

Abstract
Neuroendocrine systems integrate both extrinsic and intrinsic signals to regulate virtually all aspects of an animal’s physiology. In aquatic toxicology, studies have shown that pollutants are capable of disrupting the neuroendocrine system of teleost fish, and many chemicals found in the environment can also have a neurotoxic mode of action. Omics approaches are now used to better understand cell signaling cascades underlying fish neurophysiology and the control of pituitary hormone release, in addition to identifying adverse effects of pollutants in the teleostean central nervous system. For example, both high throughput genomics and proteomic investigations of molecular signaling cascades for both neurotransmitter and nuclear receptor agonists/antagonists have been reported. This review highlights recent studies that have utilized quantitative proteomics methods such as 2D differential in-gel electrophoresis (DIGE) and isobaric tagging for relative and absolute quantitation (iTRAQ) in neuroendocrine regions and uses these examples to demonstrate the challenges of using proteomics in neuroendocrinology and neurotoxicology research. To begin to characterize the teleost neuroproteome, we functionally annotated 623 unique proteins found in the fish hypothalamus and telencephalon. These proteins have roles in biological processes that include synaptic transmission, ATP production, receptor activity, cell structure and integrity, and stress responses. The biological processes most represented by proteins detected in the teleost neuroendocrine brain included transport (8.4%), metabolic process (5.5%), and glycolysis (4.8%). We provide an example of using sub-network enrichment analysis (SNEA) to identify protein networks in the fish hypothalamus in response to dopamine receptor signaling. Dopamine signaling altered the abundance of proteins that are binding partners of microfilaments, integrins, and intermediate filaments, consistent with data suggesting dopaminergic regulation of neuronal stability and structure. Lastly, for fish neuroendocrine studies using both high-throughput genomics and proteomics, we compare gene and protein relationships in the hypothalamus and demonstrate that correlation is often poor for single time point experiments. These studies highlight the need for additional time course analyses to better understand gene–protein relationships and adverse outcome pathways. This is important if both transcriptomics and proteomics are to be used together to investigate neuroendocrine signaling pathways or as bio-monitoring tools in ecotoxicology.
Keywords: Neuroproteome, Neurotoxicology, Hypothalamus, Microarray, Gene–protein relationship, Network analysis
1. Neuroendocrine and neurotoxicity research in teleost fishes using Omics technology
Neuroendocrine research in teleost models has yielded novel insight into the molecular pathways underlying synaptic transmission and perturbation of signal transduction by environmental pollutants. The neural signaling cascades originating in the hypothalamus and telencephalon, the major neuroendocrine regions of the brain that control pituitary hormone release, are also affected by aquatic contaminants [40]. In recent years, microarrays have been used to investigate transcriptomics changes in the neuroendocrine brain, and molecular responses to neurotransmitter agonists and antagonists have been identified in fish neuroendocrine tissues [30]. In the area of aquatic toxicology, there are a growing number of reports on the effects of neuroactive pharmaceuticals and pesticides on gene responses in both the hypothalamus and telencephalon of fish. Efforts to better understand the impact of pharmaceuticals and pesticides on the transcriptome in neuroendocrine regions of the teleost brain have included studies using fluoxetine, the active ingredient of Prozac in goldfish (Carassius auratus) [24], 17α-ethinylestradiol (EE2), a major component of the birth control pills, in goldfish and zebrafish (Danio rerio) [21,22], and the γ-aminobutyric acid A (GABA-A) receptor antagonist, dieldrin, in largemouth bass (LMB; Micropterus salmoides) [17]. These studies and others have contributed to increased knowledge of how the hypothalamus and telencephalon respond on a molecular level to aquatic pollutants.
In addition to transcriptomics, characterizing the neuroproteome for quantitative studies is important for toxicology and physiology because fish possess orthologs to most human gene drug targets [12] and in general are predicted to show conserved molecular responses to neuroactive compounds. Thus, the fields of fish neuroendocrinology and neurotoxicology will benefit from information about the neuroendocrine proteome and this will improve the value of fish as models for human disease research [2,41]. In addition, data about the presence and relative abundance of neuroendocrine proteins will facilitate more focused proteomics studies using, for example, absolute quantitation (AQUA) to measure specific proteins in the brain [20]. Currently, a limitation in fish neuroendocrine research is the lack of specific antibodies for fish neuropeptides and their associated receptors. These low abundant proteins can be a challenge to quantify and a priori knowledge about one’s likelihood to detect proteins in the brain using mass spectrometry can be beneficial, especially in non-model fish species.
To characterize the fish neuroproteome, all unique proteins identified in previous studies in the LMB hypothalamus [17], goldfish hypothalamus [29], and fathead minnow (FHM) telencephalon [18] were functionally annotated using AgBase-GOanna [24]. Protein sequences were uploaded into GOanna and a BLASTP search conducted to obtain annotated teleost proteins. The databases selected for protein annotation were UniProt and SwissProt. The substitution matrix used in the analysis was BLOSUM62 [13] and the Gap Costs was set at “Existence: 11 Extension: 1”. All other parameters for the protein searches in GOanna were set to default.
Proteins were functionally annotated using the highest BLAST score in the databases. Protein annotations from GOanna contained multiple Gene Ontology (GO) identifiers for each protein. Data were then manually filtered to remove any redundant GO categories for a single protein (due to searching both UniProt and Swiss-Prot). The GO categories for biological processes (P), molecular functions (F) and cellular components (C) were individually sorted to determine the most common GO categories represented by proteins identified in the teleost neuroendocrine brain.
There were 623 unique hypothalamic and telencephalic proteins that were functionally annotated with high confidence (Appendix 1). There was a broad range of biological functions represented by these proteins and the number of unique biological processes represented by the proteins was 457. The biological processes most represented by proteins detected in the teleost neuroendocrine brain included transport (8.4%), metabolic process (5.5%), and glycolysis (4.8%). The top ten biological processes and their relative proportion within the top ten are shown in Fig. 1A. Selected biological functions that are of interest to neurotoxicology include proteins involved in stress and human disease (Table 1). Stress proteins identified included Hsp isoforms and Gst pi and Gst rho. Synaptic-related proteins identified in the hypothalamus and telencephalon included synapsin-1 (Syn1), synaptosomal-associated protein 25-A/B (Snap25) and synaptotagmin-2 (Syt2). These proteins play a predominant role in packaging and secretion of neuropeptides and neurohormones.
