


Abstract
Recent advances have demonstrated the use of RNA-based control devices to program sophisticated cellular functions; however, the efficiency with which these devices can be quantitatively tailored has limited their broader implementation in cellular networks. Here, we developed a high-efficiency, high-throughput and quantitative two-color fluorescence-activated cell sorting-based screening strategy to support the rapid generation of ribozyme-based control devices with user-specified regulatory activities. The high-efficiency of this screening strategy enabled the isolation of a single functional sequence from a library of over 106 variants within two sorting cycles. We demonstrated the versatility of our approach by screening large libraries generated from randomizing individual components within the ribozyme device platform to efficiently isolate new device sequences that exhibit increased in vitro cleavage rates up to 10.5-fold and increased in vivo activation ratios up to 2-fold. We also identified a titratable window within which in vitro cleavage rates and in vivo gene-regulatory activities are correlated, supporting the importance of optimizing RNA device activity directly in the cellular environment. Our two-color fluorescence-activated cell sorting-based screen provides a generalizable strategy for quantitatively tailoring genetic control elements for broader integration within biological networks.
INTRODUCTION
The engineering of biological systems that exhibit complex functions has significant potential to develop solutions addressing global challenges spanning health and medicine, sustainability, and the environment (1–4). Core to the engineering of biological systems is the ability to process information within cellular networks and link this information to new cellular behaviors. Synthetic biology is a rapidly growing interdisciplinary field that applies engineering principles to support the scalable construction and design of complex biological systems. One key focus of synthetic biology is to develop engineering frameworks for the reliable construction of genetic control devices that process and transmit information within living systems. Such information processing capabilities will allow researchers to implement diverse cellular control strategies, thus laying a critical foundation for designing genetic systems that exhibit sophisticated biological functions.
RNA control devices represent an important class of synthetic genetic control devices, whose development has been fueled by advances in RNA biology, engineering, and nucleic acid computing (5–9). We recently described a modular ligand-responsive ribozyme-based platform that supports efficient tailoring of RNA device function (10). The ribozyme device platform specifies physical linkages between three functional RNA components: a sensor encoded by an RNA aptamer, an actuator encoded by a hammerhead ribozyme, and a transmitter encoded by a sequence that undergoes a strand-displacement event (Figure 1). The dynamic range of an RNA control device, defined as the difference between gene expression levels in the presence and absence of ligand, is commonly described by two key parameters: basal activity and activation ratio. The basal activity is a measure of regulatory stringency, which is set by the gene expression level in the absence of the input ligand, whereas the activation ratio is a measure of input responsiveness, which is set by the ratio of expression level in the presence to that in the absence of input ligand. The dynamic range exhibited by an RNA control device is important for its effective integration within biological systems. We have previously demonstrated a rational device tuning strategy guided by secondary structure predictions from an RNA folding program to direct the partitioning between the two functional device conformations (10). Although we demonstrated the generation of a series of devices that span wide regulatory ranges, this strategy relies on predictions made by RNA folding programs, which base their computations on parameters acquired in vitro, and thus do not fully capture the complex RNA folding process in vivo. Therefore, an alternative strategy that enables direct engineering of device dynamic ranges in the cellular environment is desired to increase the efficiency with which RNA devices can be tailored to cellular networks.
Figure 1.
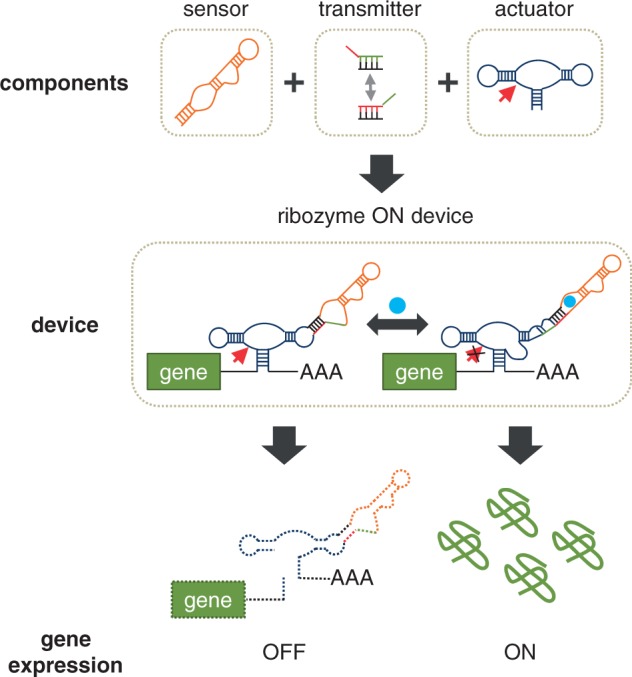
Schematic representation of modular assembly and mechanism of an RNA control device based on a ribozyme actuator. Ribozyme-based devices are constructed by modular assembly of three functional RNA components. A sensor (RNA aptamer) is linked to an actuator (hammerhead ribozyme) through a distinct information transmitter sequence (which directs a strand-displacement event and insulates the sensor and actuator components). Ribozyme-based devices are integrated into the 3′ untranslated region of the target gene and can adopt at least two functional device conformations, where each conformation is associated with different actuator and sensor activities. In the depicted example, a ribozyme ON device (upregulation of gene expression in response to increased input ligand concentration) adopts a ribozyme-active conformation associated with an aptamer ligand-unbound state, where ribozyme cleavage results in an unprotected transcript that is subject to rapid degradation by ribonucleases, thereby leading to a decrease in gene expression. The ribozyme-inactive conformation is associated with an aptamer ligand-bound state, such that ligand binding to the aptamer stabilizes the ribozyme-inactive conformation, thereby leading to an increase in gene expression in response to ligand.
Significant research efforts have been directed toward developing high-throughput in vitro and in vivo selection and screening strategies to generate RNA control devices with user-specified regulatory activities. In vitro selection strategies were applied to allosteric ribozymes to select for variants that exhibit enhanced ligand-responsive cleavage activities (11,12). However, in one case the selected variants did not retain their enhanced activities in vivo, suggesting that the observed activities were sensitive to the environment in which the selection was performed (11). To address potential loss of activities in transitioning between in vitro and in vivo environments, researchers have developed cell-based selection and screening strategies to support direct generation of RNA control devices and regulatory components in the relevant environment.
A cell-based selection strategy was developed to select for ribozyme variants that exhibit in vivo activity by directly recovering cleaved ribozyme fragments from cells (13). However, this in vivo method decoupled catalytic activity from gene-regulatory activity, resulting in a less quantitative selection strategy that did not support effective identification of sequence variants with varying gene-regulatory activities. Cell-based screening and selection strategies have been developed for RNA control devices by introducing a randomized region into the device and linking the regulatory function of the resulting device library to a single measurable gene expression output, such as colorimetric and fluorescence levels (14–16), motility (17) or viability (18). However, the majority of these methods are most effective at enriching devices from the population exhibiting extreme (either low or high) gene expression levels, limiting the recovery of devices with specific quantitative activities. In addition, the enrichment efficiencies (or, alternatively, the throughput) of screening methods based on a single output can be negatively impacted by the stochastic nature of gene expression. In particular, when utilizing high-throughput methods that measure activities within single cells, such as fluorescence-activated cell sorting (FACS), the inability to resolve changes in gene expression levels due to the genetic control device from cellular noise makes single-output strategies less efficient and quantitative.
