lifespan. Neuroimage. 2011;54:602–610. doi: 10.1016/j.neuroimage.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 27.Lancaster TM, Baird A, Wolf C, Jackson MC, Johnston SJ, Donev R, et al. Neural hyperactivation in carriers of the Alzheimer's risk variant on the clusterin gene. Eur Neuropsychopharmacol. 2011 doi: 10.1016/j.euroneuro.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Dickerson BC, Salat DH, Bates JF, Atiya M, Killiany RJ, Greve DN, et al. Medial temporal lobe function and structure in mild cognitive impairment. Ann Neurol. 2004;56:27–35. doi: 10.1002/ana.20163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sperling RA, Laviolette PS, O'Keefe K, O'Brien J, Rentz DM, Pihlajamaki M, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaishnavi SN, Vlassenko AG, Rundle MM, Snyder AZ, Mintun MA, Raichle ME. Regional aerobic glycolysis in the human brain. Proc Natl Acad Sci U S A. 2010;107:17757–17762. doi: 10.1073/pnas.1010459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grober E, Hall CB, Lipton RB, Zonderman AB, Resnick SM, Kawas C. Memory impairment, executive dysfunction, and intellectual decline in preclinical Alzheimer's disease. J Int Neuropsychol Soc. 2008;14:266–278. doi: 10.1017/S1355617708080302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chibnik LB, Shulman JM, Leurgans SE, Schneider JA, Wilson RS, Tran D, et al. CR1 is associated with amyloid plaque burden and age-related cognitive decline. Ann Neurol. 2010 doi: 10.1002/ana.22277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.