Fig. 1.

Most abundant ontologies for proteins detected in the neuroendocrine brain for (A) biological process (B) molecular function (C) cellular compartment. Fractionation of peptides was achieved using strong cation exchange followed by reverse phase HPLC. Peptides were detected by LC MS/MS using a QSTAR XL (Applied Biosystems) over a 2 h gradient (details in reference 19). Gene ontology categories depicted in the figure represent the ten most abundant categories and their relative percentages within the top ten. For biological processes, the most abundant categories comprised 37% of all GO terms, while molecular function and cellular compartment comprised 38% and 52%, respectively.
Table 1.
Some examples of proteins detected in the fish hypothalamus that are related to neurotoxicity. % coverage refers to the percentage of the protein detected from peptides. Shown are proteins that are associated with human neurological and neurodegenerative diseases, proteins involved in general/oxidative stress response, and proteins involved in protein folding and degradation.
| NCBI identifier | Protein | Species | % Cov. | Category |
|---|---|---|---|---|
| gi|76253753 | Alpha-synuclein | LMB | 47.24 | Disease biomarkers |
| gi|62199406 | Annexin | Goldfish | 9.44 | Disease biomarkers |
| gi|46329651 | Annexin A11 | Goldfish | 4.12 | Disease biomarkers |
| gi|32451741 | Annexin A4 | Goldfish | 8.41 | Disease biomarkers |
| gi|37778988 | Apolipoprotein E | LMB | 36.60 | Disease biomarkers |
| gi|3907633 | Beta-1 tubulin | LMB | 48.99 | Disease biomarkers |
| gi|47211399 | Beta-synuclein | LMB | 30.34 | Disease biomarkers |
| gi|82264543 | Cytochrome c | LMB | 20.19 | Disease biomarkers |
| gi|37590349 | Enolase 1, (alpha) | LMB | 51.16 | Disease biomarkers |
| gi|76253695 | Gamma1-synuclein | LMB | 34.51 | Disease biomarkers |
| gi|67772081 | Copper/zinc superoxide dismutase | LMB | 31.03 | Stress |
| gi|47086689 | Glutathione S-transferase mu | Goldfish | 31.96 | Stress |
| gi|112901127 | Glutathione S-transferase rho | Goldfish | 36.28 | Stress |
| gi|95832164 | Glutathione S-transferase pi | Goldfish | 55.29 | Stress |
| gi|112950077 | Heat shock cognate 60 kDa | Goldfish | 20.35 | Stress |
| gi|28569550 | Heat shock cognate 70 kDa | Goldfish | 39.29 | Stress |
| gi|56207785 | Heat shock protein 90 kDa-alpha | LMB | 7.86 | Stress |
| gi|50540284 | Proteasome (prosome, macropain) subunit, beta type, 2 | Goldfish | 19.10 | Protein degradation |
| gi|51242139 | Proteasome (prosome, macropain) subunit, beta type, 3 | Goldfish | 18.05 | Protein degradation |
| gi|37595366 | Ubiquitin C | Goldfish | 94.47 | Protein degradation |
| gi|115361552 | Ubiquitin C-terminal hydrolase L1 | Goldfish | 7.91 | Protein degradation |
| gi|47085781 | Ubiquitin-activating enzyme E1 | Goldfish | 10.78 | Protein degradation |
| gi|47087259 | Ubiquitin-conjugating enzyme E2 variant 2 | Goldfish | 47.59 | Protein degradation |
| gi|94734326 | Ubiquitin-conjugating enzyme E2N | Goldfish | 31.17 | Protein degradation |
The number of unique molecular functions represented by the neuroendocrine proteins was 289. The molecular functions most represented included nucleotide binding (6.5%), catalytic activity (5.2%), and protein binding (4.6%). The top ten molecular functions and their relative proportion within the top ten are shown in Fig. 1B. The number of unique cellular compartments represented by neuroendocrine proteins was 169. The cellular compartments containing most of the proteins detected were the cytoplasm (12.6%), plasma membrane (8.5%), and proteins integral to the plasma membrane (5.7%). The top ten cellular compartments and their relative proportion within the top ten are shown in Fig. 1C. Based upon the frequency and numbers of unique peptides detected by mass spectrometry, some of the more abundant proteins in the neuroendocrine brain included beta 2 tubulin (Tubb2), beta-actin (Actb), calmodulin (Cam), fructose-bisphosphate aldolase C (brain-type) (Aldoc), and enolase 1 (Eno1). This is based on the numbers of peptides detected for each protein but these data should be interpreted with caution as the entire protein complement of the hypothalamus and telencephalon was not detected. Nevertheless, the assumption is that the number and frequencies of peptides detected are positively correlated to relative protein abundance. It is noted that protein detection will depend upon a number of factors that include sample preparation, peptide separation techniques, and the capabilities of mass spectrometers (these studies utilized a QSTAR, Applied Biosystems). It is important to categorize the proteins that are most prominently identified in LC MS/MS based proteomics experiments because these abundant proteins may be related to important biochemical processes in the CNS.
To obtain a wider view of the neuroproteome, it will be necessary to apply additional fractionation techniques or deplete the most abundant proteins using antibody affinity columns, similar to techniques suggested by others [3]. Increased sensitivity in mass spectrometers in terms of scan time will also greatly enhance protein detection. In a recent study of the zebrafish whole brain, Singh et al. [36] were able to identify 8475 proteins by using sophisticated protein pre-fractionation techniques and an ESI-mass spectrometer with a linear ion trap mass analyzer (LTQ-IT; Thermo Fisher). Using network pathway analysis, it was found that zebrafish proteins were involved in functions that include cell motion and cell organization, activation of adenylate cyclase, apoptosis and programmed cell death, and regulation of cell proliferation. Of interest to neurotoxicology research, many proteins detected by mass spectrometry were involved in human mental disorders such as Schizophrenia and Parkinson’s disease (PD). Proteins with a role in PD included Parp1, Caspase 3 and 9, and alpha synuclein. Of interest to neuroendocrinology, Singh et al. [36] detected proteins that included hormones (thyrotropin-releasing hormone, growth hormone 1, corticotropin releasing hormone) and hormone receptors (luteinizing hormone receptor, thyroid hormone receptor alpha, ghrelin receptor, vasopressin V2 receptor, gonadotropin releasing hormone receptor 1, and growth hormone receptor). As Singh and colleagues point out, this study is a significant step forward in characterizing the brain proteome for biomarkers that underlie neurological function and disease states.