In this work, we developed a new screening strategy that utilizes two measurable outputs to resolve device regulatory activity from cellular noise. A high-efficiency, high-throughput and quantitative two-color FACS-based screening strategy was developed to support rapid generation of RNA control devices with user-specified regulatory activities. We validated the high-efficiency of our screening strategy by performing a control sort to isolate the original functional sequence from a library of over 106 variants within just two sorting cycles. We demonstrated the versatility of our two-color FACS-based approach by screening large libraries generated from randomizing individual components within the ribozyme device platform to efficiently isolate devices with improved regulatory properties. Using this screening strategy, we effectively isolated new ribozyme actuator sequences that increase in vitro cleavage rates up to 10.5-fold within the device platform and new transmitter sequences that increase in vivo activation ratios up to 2-fold. By isolating devices that exhibit specified in vivo gene-regulatory activities and quantifying their in vitro cleavage rates, we also identified a titratable window within which the two device parameters are correlated, supporting the importance of directly engineering device activity within the cellular environment.
MATERIALS AND METHODS
Plasmid and strain construction
Standard molecular biology cloning techniques were used to construct all plasmids (19). DNA synthesis was performed by Integrated DNA Technologies (Coralville, IA) or the Protein and Nucleic Acid Facility (Stanford, CA). All enzymes, including restriction enzymes and ligases, were obtained through New England Biolabs (Ipswich, MA). Ligation products were electroporated with a GenePulser XCell (Bio-Rad, Hercules, CA) into an Escherichia coli DH10B strain (Invitrogen, Carlsbad, CA), where cells harboring cloned plasmids were maintained in Luria-Bertani media containing 50 µg/mL ampicillin (EMD Chemicals, Philadelphia, PA). All cloned constructs were sequence verified by Elim Biopharmaceuticals (Hayward, CA).
The two-color screening plasmid pCS1748 (Supplementary Figure S1) was constructed by inserting an open reading frame encoding a yeast-optimized mCherry gene (ymCherry) flanked by a TEF1 promoter and a CYC1 terminator upstream of the existing open reading frame encoding a yeast-enhanced GFP gene (yEGFP3) flanked by a GAL1-10 promoter and an ADH1 terminator in the pCS321 backbone (10). The TEF1 promoter was polymerase chain reaction (PCR) amplified from pG2M (20) using forward and reverse primers SacI-TEF1-fwd (5′-GAGAGCTCAAGCTTCAAAATGTTTCTACTCC) and SacII-TEF1-rev (5′-GGCCGCGGCAAAACTTAGATTAGATTGCTATGC), respectively, and inserted into pCS321 via the unique restriction sites SacI and SacII. The ymCherry gene was PCR amplified from BBa_E2060 obtained from the iGEM parts registry (21) using forward and reverse primers SacII-mCherry-fwd (5′-GACCGCGGGAAATAATGTCTATGGTTAGTAAAGGAGAAGAAAATAACATGGC) and NotI-mCherry-rev (5′-GGGCGGCCGCTTATTATTTGTATAGTTCATCCATGCCACCAG), respectively, and inserted downstream of the cloned TEF1 promoter via the unique restriction sites SacII and NotI. The CYC1 terminator was PCR amplified from pCM159 (22) using forward and reverse primers NotI-CYC1t-fwd (5′-GAGCGGCCGCGAGGGCCGCATCATGTAATTAG) and XbaI-CYC1t-rev (5′-GGTCTAGAGGCCGCAAATTAAAGCCTTCG), respectively, and inserted downstream of the cloned ymCherry gene via the unique restriction sites NotI and XbaI. A spacer sequence was amplified from pCS745 (gift from M. Jensen, Seattle Children’s Research Institute) using forward and reverse primers XbaI-Spacer-fwd (5′-GGTCTAGACGCCTTGAGCCT GGCGAACAGTTC) and Spacer-rev (5′-AGTAAAAAAGGAGTAGAAACATTTTGAAGCTATCGATGACAGGATGAGGATCGTTTCGCATG), respectively. A TEF1 promoter fragment was amplified from pG2M using forward and reverse primers Spacer-TEF1-fwd (5′-CATGCGAAACGATCCTCATCCTGTCATCGATAGCTTCAAAATGTTTCTACTCCTTTTTTACT) and BamHI-TEF1-rev (5′-GGGGATCCCAAAACTTAGATTAGATTGCTATGCTTTCTTTC), respectively, and PCR assembled with the spacer fragment. The assembled spacer-TEF1 promoter fragment was inserted into the modified pCS321 backbone to replace the GAL1-10 promoter via the unique restriction sites XbaI and BamHI.
Two single-color plasmids harboring GFP (pCS1585) and mCherry (pCS1749) were constructed as compensation controls for FACS analysis. A fragment harboring the TEF1 promoter was PCR amplified from pGM2 using forward and reverse primers SacI-TEF1-fwd (5′-GAGAGCTCATAGCTTCAAAATGTTTCTACTCC) and EcoRI-TEF1-rev (5′-GGGAATTCTTTGTAATTAAAACTTAGATTAGA), respectively. The GFP-only plasmid pCS1585 was constructed by inserting the TEF1 promoter fragment into pCS321 to replace the GAL1-10 promoter via the unique cloning sites SacI and EcoRI. The mCherry-only plasmid pCS1749 was constructed by inserting the TEF1p-ymCherry–CYC1t cassette from pCS1748 into a modified version of pRS316 (23) via the unique restriction sites SacI and XbaI. The modified version of pRS316 (pCS4), containing no fluorescence reporter gene, was used as the negative-control construct.
Ribozyme-based devices and appropriate controls were inserted into the 3′ untranslated region of yEGFP3 in pCS1748 through appropriate restriction endonuclease and ligation-mediated cloning. DNA fragments encoding the ribozyme-based devices and controls were PCR amplified using forward and reverse primers L1-2-fwd (5′-GACCTAGGAAACAAACAAAGCTGTCACC) and L1-2-rev (5′-GGCTCGAGTTTTTATTTTTCTTTTTGCTGTTTCG), respectively, and inserted into pCS1748 via the unique restriction sites AvrII and XhoI, which are located 3 nucleotides downstream of the yEGFP3 stop codon. Cloned plasmids were transformed into the budding yeast Saccharomyces cerevisiae strain W303α (MAT α his3-11,15 trp1-1 leu2-3 ura3-1 ade2-1) through a standard lithium acetate method (24). All yeast strains harboring cloned plasmids were maintained on synthetic complete media with a uracil dropout solution containing 2% dextrose (SC-URA) and grown at 30°C. All plasmids constructed in this study are summarized in Supplementary Table S1.