It should be acknowledged that the zebrafish genome is completely sequenced, facilitating increased protein identifications. However, databases for other teleost fishes are rapidly increasing in sequence information. Our research in goldfish, largemouth bass, and fathead minnow has also identified many of the same zebrafish whole brain proteins in the hypothalamus and telencephalon. The potential for extending deeper into the teleostean neuroendocrine proteome is high and this new information on peptide-protein identification can now be used to design more sophisticated proteomics experiments in fish.
2. Quantitative proteomics studies in fish
There have been recent comprehensive reviews on the incorporation of proteomics methodology into fish physiology [11] and aquatic toxicology [34]. Therefore our focus is to highlight the use of proteomics in studies of the teleostean brain, including both neurophysiology and neurotoxicity studies. Protein quantitation methods such as 2D-PAGE have offered unique insight into cell signaling cascades in the brain that are stimulated or inhibited by environmental contaminants. Cadmium (Cd) is a heavy metal that is released into the environment during metal mining and is toxic at low concentrations. Zhu et al. [43] used a 2D gel approach to identify protein biomarker candidates for Cd toxicity in the whole brain of the Japanese flounder (Paralichthys olivaceus). Proteins such as creatine kinase, actin 1, putative xylose repressor, mitochondrial transcriptional regulator, and transferrin were differentially affected by Cd. In another study in Japanese flounder treated with the insecticide methyl parathion, whole brain proteins were quantified using 2D PAGE. Stress proteins such as Hsp70 and Gst showed differences in abundance compared to controls and the authors concluded that the application of multiple biomarkers is more advantageous than that of a single biomarker for monitoring methyl parathion pollution [27]. Using the 2D-DIGE approach, Damodaran and colleagues [5] investigated the zebrafish brain proteome after ethanol treatment. Proteins such as voltage-dependent anion channels 1 and 2 (Vdac1 and Vdac2), apolipoprotein-A1 (Apoa1), Hsp70, and glutamic-oxaloacetic acid transaminase-1 (Got1) were altered in abundance by ethanol. It is noted that these studies examined whole brain responses to aquatic contaminants and results obtained in whole brain will not be representative of effects in specific neuroendocrine tissues. However, these studies have provided valuable mechanistic insight into the basis of neurotoxicity and provided data on putative protein biomarkers of adverse effect in the fish brain.
Non-gel based proteomics approaches can also offer novel mechanistic insight into disruptions in neurotransmitter signaling in fish by environmental pollutants. Isobaric tagging for relative and absolute quantitation (iTRAQ) [31] has been used to identify hypothalamic proteins that were responsive to the neuroactive organochlorine pesticide, dieldrin [17]. A number of potential protein biomarkers for dieldrin-induced neurotoxicity were identified and interestingly, many of these proteins were implicated in human neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease. Proteins affected in the LMB hypothalamus by dieldrin included apolipoprotein E (ApoE), microtubule-associated protein Tau (Mapt), beta-actin (Actb), and enolase 1 (Eno1). This study offers novel mechanistic insight into possible relationships between environmental exposures to contaminants and human neurological diseases. Moreover, well-characterized biomarkers for general and oxidative stress are readily detected in the fish brain, for example heat shock proteins (Hsp) and glutathione-s-transferase (Gst) and these proteins can be quantified after toxicant treatments [17,29]. Other protein biomarkers such as alpha II spectrin (Spna2), a biomarker for traumatic brain injury in juvenile Chinook salmon (Oncorhynchus tshawytscha) encountering spill ways during migration [26], can also be quantified using LC MS/MS. Fish have been utilized as vertebrate models for neuroendocrine research and human neurodegenerative disease models [23,28,30]. Therefore, these aforementioned proteomics studies generate valuable information about the relationship between environmentally relevant waterborne exposures and dysfunction within the CNS.
3. Protein network identification in the neuroendocrine brain using bioinformatics
Bioinformatics approaches allow for the discovery of unique processes underlying neuroendocrine physiology and toxicology. In microarray analysis, functional enrichment of gene ontology categories, gene set enrichment, and pathway analysis can be common methods for identifying biological processes in fish impacted by a hormone or aquatic pollutant in tissues such as the neuroendocrine brain. Marlatt et al. [16] used gene ontology and functional enrichment analysis to evaluate the effects of estradiol treatment (100 μg/g body wt in silastic implants; 1 day) on the expression of genes in the goldfish hypothalamus and associated transcriptomics changes to the processes of protein metabolism, nucleobase or nucleotide metabolism, and cellular metabolism. Fluoxetine injection (5 μg/g body wt; 5 times, every 3 days) affected expression of goldfish hypothalamic genes involved in signal transduction, metabolic pathways, organogenesis, and reproduction differentially in the hypothalamus [25]. These approaches have improved our understanding of the pathways underlying hormone feedback in the neuroendocrine brain and responses to pharmaceuticals found in aquatic environments.
However, there are obstacles in using similar bioinformatics approaches for quantitative proteomics datasets. In order to perform gene set enrichment analysis, annotation for a gene/protein must be known a priori. This facilitates statistical analysis, e.g., the Fisher’s Exact Test, to determine the proportions of regulated genes with a given biological process that are over represented in comparison to all gene probes on the microarray. In proteomics experiments, this is not possible as proteins may only be identified in a single biological replicate due to the instrument scan speed, sensitivity of the mass spectrometer, or biological changes in the individual that render the protein below detection. As demonstrated above, functional annotation can characterize the neuroendocrine proteome but regulated proteins cannot be functionally compared to all non-regulated proteins because the number of proteins in a fish tissue at a given time is not known.
There are some bioinformatics approaches that can be quite useful such as comparing regulated proteins to known pathways or cell signaling cascades to determine whether a pathway is over-represented in a known network. It is well established that the use of network methodologies improve predictive ability and discriminant analysis for gene/protein responses by identifying common regulatory pathways [4,35]. To demonstrate the utility of this approach for proteins quantified in the teleost hypothalamus, sub-network enrichment analysis (SNEA) was performed on proteomic data obtained from Popesku et al. [29] using Pathway Studio 7.1 (Ariadne, Rockville, MD, USA). The experiment involved injections of D1 and D2 dopamine agonists; LY 171555 (D2 agonist; label 115) and SKF 38393 (D1 agonist; label 117). A total of 621 proteins were identified in the goldfish hypothalamus and of these detected proteins, 207 proteins for LY 171555 treatment and 185 proteins for SKF 38393 treatment met criteria for quantitation using iTRAQ. Of the proteins quantified in each treatment group, a total of 335 proteins were successfully mapped to human homologs using the NCBI GenBank protein ID. In order to identify protein networks that are differentially affected by DA receptor agonists, SNEA was performed using expression targets, binding partners and post-translational modification targets as options in the analysis. SNEA identifies a central molecule or “seed” that shares common molecular targets with a particular treatment. This approach statistically identified protein networks that were differentially regulated via the two dopamine receptors. The enrichment p-value for gene seeds was set at p < 0.05.
In the case of binding targets, SNEA analysis determined that the D2 receptor agonist LY 171555 decreased a network of proteins that bind integrin while the D1 receptor agonist SKF 38393 increased a network of proteins that bind integrin (Table 2). SNEA therefore provides novel insight into dopamine signaling and the involvement of integrins. Integrins play a key role in cell attachment and cell structure/mobility. There are other studies suggesting that integrins are regulated via dopamine receptor signaling. For example, DA has been shown to activate integrins via D3 receptors in human T cell fractions. DA activation stimulated adhesion of naive CD8 + T cells to fibronectin, providing partial mechanisms for the role of dopamine in migration of immune related cells [39]. Using specific agonists and antagonists, Levite et al. [14] showed that the interaction of dopamine with T cells via integrins (β1 subtype) is regulated by both D3 and D2 receptor subtypes. Interestingly, the D2 receptor agonist in the Popesku study decreased binding partners of integrins, while D1 increased this binding network. The role of D1 receptor mediated action on integrins is not well characterized and this analysis provides an example of how SNEA can extract additional information from proteomics data to generate new hypotheses in the goldfish about DA signaling and regulation.
Table 2.
Sub-network enrichment analysis (SNEA) for proteins in the goldfish hypothalamus affected by LY 171555 and SKF 38393 that share a given binding partner.
| Name | # Of measured neighbors |
Median change |
p-Value |
|---|---|---|---|
| LY 171555 (D2 agonist) | |||
| Binding partners of integrin | 5 | −1.518 | 0.017 |
| Binding partners of type IV IF protein | 5 | 1.356 | 0.045 |
| SKF 38393 (D1 agonist) | |||
| Binding partners of integrin | 5 | 1.452 | 0.007 |
| Binding partners of microtubule | 27 | −1.072 | 0.013 |
| Binding partners of type IV IF protein | 5 | 1.224 | 0.026 |
| Binding partners of Caveolin-1 | 8 | 1.310 | 0.045 |
| Binding partners of amyloid protein | 5 | −1.254 | 0.047 |
4. Relationship between transcript and protein in the fish neuroendocrine brain
For fish ecotoxicogenomics and physiological genomics studies, an important concept that requires further study is that of geneprotein relationships. Regulation (synthesis and breakdown) of both mRNAs and proteins have different kinetics and turnover rates. In aquatic toxicology, this information can be useful for determining the type of molecular biomarker (gene, protein, metabolite, or others) that might be most informative in predicting adverse outcome pathways over a given time period. The question arises of whether gene expression data are more informative in acute exposures and protein data are more informative in sub-chronic exposures for predicting downstream physiological changes. Gene–protein relationships are an important consideration in ecotoxicology studies because many laboratory toxicity assays are acute exposures for 48 h or 21 d so the timing of molecular changes is critical. It is not known whether changes in steady state mRNA levels are more informative in determining a chemical’s mode of action at 48 h of exposure and proteins are more informative at 21 d of exposure. A good example of this is illustrated with vitellogenin, the classical biomarker for estrogens. Sheepshead minnows (Cyprinodon variegatus) injected with 2.5 mg/kg E2 had elevated vtg mRNA levels in the liver at 24 h, showed mRNA reaching a plateau by 48 h, and then showed vtg mRNA expression returned to background levels after 6 days. Conversely protein levels did not significantly increase until 4–5 days after the injection and remained consistently elevated over a longer time period (>10 days) [8]. Thus, gene–protein relationships can provide different information depending on the temporal scale.
The neuroendocrine brain presents a particular challenge for transcriptional regulation studies, and especially in relating transcript level with protein. Firstly, mRNA levels in many studies are thought not to change very much in comparison to tissues such as the liver but this hypothesis must be rigorously tested. Fine regulatory control of transcripts in the brain or protection provided by the brain-blood barrier may be the reason why the brain transcriptome appears not to respond as dramatically as other tissues. Secondly, there can be experimental reasons for a disconnect such as the time lag between mRNA changes and protein level changes, such as the example with vitellogenin. There are also complex biological regulatory factors involved in transcription and translation that are not well understood in fish, such as miRNA, siRNA, alternative splicing of mRNA, and post-translational modification of proteins. These regulatory pathways will also contribute significantly to gene–protein relationships.