Library-scale yeast transformation
Device libraries (Supplementary Figure S2) were amplified using forward and reverse primers GAP-L1-2-fwd (5′-TCCATGGTATGGATGAATTGTACAAATAAAGCCTAGGAAACAAACAAAGCTGTCACC) and GAP-L1-2-rev (5′-AAGAAATTCGCTTATTTAGAAGTGGCGCGCCCTCTCGAGTTTTTATTTTTCTTTTTGCTGTTTCG), respectively. The library was inserted into pCS1748 through homologous recombination-mediated gap-repair during transformation into yeast strain W303 (25). Briefly, an 800 µL library PCR reaction was performed with 160 pmol of each primer and 16 pmol of the library template. Eight micrograms of the plasmid pCS1748 were digested with AvrII and XhoI. The digested vector was combined with the library PCR product, extracted with phenol chloroform, and precipitated into a dry pellet with ethanol. A Tris–DTT buffer (2.5 M DTT, 1 M Tris, pH 8.0) was added to a 50 mL yeast culture (OD600 1.3–1.5) and incubated at 30°C for 10–15 min. The yeast were pelleted, washed with chilled Buffer E (10 mM Tris, pH 7.5, 2 mM MgCl2, 270 mM sucrose), and resuspended in Buffer E to a final volume of 300 µL. The yeast mixture was directly added to the precipitated DNA pellet and 50 µL of the mixture was transferred to a chilled 2 mm gap cuvette for electroporation (540 V, 25 µF, 1000 Ω). Following transformation, the cells were resuspended in 1 mL warmed YPD media and incubated at 30°C for 1 h. Multiple transformations (∼5) were performed to cover the desired library diversity (∼106–107). Transformation efficiencies were determined by plating serial dilutions of the transformants, and transformed cells were propagated in SC-URA media for FACS.
Two-color FACS-based screen
Cells harboring the RNA device libraries and control constructs were washed, resuspended in FACS buffer (1% BSA in PBS) and stained with a DAPI viability dye (Invitrogen). The cell suspension was filtered through a 40 µm cell strainer (BD Systems, San Jose, CA) prior to analysis on a FACSAria II cell sorter (BD Systems). GFP was excited at 488 nm and measured with a splitter of 505 nm and bandpass filter of 525/50 nm. mCherry was excited at 532 nm and measured with a splitter of 600 nm and a bandpass filter of 610/20 nm. DAPI was excited at 355 nm and measured with a bandpass filter of 450/50 nm. A scatter gate was set based on the forward and side-scatter area of cells harboring the negative-control plasmid (pCS4) to exclude debris, followed by a DAPI-(–) viability gate to exclude dead cells in the DAPI-(+) gate from the analysis. Cells harboring the single-color control plasmids (pCS1585, pCS1749) were analysed to compensate spillover from GFP to the mCherry detector. A general sorting strategy was followed in which cells harboring devices with targeted gene-regulatory activities were analysed to set a sorting gate on a two-dimensional scatter plot that correlates GFP and mCherry fluorescence. Cells within this gate were collected into SC-URA media and propagated to sufficient density for further screening or analysis.
Ribozyme-based device characterization through flow cytometry analysis
Enriched device libraries from FACS were directly plated onto SC-URA solid medium. Individual colonies were screened and characterized for gene-regulatory activity of the devices based on flow cytometry analysis. The GFP fluorescence was measured on a Quanta flow cytometer (Beckman Coulter, Fullerton, CA). GFP was excited at 488 nm and measured with a splitter of 488 nm and a bandpass filter of 525/40 nm. Cells harboring the negative-control plasmid (pCS4) were analysed to set the GFP-(−) and GFP-(+) gate. Gene-regulatory activities were reported as relative GFP expression levels and were determined as the geometric mean of the GFP fluorescence based on the GFP-(+) population using FlowJo (Tree Star), and normalized to the geometric mean of the GFP fluorescence of a positive control (satellite tobacco ring spot virus [sTRSV] Contl, a noncleaving sTRSV ribozyme with a scrambled core) that is grown under identical ligand conditions, run in the same experiment and set to 100%.
Devices that exhibited desired activities were amplified by colony PCR using forward and reverse primers CS653 (5′-GGTCACAAATTGGAATACAACTATAACTCT) and CS654 (5′-CGGAATTAACCCTCACTAAAGGG), respectively, and sequenced. The recovered devices were resynthesized and recloned into the vector backbone to confirm the observed activity. DNA oligos were synthesized and amplified for insertion into pCS1748 using forward and reverse primers L1-2-fwd (5′-GACCTAGGAAACAAACAAAGCTGTCACC) and L1-2-rev (5′-GGCTCGAGTTTTTATTTTTCTTTTTGCTGTTTCG), respectively. The resynthesized devices were inserted into pCS1748 via the unique restriction sites AvrII and XhoI. The reconstructed device plasmids were transformed into the W303 yeast strain through a standard lithium acetate method (24). Cells harboring the selected devices and appropriate controls were prepared as described above for the sorting experiments and analysed on an LSRII flow cytometer (BD Systems) to characterize the gene-regulatory activity of each device. GFP was excited at 488 nm and measured with a splitter of 505 nm and a bandpass filter of 525/50. mCherry was excited at 532 nm and measured with a splitter of 600 nm LP and a bandpass filter of 610/20 nm. DAPI was excited at 405 nm and measured with a bandpass filter of 450/50 nm. FlowJo was used to process all flow cytometry data. Cells harboring pCS4 and pCS1749 were analysed to set the mCherry-(−) and mCherry-(+) gates. Gene-regulatory activities in the absence or presence of ligand were determined as the geometric mean of the GFP fluorescence based on the mCherry-(+) population, and normalized to the geometric mean of the GFP fluorescence of a positive control (sTRSV Contl, a noncleaving sTRSV ribozyme with a scrambled core) in the absence or presence of ligand, respectively, to correct for any nonspecific effects of ligand on the measured fluorescence (Supplementary Figure S3). Reported gene-regulatory activities are the mean and standard deviation of at least three independent experiments.
In vitro transcription and purification of ribozyme-based devices
Selected ribozyme-based devices and ribozyme and noncleaving controls were PCR amplified to include an upstream T7 promoter site and spacer sequence and downstream spacer sequence using forward and reverse primers T7-L1-2-fwd (5′-TTCTAATACGACTCACTATAGGGA CCTAGGAAACAAACAAAGCTGTCACC) and L1-2-rev (5′-GGCTCGAGTTTTTATTTTTCTTTTTGCTGTTTCG), respectively. A total of 1–2 µg of PCR product was transcribed in a 25 µl reaction, consisting of the following components: 1×RNA Pol Reaction Buffer (New England Biolabs), 2.5 mM rATP, 2.5 mM rCTP, 2.5 mM rUTP, 0.25 mM rGTP, 1 µl RNaseOUT (Invitrogen), 10 mM MgCl2, 1 µl T7 Polymerase (New England Biolabs) and 0.5 µCi α-32P-GTP (MP Biomedicals, Solon, OH). 400 pmol of antisense DNA oligonucleotide, device-blocking (5′-GGTGACAGCTTTGTTTGTTTCCTAGGTCCCCC) and sTRSV-blocking (5′-GCTGTTTCGTCCTCACG) was added to each reaction to inhibit cleavage of the RNA devices and sTRSV hammerhead ribozyme, respectively, during transcription. After incubation at 37°C for 2 h, NucAway Spin Columns (Ambion, Austin, TX) were used to remove unincorporated nucleotides from the transcription reactions according to manufacturer’s instructions. Ten microliters aliquots of the recovered RNA were mixed with three volumes of RNA stop/load buffer (95% formamide, 30 mM EDTA, 0.25% bromophenol blue, 0.25% xylene cyanol), heated at 95°C for 5 min, snap cooled on ice for 5 min, and size-fractionated on a denaturing (8.3 M urea) 10% polyacrylamide gel at 25 W for 45 min. Gels were imaged by phosphorimaging analysis on an FX Molecular Imager (Bio-Rad). Uncleaved transcripts were gel extracted and recovered with the ZR Small-RNA PAGE Recovery Kit (Zymo Research, Irvine, CA) according to manufacturer’s instructions. Samples were stored in sterile, nuclease-free, deionized water at −80°C to limit the extent of RNA self-cleavage prior to performing the cleavage assays.