There are some microarray and iTRAQ data in fish that can be used to explore gene–protein relationships on a larger scale. In the goldfish, exposure to the dopamine D1 receptor agonist for 5 h showed that 57% of the regulated proteins corresponded to similar increases in mRNA levels while the D2-agonist-treated fish showed that only 14% of the proteins corresponded in directional change with the mRNA [29]. These data are comparable to studies in both unicellular and multi-cellular organisms. In a meta-analysis of mRNA and protein relationships, De Sousa et al. [6] demonstrated that squared Pearson’s correlation coefficients for gene–protein studies range from 0.20 to 0.47 in bacteria, 0.34–0.87 in yeast, and 0.09–0.46 in multi-cellular organisms. Based upon regression analyses, approximately 30–80% of variation in yeast and bacteria protein levels can be attributed to variation in mRNA expression. These data were collected by methods that included LC-MS/MS, 2D gel electrophoresis, SAGE, and microarrays. It is apparent that the kinetics of gene expression, mRNA stability, protein translation, and protein degradation is highly specific and diverse and can result in poor relationships among molecules. Careful consideration of experimental design and new studies will improve the ability to assess the relationship between mRNA and protein levels in fish neuroendocrine regions.
A correlation approach by Popesku et al. [29] is one method to better understanding mRNA–protein relationships. In this case for the D1 agonist study mentioned above, there was relative agreement in directional change between mRNA and protein in the hypothalamus. To investigate whether there are cause and effect relationships between mRNA and protein changes in the fish neuroendocrine brain, we performed a regression analysis using combined data from Popesku et al. [29] and Martyniuk et al. [17]. There were 39 data points analyzed using linear regression but in the presentation of the data, only those points that had protein fold changes below 4-fold are shown (Fig. 2). The two points with relatively high protein fold changes (8 and 16-fold change) were removed to accommodate the graphical representation. The inclusion of these two points in the regression did not change the overall conclusions of the data. In the fish hypothalamus, there was no significant relationship between protein and mRNA fold changes as determined by iTRAQ and microarray analysis, respectively (R2 = 0.07) (d.f. = 1, F = 2.9, p = 0.10). It is pointed out that gene–protein relationships will depend upon the nature of the treatment or contaminant and it may be expected that over an exposure time gene–protein relationships may become more apparent. Conversely, there may be a critical window of time in which genes and protein changes highly correlate in response to a neurotoxicant or neurotransmitter receptor agonist.
Fig. 2.
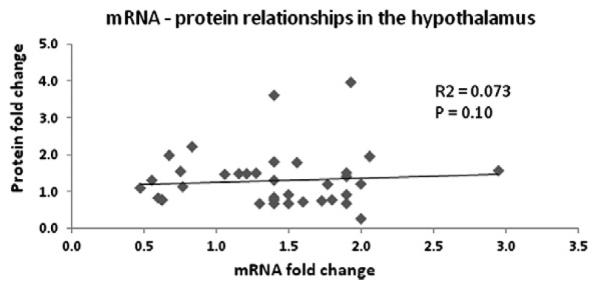
Regression analyses of mRNA steady state levels (generated through microarray data) and protein abundance (generated by iTRAQ) in the hypothalamus of female teleost fish. The transcripts and proteins used in the analysis showed significant changes in steady state levels. It is important to point out that not all proteins had corresponding probes on the LMB microarray and not all significantly altered proteins had corresponding changes in mRNA levels.
Low agreement of gene–protein expression in the neuroendocrine brain described above is typical of what others have found in various fish tissues. In a study of the liver from male and female zebrafish exposed to 30 ng EE2/L, De Wit et al. [7] performed both microarrays and DIGE on samples after 4 and 28 days. The authors were only able to correlate the abundance of 10 differentially expressed proteins to their corresponding mRNA expression level that were present on the microarray, despite the fact that over 70 proteins were identified. There was some correspondence at both time points between gene–protein, as catalase and apolipoprotein A1 (Apoa1) mRNA and protein expression were decreased in male liver after 28 d exposure while vitellogenin (Vtg) gene and protein were increased in the liver [7]. The aforementioned studies highlight the need for more elaborate studies at the molecular level (genes and proteins) to better understand changes in whole animal physiology. A significant challenge will be to integrate different sources of unique molecular data in a framework to characterize cell signaling cascades on a temporal scale. Furthermore, gel-based and non-gel based proteomics methods identify the most abundant proteins while microarrays only detect transcripts for which there is a corresponding probe on the platform. The implementation of “next-generation sequencing” such as RNA-Seq may improve sensitivity and accuracy in quantifying mRNA steady state levels compared to microarray data, but detailed studies using RNA-Seq are currently cost prohibitive.
In addition to microarray data, another approach is to compare real-time PCR data to proteomics data and one might expect, due to the increased sensitivity and specificity of real-time PCR that gene–protein relationships would be more apparent. In the liver of rare minnow (Gobiocypris rarus) after treatment with pentachlorophenol (PCP) at three different doses (0.5, 5.0 and 50 μg/L), Fang et al. [9] demonstrated that gene–protein correspondence from data collected with real-time PCR data and 2D gel electrophoresis was highly correlated for some genes and proteins (fructose-1,6-bisphosphatase) but not for others. For example, glutamate dehydrogenase (GDH) mRNA levels were increased at concentrations of 0.5 μg/L and 5 μg/L and decreased at 50 μg/L while GDH protein was significantly increased at the highest concentration. This may reflect negative feedback loops between gene and protein abundance, similar to feedback loops with hormones. Martyniuk et al. [19] report similar findings in the liver of FHMs after treatment with androgenic and anti-androgenic compounds. In that study, proteomics data was collected using iTRAQ and a focused gene expression approach (real-time PCR) was used to measure transcript levels and gene–protein data did not always correspond. It appears that, regardless of the methodology, the transcriptional and translational control mechanisms are complex and there is a “disconnect” in Omics data due to the underlying biology. This will be a significant challenge for the integration of Omics data into a system biology framework.
5. Challenges and directions in proteomics for fish neuroendocrine/neurotoxicology research
In fish, it is not uncommon to have approximately 40–50% of the peptide spectra unidentified and not assigned confidently to specific proteins (Martyniuk et al., unpub.). In the case of proteins that may be involved in pituitary hormone release, it becomes increasingly important to decipher protein function and protein prediction tools such as Pfam [10,33], which identifies conserved protein family domains, can be applied to de-orphanize unknown proteins.