In vitro ribozyme cleavage assays
The purified uncleaved transcripts were incubated in 100 mM NaCl, 50 mM Tris–HCl (pH 7.5) at 95°C for 5 min, cooled at a rate of −1.3°C/min to 37°C and held there for 10 min to allow equilibration of secondary structure. A zero time-point aliquot was taken prior to initiating the self-cleavage reaction at 37°C with the addition of MgCl2 to a final concentration of 500 µM. Reactions were quenched at specified time points with addition of three volumes of RNA stop/load buffer on ice. Samples were heated at 95°C for 5 min, snap cooled on ice for 5 min, and size-fractionated on a denaturing (8.3 M urea) 10% polyacrylamide gel at 25 W for 45 to 60 min. Gels were exposed overnight on a phosphor screen and analysed for relative levels of the full-length transcript and cleaved products by phosphorimaging analysis on an FX Molecular Imager (Bio-Rad). The cleaved product fraction at each time point (Ft) was fit to the single exponential equation Ft = F0 + (F∞ − F0) × (1 – e−kt) using Prism 5 (GraphPad), where F0 and F∞ are the fractions cleaved before the start of the reaction and at the reaction endpoint, respectively, and k is the first-order rate constant of self-cleavage. Reported cleavage rate values are the mean of at least three independent experiments.
RESULTS
A two-color reporter construct supports a high-efficiency, high-throughput, quantitative, cell-based screening strategy
Genetic control devices with desired gene-regulatory activities are often generated through cell-based screening strategies by coupling the regulatory function to a measurable output. However, the efficiencies of high-throughput cell-based screening strategies based on measuring activities in single cells are negatively impacted by gene expression noise that arises from both extrinsic (e.g. cell size and shape, cell cycle stage, plasmid copy number) and intrinsic factors (e.g. fluctuation in numbers of DNA, RNA, transcription factors, environmental stimuli) (26,27). To develop a more effective and quantitative in vivo screening strategy for gene-regulatory devices, we constructed a screening plasmid composed of two independent functional modules (Figure 2A). The first module, the device activity reporter, utilizes a GFP reporter to measure the gene expression level associated with the regulatory device, whereas the second module, the noise reporter, utilizes a mCherry reporter to provide a measure of cellular gene expression level that is independent of the regulatory device. Previous studies in yeast have suggested that extrinsic noise is the predominant source of gene expression noise (27). Therefore, by incorporating two independent expression modules on the same plasmid, we aimed to use the mCherry expression level as an internal standard to normalize the corresponding GFP expression level within a single cell. We used the same constitutive promoter (TEF1) in each module, although other promoter pairs may be substituted into the construct as previous studies in yeast suggest that extrinsic noise is not promoter specific (27). In addition, different terminators for each module were used to decrease the frequency of homologous recombination between the two modules.
Figure 2.

A high-efficiency, quantitative cell-based screening strategy for genetic devices based on a two-color screening construct. (A) The two-color screening construct is composed of two independent activity reporters. The device activity reporter measures the gene-regulatory activity associated with the device from GFP fluorescence, whereas the noise reporter measures the variation in cellular gene expression level that is independent of the regulatory device from mCherry fluorescence. (B) Single-color (GFP) scatter plots of three ribozyme-based devices that span a wide range of gene-regulatory activities, as measured by their mean values, and cellular autofluorescence from a construct containing no fluorescence reporter gene exhibit significant overlap due to noise associated with gene expression. (C) Single-color (GFP) histograms illustrate that isolation of a device with a specific gene-regulatory activity based on a single reporter output is inefficient due to overlapping population distributions. (D) The gene expression levels of individual cells can be normalized by correlating the device and noise reporter outputs from the two-color screening construct. Cell populations harboring the three ribozyme-based devices in (B) can be cleanly resolved on a two-color scatter plot, where each population exhibits a tight linear relationship between the two outputs. (E) The two-color screening strategy is based on the output correlation between the two reporter modules. A library of control devices can be integrated in the two-color screening construct and transformed into the target cell host. The sorting gate is set by the two-color correlation (slope) associated with a control device that exhibits a target gene-regulatory activity and applied to the library to specifically isolate a cell population that exhibits similar activity (slope).
We first used the two-color screening construct to examine the basal activities of three previously characterized theophylline-responsive ribozyme-based devices (10) based on the output from the device activity reporter (GFP): L2bulge1 (L2b1), 40%; L2bulge5 (L2b5), 70%; and L2bulge8 (L2b8), 11%. The reported basal activities refer to the GFP expression levels relative to that of the inactive ribozyme control, which does not exhibit catalytic activity or respond to theophylline. Although these three devices exhibit a wide range of gene-regulatory activities, cellular noise results in a broad distribution of GFP fluorescence levels, making it challenging to cleanly isolate members with target activities from a library through a FACS-based sorting strategy based solely on one output (Figure 2B and C). To provide a filter for variations in gene expression level due to cellular noise, we used the noise reporter in the two-color screening construct to assess activity that is independent of the regulatory device. By using the output from the noise reporter (mCherry), we were able to normalize the gene expression levels of individual cells by correlating the outputs between the two reporters and cleanly resolve the cell populations harboring three different devices (Figure 2D). For each device, the cell population exhibits a tight linear relationship between the two outputs, such that the device gene-regulatory activity is associated with a distinct slope (Supplementary Figure S4). The increased resolution between cell populations harboring devices with different gene-regulatory activities enables the development of a high-efficiency, quantitative, two-color FACS-based screening strategy based on the output correlation between the two reporter modules (Figure 2E).
We demonstrated the high-enrichment efficiency of our FACS-based two-color screening strategy by performing a control screen on a sensor library (sN10). The sN10 sensor library was generated by randomizing 10 nucleotide (nt) positions in the theophylline aptamer binding core (sensor component) within the L2b8 device (∼1 × 106 variants) (28) (Figure 3A). The randomized positions were chosen based on previous characterization of the theophylline aptamer, in which 14 conserved residues were identified to be essential for theophylline binding (29). We first assessed the basal activity of the sN10 sensor library through two-color flow cytometry analysis and observed that the majority of the library exhibits higher activity (greater slope) than the parent device (Figure 3B). We set a sorting gate based on the basal activity of the parent device (negative gate) and performed a negative sort to enrich cells (∼10% of the initial library) that exhibit similar basal activities (same slope). We calculated the fold enrichment based on the percentage of the cell population isolated from each sort. In the initial negative sort, we collected 9.53% of the cell population and therefore this population was enriched ∼11 times (100/9.53, or ∼11-fold) for the subsequent screen. We performed a positive sort on the recovered cells, in which the lower bound of the sorting gate was set by the gene-regulatory activity of the L2b8 device in the presence of 5 mM theophylline. The positive sorting gate was expanded further to enrich members of the library (∼7% of the enriched library) that exhibit equal or increased activities in response to theophylline relative to the parent device by ∼15-fold. After one sorting cycle, a distinct small sub-population of cells that exhibit comparable basal activity with the parent device was observed. We then performed another round of sorting to specifically enrich this sub-population (∼1% of the enriched library) through a negative sort by ∼143-fold, followed by a positive sort on the recovered cells to enrich a clearly distinct cell population that exhibits increased gene-regulatory activity (∼4% of the enriched library) in response to theophylline by ∼25-fold. After two sorting cycles, ∼98% of the enriched library exhibits a clear population shift in response to theophylline (Figure 3C).
Figure 3.

Screening of a sensor library within the device platform demonstrates the high enrichment efficiency of the two-color sorting strategy. (A) A sensor library, sN10, is generated by randomizing 10 nucleotides at key positions within the aptamer component in a previously engineered theophylline-responsive ribozyme-based device, L2b8. (B) The sN10 library is subjected to two sorting rounds. Each round consists of one negative sort in the absence of theophylline (light green gate set by the activity of the parent L2b8 device in the absence of theophylline), followed by one positive sort in the presence of theophylline (dark green gate set by the activity of the parent L2b8 device in the presence of 5 mM theophylline). Percentage of cells collected in the sorting gate is indicated on each plot. (C) After two sorting rounds, the enriched pool was analysed by flow cytometry in the absence and presence of theophylline (theo). A gate was set by the library population in the absence of theophylline. Upon addition of theophylline, ∼98% of the population within the gate shifted, suggesting that the majority of the population exhibits switching activity.
As the two rounds of sorting yield an overall ∼6 × 105-fold enrichment, given an initial library diversity of ∼106 variants, we expected to recover the original parent device from the enriched library by screening a small number of individual clones. We characterized 48 individual colonies from the enriched library for theophylline-responsive gene-regulatory activities through flow cytometry. All 48 colonies exhibited similar increases in GFP expression levels in response to theophylline as the parent L2b8 device. We sequenced the recovered devices from 10 colonies, and all were verified to be the parent sequence. In contrast, after performing two similar sorting cycles on the sensor library using the more traditional single-color sorting strategy, the resulting enriched library does not exhibit any observable population shift in response to theophylline (Supplementary Figure S5). These results demonstrate the high efficiency of our two-color FACS-based screening strategy, which can enrich a single sequence from a very large number of variants, such that very few isolates need to be screened to identify true positives.
Screening of an actuator library results in RNA control devices with improved regulatory stringencies
The implementation of RNA control devices in biological systems requires flexible tailoring of regulatory functions, where regulatory stringency of a device can be an important property for crossing phenotypic thresholds (1). The basal activity of a single ribozyme-based device depends on both the actuator and transmitter components. The catalytic activity of the hammerhead ribozyme (actuator) within the context of the device platform sets a lower limit to the minimal gene expression level a device exhibits in the absence of ligand, whereas the transmitter directs the partitioning between the functional conformations, which in turn impacts the basal activity (10). The device platform specifies the integration of a sensor-transmitter element through loop II (or loop I) of the sTRSV ribozyme actuator. Previous sTRSV ribozyme characterization studies have demonstrated that a tertiary interaction between loop I and loop II is essential for the catalytic activity of the ribozyme in cellular environments (30). The integration of additional structural elements through the ribozyme loops may negatively impact the required loop–loop interaction, thus resulting in a less active ribozyme within the device platform. The previously described theophylline-responsive ribozyme devices (L2b1, 40%; L2b5, 70%; L2b8, 11%) exhibit a basal activity higher than that exhibited by the ribozyme alone (1%), suggesting that the current natural ribozyme sequence in the device platform may present a lower limit on the basal activity of the resultant RNA control devices.
To engineer RNA control devices that exhibit tighter regulatory stringencies, we focused initially on optimizing the sTRSV ribozyme sequence in the L2b8 device as it has the lowest basal activity among the series of previously engineered theophylline-responsive devices (10). As prior sTRSV ribozyme characterization studies have demonstrated that mutations to either loop I or II sequences can enhance or hinder ribozyme catalytic activity (30), we designed a device library by randomizing loop sequences and applied our two-color FACS-based screening approach to search for improved loop–loop interactions that support lower basal activity. We generated an actuator library (aN7) by randomizing 7 nucleotides in loop I within the L2b8 device (∼2 × 104 variants) (Figure 4A). Loop I was targeted for randomization as it is isolated from the conformational change that is facilitated by the transmitter component, which is integrated via loop II. We analysed the initial aN7 actuator library by two-color flow cytometry analysis and observed that ∼99% of the library exhibits a higher basal activity (greater slope) than the parent L2b8 device, indicating that the majority of loop I library sequences have deleterious effects on ribozyme catalytic activity (Figure 4B). The two-color approach also enables us to clearly distinguish cells with low gene expression levels from cells that have lost or mutated the plasmid, whereas the two cell populations are almost indistinguishable if GFP is used as the only measure of device regulatory activity (Supplementary Figure S6). We set a sorting gate based on cells harboring the parent L2b8 device and collected cells that exhibit a decreased slope relative to the parent device (∼0.4% of the initial library) to enrich library members with enhanced loop–loop interactions that support lower device basal activity.
Figure 4.

Screening of an actuator library within the device platform results in ribozyme variants that exhibit improved regulatory stringencies and cleavage rates. (A) An actuator library, aN7, is generated by randomizing 7 nucleotides at key positions within the loop I region of the ribozyme actuator in the L2b8 device. (B) The aN7 library is subjected to a single sort to enrich for devices that exhibit lower basal activities than the parent L2b8 device. The majority (∼99%) of the aN7 library exhibits a greater slope than that of the parent L2b8 device, such that one sort is sufficient to isolate members that exhibit improved regulatory stringency. (C) Ribozyme variants isolated from the aN7 library screen exhibit lower basal activities relative to the parent L2b8 device. Gene-regulatory activities are reported as the geometric mean of the GFP fluorescence of the indicated sample normalized to that of a positive control (sTRSV Contl, a noncleaving sTRSV ribozyme with a scrambled core) that is grown under identical ligand conditions and is set to 100%. Reported values are the mean and standard deviation of at least three independent experiments. (D) The recovered ribozyme variants (L2b8-al, -a14) exhibit faster in vitro cleavage rates than the parent device (L2b8). Cleavage assays were performed at 37°C with 500 µM MgCl2, 100 mM NaCl, 50 mM Tris-HCl (pH 7.5). Cleavage rate constants (k) and errors are reported as the mean and standard deviation from at least three independent assays. (E) In vitro cleavage kinetics of the ribozyme variants (L2b8-al, -a14) and the parent device (L2b8). The projected cleavage kinetics are generated from the single-exponential equation Ft = F0 + (F∞ − F0) × (1 – e−kt), setting the fraction cleaved before the start of the reaction (F0) and at reaction endpoint (F∞) to 0 and 1, respectively, and k to the experimentally determined value for each RNA device.
Because of the relatively small sequence space of the library and high-efficiency of our screen, we directly plated the sorted cells to obtain single colonies for further characterization. We recovered a total of 22 clones and identified 22 unique device sequences from the recovered clones. However, only eight of the recovered devices maintained low basal activities upon re-cloning (Figure 4C). We believe that the majority of the identified false positives likely had mutations within the recovered plasmid backbone that resulted in low GFP expression levels, although we cannot rule out the possibility of some fraction of these false positives being recovered due to remaining noise in the system that is not captured in our two-color correlation. Additional sorts (positive and negative) could be incorporated to reduce this false positive rate. The basal activities exhibited by the recovered devices are up to 3.5-fold lower than that of the parent device, while six of the devices retained functional switching (i.e. responsiveness to ligand) (Supplementary Figure S7).