Functional assays are also required to determine whether the protein is a novel receptor, enzyme, or has another role in the teleostean brain. Some avenues of exploration include developing recombinant proteins to perform pull down assays using nickel-NTA (Ni-NTA) resins in a column for high-yield His-tagged protein purification (Fig. 3). The idea is to immobilize the unknown recombinant protein on the column and pass protein eluant from cell fractions over the protein. Proteins that bind and interact with the recombinant protein can be eluted with imidazole and can further be identified with mass spectrometry. This approach is feasible in many laboratories but there can be false positives due to non-specific interactions between proteins. New approaches in tandem affinity purification [38] can reduce the number of false protein interactions dramatically. It is also recommended that other assays be performed to validate putative protein–protein interactions such as co-localization approaches using immunohistochemistry or yeast two hybrid systems.
Fig. 3.
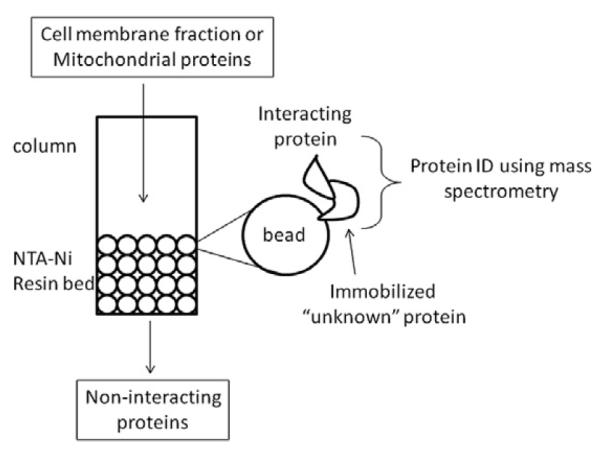
Recombinant protein strategies can be used to reconstruct the protein for column affinity purification and initial screening for putative protein–protein interactions using immobilization beads. This approach will provide information on regulatory interactions amongst proteins and insight into the relationships among corresponding transcripts.
Other approaches to decipher protein function are to localize the mRNA (in situ hybridization) or protein immunohistochemistry to a particular region of the neuroendocrine brain. This localization can infer functionality, for example, if the unknown protein is localized to the Golgi apparatus (and is not a secreted protein) then a function of the unknown protein may be protein processing (i.e. involved in post translational modification), packaging or transport. In the case of zebrafish and medaka, morpholino knock down in embryos have been invaluable in determining functions of proteins found in neuroendocrine regions and throughout the CNS [1,42].
The transcriptional and translational regulation of molecules is not well described in fish and it is becoming increasingly apparent that genes and proteins will not show a correlation in many studies, regardless of the tissue examined. Protein data collected by both gel-based and non-gel based approaches and mRNA expression data generated by microarrays and real-time PCR are verifying few correlated gene–protein relationships. Studies in fish gonad tissue have considered temporal changes at the transcriptomics level [37]. However, more cost-effective time course studies in proteomics and full-genome microarray platforms, focused real-time PCR studies, or RNA-Seq are expected to provide new insight into these molecular relationships in fish in future studies. A second approach to explore these relationships in more detail and with more accuracy is to utilize methods such as immunohistochemistry and in situ hybridization in a quantitative manner. This will allow for regional discrimination of genes and proteins in specific nuclei and overcome the limitation of current genomics and proteomics studies in the fish CNS that focus on whole brain or tissues such as the hypothalamus and telencephalon. It is well documented that estrogens regulate gene expression differentially in hypothalamic nuclei of rats [15], and micro-dissections followed by microarray analysis would be of great benefit. This may be especially relevant for environmental estrogens and it is reasonable to expect gene expression patterns within the hypothalamus might show region-specific regulation. Also, in the case of E2 and estrogenic chemicals such as 4-nonophenol, there can be different pattern of expression, suggesting that the mode of action of aquatic pollutants can be varied which also contributes to the challenge of deciphering gene responses [32].
In summary, the molecular signaling cascades that are induced or inhibited in neurons and supporting glia involve complex gene regulatory networks, multiple protein–protein interactions, and feedback at both the level of transcription and translation. The continued characterization of the neuroendocrine proteome in fish will advance understanding of the molecular signaling cascades that underlie hormone release and neural function. This is important because a significant number of aquatic contaminants are neuroactive by design (i.e. human pharmaceuticals and pesticides), modulating neurotransmitters receptors in the CNS. Further studies are required to better characterize protein networks underlying neuroendocrine regulation of hormone release, feedback to the CNS, and neurotoxicity pathways in fish.
Supplementary Material
Acknowledgments
The current and former members of the Proteomics Research Group (C. Diaz, S. Chen, and S. McClung) at the University of Florida are acknowledged and appreciated for their expertise in quantitative proteomics. We would also like to acknowledge and thank D. Barber for his expertise and advice in quantitative proteomics. This research was supported and funded by the Canada Research Chair program (CJM), NIH RO1 ES015449 Grant and EPA STAR Grant (R831848) (NDD) and NSERC Discovery Grant program (CJM and VLT).
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.ygcen.2011.12.006.