We performed further characterization by mutagenesis of the newly identified ribozymes recovered from the aN7 library screen to identify conserved motifs that contribute to decreased basal activities. A series of point mutations to the recovered loop I sequences were designed, and the activities of these loop I sequences were characterized through flow cytometry analysis (Supplementary Figure S8). Two distinct consensus sequences were identified that support lower basal activities than the parent L2b8 device. RNA devices with a loop I heptaloop adhering to the consensus sequence AUNNARG, where N is any nucleotide base and R is a purine base, exhibit basal activities that are less than or equal to that of the parent (11%). A subset of these devices with the loop I consensus sequence AUCNARG exhibit basal activities less than 40% of that of the L2b8 parent device. RNA devices with a second, distinct loop I consensus sequence predicted to form a triloop N1UN2GGGN1|, where N1 and N1| are any Watson–Crick base pair, exhibit improved basal activities less than 70% of that of the L2b8 parent.
Engineered actuator components result in optimized ribozyme-based devices exhibiting faster cleavage rates
In vitro kinetic analysis of select ribozyme-based devices harboring loop I sequence modifications was performed to assess whether the improved in vivo basal activities were a result of increased cleavage rates relative to the parent device. Cleavage rates (k) were determined at physiologically relevant reaction conditions [500 μM MgCl2, 100 mM NaCl, and 50 mM Tris–HCl (pH 7.5)] at 37°C, where the submillimolar magnesium concentration is within the range estimated for intracellular magnesium concentration (31). Cleavage rate constants were obtained for the L2b8 parent device and the L2b8-a1 and -a14 engineered devices (Figure 4D and E) by quantifying relative levels of full-length and cleaved radiolabeled transcripts through polyacrylamide gel electrophoresis analysis (Supplementary Figure S9). Compared with the cleavage rate constant of L2b8 (0.14 min-1), the cleavage rates associated with the engineered devices are increased 5.2- and 10.5-fold for L2b8-a1 (0.70 min−1) and -a14 (1.4 min-1), respectively. The cleavage rate of the sTRSV hammerhead ribozyme (4.3 min-1) was determined at these conditions to be 32-fold greater than L2b8 (Supplementary Figure S9), in agreement with prior analyses on this ribozyme (30) and other well-characterized natural ribozymes under physiologically relevant conditions (32). The results indicate that the modified loop I sequences produce ribozyme-based devices exhibiting faster cleavage rates and thus lower in vivo basal activities.
Screening of transmitter libraries results in RNA devices exhibiting improved activation ratios
The dynamic range of an RNA control device depends on many parameters, including irreversible rate (e.g. ribozyme cleavage rate), intracellular ligand concentration, and mechanism by which binding information at the sensor is transmitted to an activity change in the actuator (e.g. transmitter design). We applied our two-color FACS-based screening strategy to explore a greater functional space to search for new transmitter sequences that support improved activation ratios given the designated sensor and actuator component pairs. We generated two transmitter libraries, tN11 and a1-tN11, based on two theophylline-responsive ribozyme-based devices, L2b8 and L2b8-a1, respectively, with ribozyme components that exhibit varying catalytic activities and thus in vivo basal activities. Both libraries were generated by randomizing 11 nucleotides in the transmitter component within the corresponding parent device (∼4 × 106 variants) (Figure 5A). We limited the number of randomized nucleotides in the transmitter components to result in libraries that could be completely searched based on the efficiency of our yeast library transformation method (see ‘Materials and Methods’). Both libraries were screened to identify devices with comparable basal activities and greater activation ratios relative to the corresponding parent devices (Figure 5B and D). We first performed a negative sort on both libraries by setting the sorting gates based on the basal activity of the respective parent device. The negative sort enriched members of the library (∼3% of the initial tN11 library; ∼5% of the initial a1-tN11 library) that exhibit lower or comparable basal activities as the parent device. A subsequent positive sort was performed on the recovered cell populations by setting sorting gates based on the gene-regulatory activity of the respective parent device in the presence of 5 mM theophylline. To enrich members of the library that exhibit improved activation ratios, cells that exhibit equal or higher gene-regulatory activities (∼0.3% of the enriched tN11 library; ∼0.4% of the enriched a1-tN11 library) were isolated. One additional positive sort was performed on the enriched tN11 library to further enrich for cells that exhibit activation ratios greater than or equal to the parent device (∼24% of the enriched library).
Figure 5.
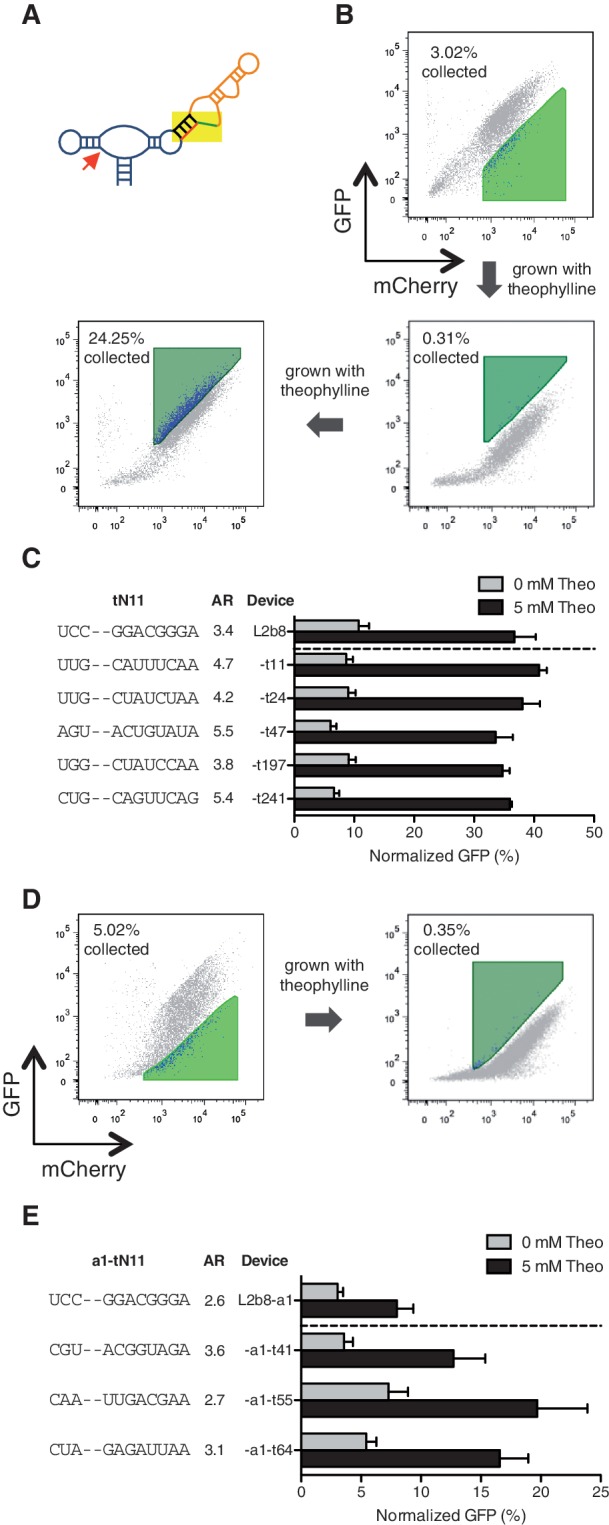
Screening of transmitter libraries within the device platform results in transmitter variants that exhibit improved activation ratios. (A) Two transmitter libraries, tN11 and a1-tN11, are generated by randomizing 11 nucleotides within the transmitter components in the L2b8 (wild-type ribozyme actuator) and L2b8-a1 (enhanced ribozyme actuator) devices, respectively. (B) The tN11 library is subjected to one sorting round (negative and positive sort), followed by an additional positive sort to further enrich members of the library that exhibit equal or greater increases in gene-regulatory activities in response to theophylline. The negative (light green) and positive (dark green) sorting gates are set based on the activity of the parent L2b8 device in the absence and presence of 5 mM theophylline, respectively. (C) Transmitter variants isolated from the tN11 library screen exhibit improved activation ratios (AR), which are determined as the ratio of gene expression levels in the presence and absence of theophylline (theo). (D) The a1-tN11 library is subjected to one sorting round to enrich members of the library that exhibit equal or greater increases in gene-regulatory activities in response to theophylline. The negative (light green) and positive (dark green) sorting gates are set based on the activity of the parent L2b8-a1 device in the absence and presence of 5 mM theophylline, respectively. (E) Transmitter variants isolated from the a1-tN11 library screen exhibit improved activation ratios. Gene-regulatory activities are reported as described in Figure 4C.
We plated the recovered cells from both final enriched libraries and screened 287 and 207 individual colonies from the tN11 and a1-tN11 libraries, respectively, through flow cytometry in the absence and presence of 5 mM theophylline and verified improved device activation ratios by re-cloning. We identified a total of five (t11, t24, t47, t197 and t241) and three (a1-t41, a1-t55 and a1-t64) transmitter variants from the tN11 and a1-tN11 library screens, respectively, that exhibit moderately improved activation ratios (up to 1.6-fold) relative to the respective parent devices at 5 mM theophylline (Figure 5C and E). We speculated that the intracellular theophylline concentration in yeast might limit the maximum activation ratio achievable by a device, as previous studies have estimated a 1000-fold drop in theophylline concentration across the E. coli cell membrane (33). We tested this hypothesis by characterizing transmitter variants at a higher theophylline concentration (40 mM). All characterized transmitter variants exhibited increased activation ratios at 40 mM theophylline relative to 5 mM theophylline (Figure 6, Supplementary Figure S10). For example, the activation ratio of the L2b8-t47 device increased from 5.5-fold at 5 mM theophylline to 10.7-fold at 40 mM theophylline, a 2-fold improvement relative to the parent L2b8 device at 40 mM theophylline. These results suggest that the intracellular theophylline concentration is limiting the device gene-regulatory activity. No difference in cell viability was observed under all theophylline conditions tested (Supplementary Figure S3A), although the higher theophylline concentration resulted in slower cell growth, possibly due to the ligand cytotoxicity, thus potentially rendering the implementation of these devices at higher ligand concentrations less effective for cellular applications.
Figure 6.

Component swapping demonstrates modularity of actuator components. For devices isolated from the tN11 sort (L2b8-t11 and L2b8-t47), replacement of the wild-type actuator loop I sequence with the a1 sequence results in devices exhibiting lower basal activities (shaded). For L2b8-a1-t41, which was isolated from the a1-tN11 sort, replacement of the a1 sequence with the wild-type actuator loop I sequence results in a device exhibiting higher basal activity (shaded). Gene-regulatory activities and activation ratios (AR) are reported as described in Figures 4C and 5C, respectively.
Modular assembly of optimized actuator components results in devices exhibiting improved regulatory stringencies
The modular composition framework of our ribozyme device platform supports modular assembly of functional RNA components to generate new device functions without significant redesign (10,34). We examined the ability of the optimized ribozyme sequences to be used to build new device regulatory functions, by coupling two of the recovered ribozyme variants (a1 and a14) with different transmitter and sensor components. We coupled the new actuator sequences with the transmitter and sensor components from two theophylline-responsive ribozyme ON devices (L2b1 and L2b5), one theophylline-responsive OFF device (L2bOFF1) and one tetracycline-responsive ON device (L2b8tc) to generate eight new devices. All newly generated devices exhibit lower basal activities relative to their respective parent, and the majority of them retain switching activity (Supplementary Figure S11A–D). The ribozyme variants were also coupled with selected transmitter variants to generate new functional devices with varying regulatory stringencies and activation ratios (Figure 6). The results indicate that the improved ribozyme variants are relatively insensitive to the sequence identities of the other device components (transmitter, sensor) and highlight that the modular device platform can be used in combination with improved components to program RNA devices exhibiting desired activities. Finally, we examined the activities of the loop sequences outside of the device context by replacing the wild-type loop I sequence of sTRSV to generate two ribozyme variants (Supplementary Figure S11E). The gene-regulatory activities of the resulting ribozyme variants were substantially higher than that of wild-type sTRSV, indicating that the improvement in regulatory stringencies associated with these loop sequences were specific to the context of the device platform, and thus supporting the importance of optimizing component functions directly within the device platform.
In vitro cleavage kinetics reflect in vivo RNA device gene-regulatory activities
The observation that loop I modified ribozyme-based devices exhibit improved in vivo basal activities and in vitro cleavage rates relative to the L2b8 parent device suggests a correlation between these two measures of activity. However, previous studies had shown that allosteric ribozymes exhibiting enhanced in vitro cleavage rates through in vitro selections failed to exhibit enhanced in vivo gene-regulatory activities (11). Therefore, we examined the relationship between in vitro cleavage rates and in vivo gene-regulatory activities for a set of RNA devices generated through the two-color screening and rational tuning strategies. Cleavage assays were performed under the previously described physiologically relevant reaction conditions in the absence and presence of 5 mM theophylline (Supplementary Figure S9). The ribozyme-based devices exhibit cleavage rates that span approximately two orders of magnitude from 0.01 min-1 (L2b5) to 1.0 min-1 (L2b8-a1, -a14) (Figure 7A and B). All the examined RNA devices exhibit reduced cleavage rates and increased gene-regulatory activities in response to theophylline, except for L2b8-a14, which exhibits slightly decreased gene-regulatory activity in the presence of theophylline (Supplementary Figures S7 and S9). The rank ordering of the examined RNA devices and the sTRSV ribozyme control by in vitro-determined cleavage rates matches the inverse ranking by in vivo gene-regulatory activities (Figure 7C). A correlation analysis between these two measures of activity indicates a strong negative correlation (Pearson coefficient, r = −0.9018) between the cleavage rates 0.007 and 0.16 min-1, where at a cleavage rate of 0.16 min-1 near background levels of gene expression are reached such that further decreases in expression levels are not observed with faster cleavage rates.
Figure 7.

In vitro cleavage kinetics of selected ribozyme-based devices and controls. Cleavage assays were performed at 37°C with 500 µM MgCl2, 100 mM NaCl and 50 mM Tris-HCl (pH 7.5) and in the presence of 5 mM theophylline when indicated. (A) Ribozyme-based devices exhibit decreased cleavage rates in the presence of ligand. Cleavage assays were performed as described in Figure 4D in the absence or presence of 5 mM theophylline. (B) In vitro cleavage kinetics of ribozyme-based devices in the absence and presence of ligand. Projected cleavage kinetics are generated as described in Figure 4E. Solid lines: 0 mM theophylline; dashed lines: 5 mM theophylline. (C) Correlation analysis of normalized gene expression levels and cleavage rate constants indicates a strong correlation between the in vivo gene-regulatory activity and in vitro cleavage rate; Pearson correlation coefficient (r) of −0.9018. A semi-log line is well fit (R2 = 0.94) for cleavage rates less than or equal to 0.16 min-1 (black data points). Devices excluded from this analysis are indicated in red.