References
- [1].Ando H, Kobayashi M, Tsubokawa T, Uyemura K, Furuta T, Okamoto H. Lhx2 mediates the activity of Six3 in zebrafish forebrain growth. Dev. Biol. 2005;287:456–468. doi: 10.1016/j.ydbio.2005.09.023. [DOI] [PubMed] [Google Scholar]
- [2].Bassett D, Currie PD. Identification of a zebrafish model of muscular, dystrophy. Clin. Exp. Pharmacol. Physiol. 2004;31:537–540. doi: 10.1111/j.1440-1681.2004.04030.x. [DOI] [PubMed] [Google Scholar]
- [3].Boschetti E, Righetti PG. The art of observing rare protein species in proteomes with peptide ligand libraries. Proteomics. 2009;9:1492–1510. doi: 10.1002/pmic.200800389. [DOI] [PubMed] [Google Scholar]
- [4].Chuang HY, Lee E, Liu YT, Lee D, Ideker T. Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 2007;3:140. doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Damodaran S, Dlugos CA, Wood TD, Rabin RA. Effects of chronic ethanol administration on brain protein levels: a proteomic investigation using 2-D DIGE system. Eur. J. Pharmacol. 2006;547:75–82. doi: 10.1016/j.ejphar.2006.08.005. [DOI] [PubMed] [Google Scholar]
- [6].de Sousa Abreu R, Penalva LO, Marcotte EM, Vogel C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009;5:1512–1526. doi: 10.1039/b908315d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].De Wit M, Keil D, van der Ven K, Vandamme S, Witters E, De Coen W. An integrated transcriptomic and proteomic approach characterizing estrogenic and metabolic effects of 17 alpha-ethinylestradiol in zebrafish (Danio rerio) Gen. Comp. Endocrinol. 2010;167:190–201. doi: 10.1016/j.ygcen.2010.03.003. [DOI] [PubMed] [Google Scholar]
- [8].Denslow ND, Knoebl I, Larkin P. Mommsen TP, Moon TW, editors. Approaches in proteomics and genomics for eco-toxicology. Biochemistry and Molecular Biology of Fishes. 2005;(vol. 6):85–116. Chap. 3. [Google Scholar]
- [9].Fang Y, Gao X, Zha J, Ning B, Li X, Gao Z, Chao F. Identification of differential hepatic proteins in rare minnow (Gobiocypris rarus) exposed to pentachlorophenol (PCP) by proteomic analysis. Toxicol. Lett. 2010;199:69–79. doi: 10.1016/j.toxlet.2010.08.008. [DOI] [PubMed] [Google Scholar]
- [10].Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Forné I, Abián J, Cerdà J. Fish proteome analysis: model organisms and nonsequenced species. Proteomics. 2010;10:858–872. doi: 10.1002/pmic.200900609. [DOI] [PubMed] [Google Scholar]
- [12].Gunnarsson L, Jauhiainen A, Kristiansson E, Nerman O, Larsson DG. Evolutionary conservation of human drug targets in organisms used for environmental risk assessments. Environ. Sci. Technol. 2008;42:5807–5813. doi: 10.1021/es8005173. [DOI] [PubMed] [Google Scholar]
- [13].Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Levite M, Chowers Y, Ganor Y, Besser M, Hershkovits R, Cahalon L. Dopamine interacts directly with its D3 and D2 receptors on normal human T cells, and activates beta1 integrin function. Eur. J. Immunol. 2001;31:3504–3512. doi: 10.1002/1521-4141(200112)31:12<3504::aid-immu3504>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- [15].Malyala A, Pattee P, Nagalla SR, Kelly MJ, Rønnekleiv OK. Suppression subtractive hybridization and microarray identification of estrogen-regulated hypothalamic genes. Neurochem. Res. 2004;29:1189–1200. doi: 10.1023/b:nere.0000023606.13670.1d. [DOI] [PubMed] [Google Scholar]
- [16].Marlatt VL, Martyniuk CJ, Zhang D, Xiong H, Watt J, Xia X, Moon T, Trudeau VL. Auto-regulation of estrogen receptor subtypes and gene expression profiling of 17beta-estradiol action in the neuroendocrine axis of male goldfish. Mol. Cell. Endocrinol. 2008;283:38–48. doi: 10.1016/j.mce.2007.10.013. [DOI] [PubMed] [Google Scholar]
- [17].Martyniuk CJ, Kroll KJ, Doperalski NJ, Barber DS, Denslow ND. Genomic and proteomic responses to environmentally relevant exposures to dieldrin: indicators of neurodegeneration? Toxicol Sci. 2010;117:190–199. doi: 10.1093/toxsci/kfq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Martyniuk CJ, Kroll KJ, Doperalski NJ, Barber DS, Denslow ND. Environmentally relevant exposure to 17alpha-ethinylestradiol affects the telencephalic proteome of male fathead minnows. Aquat. Toxicol. 2010;98:344–353. doi: 10.1016/j.aquatox.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Martyniuk CJ, Alvarez S, McClung S, Villeneuve DL, Ankley GT, Denslow ND. Quantitative proteomic profiles of androgen receptor signaling in the liver of fathead minnows (Pimephales promelas) J. Proteome Res. 2009;8:2186–2200. doi: 10.1021/pr800627n. [DOI] [PubMed] [Google Scholar]
- [20].Martyniuk CJ, Denslow ND. Towards functional genomics in fish using quantitative proteomics. Gen. Comp. Endocrinol. 2009;164:135–141. doi: 10.1016/j.ygcen.2009.01.023. [DOI] [PubMed] [Google Scholar]
- [21].Martyniuk CJ, Gerrie ER, Popesku JT, Ekker M, Trudeau VL. Microarray analysis in the zebrafish (Danio rerio) liver and telencephalon after exposure to low concentration of 17alpha-ethinylestradiol. Aquat. Toxicol. 2007;84:38–49. doi: 10.1016/j.aquatox.2007.05.012. [DOI] [PubMed] [Google Scholar]
- [22].Martyniuk CJ, Xiong H, Crump K, Chiu S, Sardana R, Nadler A, Gerrie ER, Xia X, Trudeau VL. Gene expression profiling in the neuroendocrine brain of male goldfish (Carassius auratus) exposed to 17alpha-ethinylestradiol. Physiol. Genomics. 2006;27:328–336. doi: 10.1152/physiolgenomics.00090.2006. [DOI] [PubMed] [Google Scholar]
- [23].Matsui H, Taniguchi Y, Inoue H, Uemura K, Takeda S, Takahashi SR. A chemical neurotoxin, MPTP induces Parkinson’s disease like phenotype, movement disorders and persistent loss of dopamine neurons in medaka fish. Neurosci. Res. 2009;65:263–271. doi: 10.1016/j.neures.2009.07.010. [DOI] [PubMed] [Google Scholar]
- [24].McCarthy FM, Wang N, Magee GB, Nanduri B, Lawrence ML, Camon EB, Barrell DG, Hill DP, Dolan ME, Williams WP, Luthe DS, Bridges SM, Burgess SC. AgBase: a functional genomics resource for agriculture. BMC Genomics. 2006;7:229. doi: 10.1186/1471-2164-7-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mennigen JA, Martyniuk CJ, Crump K, Xiong H, Zhao E, Popesku JT, Anisman H, Cossins AR, Xia X, Trudeau VL. Effects of fluoxetine on the reproductive axis of female goldfish (Carassius auratus) Physiol. Genomics. 2008;35:273–282. doi: 10.1152/physiolgenomics.90263.2008. [DOI] [PubMed] [Google Scholar]
- [26].Miracle A, Denslow ND, Kroll KJ, Liu MC, Wang KK. Spillway-induced salmon head injury triggers the generation of brain alphaII-spectrin breakdown product biomarkers similar to mammalian traumatic brain injury. PLoS One. 2009;4:e4491. doi: 10.1371/journal.pone.0004491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Na H, Huang Q, Chen Y, Huang H. Differential proteins revealed with proteomics in the brain tissue of Paralichthys olivaceus under the stress of methyl parathion. Se. Pu. 2008;26:662–666. doi: 10.1016/s1872-2059(08)60030-9. [DOI] [PubMed] [Google Scholar]
- [28].Pollard HB, Adeyemo M, Dhariwal K, Levine M, Caohuy H, Markey S, Markey CJ, Youdim MB. The goldfish as a drug discovery vehicle for Parkinson’s disease and other neurodegenerative disorders. Ann. NY Acad. Sci. 1993;679:317–320. doi: 10.1111/j.1749-6632.1993.tb18314.x. [DOI] [PubMed] [Google Scholar]
- [29].Popesku JT, Martyniuk CJ, Denslow ND, Trudeau VL. Rapid dopaminergic modulation of the fish hypothalamic transcriptome and proteome. PLoS One. 2010;5:e12338. doi: 10.1371/journal.pone.0012338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Popesku JT, Martyniuk CJ, Mennigen J, Xiong H, Zhang D, Xia X, Cossins AR, Trudeau VL. The goldfish (Carassius auratus) as a model for neuroendocrine signaling. Mol. Cell. Endocrinol. 2008;293:43–56. doi: 10.1016/j.mce.2008.06.017. (2008) [DOI] [PubMed] [Google Scholar]
- [31].Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Pillai S, Dey S, Daniels S, Martin S, Barlet-Jones M, He F, Jacobson A, Pain DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using aminereactive isobaric tagging reagents. Mol. Cell. Proteomics. 2004;3:174–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- [32].Ruggeri B, Ubaldi M, Lourdusamy A, Soverchia L, Ciccocioppo R, Hardiman G, Baker ME, Palermo F, Polzonetti-Magni AM. Variation of the genetic expression pattern after exposure to estradiol-17beta and 4-nonylphenol in male zebrafish (Danio rerio) Gen. Comp. Endocrinol. 2008;158:138–144. doi: 10.1016/j.ygcen.2008.05.012. [DOI] [PubMed] [Google Scholar]
- [33].Sammut SJ, Finn RD, Bateman A. Pfam 10 years on: 10, 000 families and still growing. Briefings Bioinform. 2008;9:210–219. doi: 10.1093/bib/bbn010. [DOI] [PubMed] [Google Scholar]
- [34].Sanchez BC, Ralston-Hooper K, Sepúlveda MS. A review of recent proteomic alications in aquatic toxicology. Environ. Toxicol. Chem. 2011;30:274–282. doi: 10.1002/etc.402. [DOI] [PubMed] [Google Scholar]
- [35].Shojaie A, Michailidis G. Network enrichment analysis in complex experiments. Stat. Al. Genet. Mol. Biol. 2010;9:22. doi: 10.2202/1544-6115.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Singh SK, Rakesh KS, Ramamoorthy K, Saradhi AVP, Idris MM. Proteome profile of zebrafish brain based on Gel LC-ESI MS/MS analysis. J. Proteomics Bioinform. 2010;3:135–142. [Google Scholar]
- [37].Villeneuve DL, Garcia-Reyero N, Martinović D, Cavallin JE, Mueller ND, Wehmas LC, Kahl MD, Linnum AL, Perkins EJ, Ankley GT. Influence of ovarian stage on transcript profiles in fathead minnow (Pimephales promelas) ovary tissue. Aquat. Toxicol. 2010;98:354–366. doi: 10.1016/j.aquatox.2010.03.006. [DOI] [PubMed] [Google Scholar]
- [38].Völkel P, Le Faou P, Angrand PO. Interaction proteomics: characterization of protein complexes using tandem affinity purification-mass spectrometry. Biochem. Soc. Trans. 2010;38:883–887. doi: 10.1042/BST0380883. [DOI] [PubMed] [Google Scholar]
- [39].Watanabe Y, Nakayama T, Nagakubo D, Hieshima K, Jin Z, Katou F, Hashimoto K, Yoshie O. Dopamine selectively induces migration and homing of naive CD8+ T cells via dopamine receptor D3. J. Immunol. 2006;176:848–856. doi: 10.4049/jimmunol.176.2.848. [DOI] [PubMed] [Google Scholar]
- [40].Waye A, Trudeau VL. Neuroendocrine disruption more than hormones are upset. J. Toxicol. Environ. Health B: Crit. Rev. 2011;14:270–291. doi: 10.1080/10937404.2011.578273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Xi Y, Noble S, Ekker M. Modeling neurodegeneration in zebrafish. Curr. Neurol. Neurosci. Rep. 2011;11:274–282. doi: 10.1007/s11910-011-0182-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yamamoto T, Yao Y, Harumi T, Suzuki N. Localization of the nitric oxide/cGMP signaling pathway-related genes and influences of morpholino knockdown of soluble guanylyl cyclase on medaka fish embryogenesis. Zoolog. Sci. 2003;20:181–191. doi: 10.2108/zsj.20.181. [DOI] [PubMed] [Google Scholar]
- [43].Zhu JY, Huang HQ, Bao XD, Lin QM, Cai Z. Acute toxicity profile of cadmium revealed by proteomics in brain tissue of Paralichthys olivaceus: potential role of transferrin in cadmium toxicity. Aquat. Toxicol. 2006;78:127–135. doi: 10.1016/j.aquatox.2006.02.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.