DISCUSSION
We developed a two-color FACS-based screening approach that provides substantially increased screening resolution over more commonly used single-color strategies, enabling the enrichment of gene-regulatory elements with distinct (versus just high and low activities) quantitative gene-regulatory activities. We demonstrated the high efficiency of our screen by enriching a single sequence from a large ∼106 library to close to pure isolation in two sorting rounds. While other two-color fluorescence-based screening strategies have been described (35), these systems have generally been designed to compare distinct modes of gene expression. For example, one system was used to screen for peptides that facilitate a ribosomal frameshift event in E. coli based on a specialized bacterial ribosomal system (35). In contrast, our two-color screening construct does not depend on specialized gene expression machinery and can normalize gene expression noise that arises from both plasmid and potentially chromosomal constructs.
We applied our two-color screening approach to optimize the actuator and transmitter components directly within the context of the ribozyme device platform in the relevant cellular environment, thereby addressing challenges previously encountered with optimizing component activities through in vitro strategies (11). The highly quantitative nature of our screening approach allowed us to efficiently isolate ribozyme and transmitter variants that exhibit specified gene-regulatory activities. The actuator and transmitter screens did not provide as strong an enrichment as observed for the sensor screen (i.e. close to pure isolation of a single sequence), which is likely explained by several factors, including fewer sorts applied to the actuator and transmitter screens (resulting in a higher false positive rate) and greater number of functional sequences in the actuator and transmitter libraries that fell into the set sorting gates. Our two-color screening approach is also fully complemented by the modular composition framework of the ribozyme-based platform. We demonstrated a plug-and-play device construction strategy by linking components with varying activities to generate new RNA devices that exhibit diverse gene-regulatory activities. In comparison to the previously demonstrated computational-guided device tuning strategy, which is restricted to experimentally sampling a smaller sequence space, our two-color screening approach offers greater flexibility and higher-throughput in quantitatively tailoring device gene-regulatory activities.
We further characterized the isolated ribozyme and transmitter variants in addition to the devices generated previously by rational design through both in vivo and in vitro assays and found a strong negative correlation between in vivo gene-regulatory activities and in vitro cleavage rates, for cleavage rates within the range of 0.007 and 0.16 min−1 (measured at the specified reaction condition of 500 µM MgCl2). Gene expression levels reach background values at ∼0.16 min-1, such that increases in cleavage rate above this value have minimal impact on expression levels. In addition, our analysis (Figure 7C) indicates that below cleavage rates of ∼0.007 min-1 gene expression levels reach saturating (fully ON) levels, such that further decreases in cleavage rate below this value do not significantly impact gene expression. Our work demonstrates that there is a narrow window of in vitro cleavage rates (spanning less than two orders of magnitude) that corresponds to titratable in vivo gene-regulatory activities. This observation may explain previous work in the field that observed no changes in gene-regulatory activities among ribozyme variants exhibiting different cleavage rates, if the changes in cleavage rates occurred outside of the titratable window (13).
Our results also highlight the importance of optimizing individual components within the context of device platforms. The parent actuator component (sTRSV ribozyme) is a type-III hammerhead ribozyme, in which the 5′ and 3′ ends of viroid RNA extend from stem III. The tertiary interactions essential for cleavage activity at physiological conditions are formed by a Watson–Crick base triple between the 5′ U of loop I, next-to-last U of loop I, and the 3′ A of loop II. Recent structural analyses have shown that these essential tertiary interactions are conserved across a significant fraction of natural hammerhead ribozymes (36). As the loop I sequence of the originally designed ribozyme-based devices, including the parent device L2b8, is unchanged from the native sTRSV ribozyme (UGUGCUU), it was assumed that aptamer integration into loop II would maintain the same tertiary interactions required for cleavage activity (10). However, both the heptaloop (AUNNARG) and triloop (N1UN2GGGN1|) consensus sequences identified through the actuator library screen that result in devices exhibiting lower basal activities than the L2b8 parent device do not follow the common tertiary motif of the sTRSV ribozyme. The divergence of the improved loop I sequences from the L2b8 and sTRSV parents are not necessarily surprising, given recent work identifying thousands of new hammerhead ribozymes in all domains of life with unique tertiary motifs (36–38). In particular, the integration of the transmitter-aptamer element into loop II of the sTRSV hammerhead ribozyme in the device platform makes the overall structure of the ribozyme-based devices more similar to other, less well-characterized hammerhead ribozymes (Supplementary Figure S12). Interestingly, the improved loop I consensus sequences identified in our screen share similarities to these other natural ribozymes and do not exhibit improved gene-regulatory activities in the context of the sTRSV ribozyme alone.
Based on its increased screening resolution, our two-color FACS-based screen can provide a powerful strategy to guide future RNA device design. For example, RNA gene-regulatory devices generally exhibit moderate dynamic ranges (1,2,4). Our screen can be applied to effectively search libraries of RNA devices to provide insight into specific constraints limiting RNA device gene-regulatory activity. In this work, we performed screens on two different libraries harboring ribozyme components exhibiting different catalytic activities and randomized transmitter components. Although we successfully isolated variants from each library that exhibit improved activation ratios relative to the respective parent device, the improvements were modest. Based on our prior computational modeling work on RNA control devices (39), we speculated that the intracellular ligand concentration may place constraints on the activation ratios. Further increases in the activation ratios of transmitter variants from both libraries were observed at increased ligand concentrations (Figure 6, Supplementary Figure S10), supporting this hypothesis. However, the improvements were smaller for transmitter variants based on the more catalytically active ribozyme, possibly due to a more dominant irreversible event (faster ribozyme cleavage rate). These results suggest that RNA control devices that utilize ribozyme-based actuator components require careful balance of irreversible rates (ribozyme cleavage) and reversible rates (conformational switching, ligand binding) to optimize dynamic ranges.
Finally, recent reports highlight the importance of fine-tuning regulatory properties of gene-regulatory elements for their successful integration within biological systems (1,2). Examples to-date have relied on using low-throughput and inefficient single-color screening strategies on individual clones from libraries of control elements, including promoters and RNase cleavage sites, to identify new elements that exhibit varying activities in different organisms (40–42). In contrast, our two-color FACS-based screening approach is based on the correlation between the gene outputs from two independent functional modules. Similar dual-reporter screening plasmid systems can be constructed in other organisms, including bacteria and mammalian cells, to support the efficient generation of new gene control elements, including those acting through transcriptional and posttranscriptional mechanisms. Our strategy offers a high-throughput and high-efficiency alternative to rapidly screen through diverse libraries of control elements for members exhibiting specified quantitative activities, thereby addressing a key challenge in quantitatively tailoring these control elements to specific genetic systems.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1, Supplementary Figures 1–12, and Supplementary References [36,43–45].
FUNDING
National Institutes of Health [R01GM086663]; National Science Foundation [CBET-0917638, CCF-0943269]; Defense Advanced Research Projects Agency [HR0011-11-2-0002]; National Sciences and Engineering Research Council of Canada (fellowship to A.B.K.); Alfred P. Sloan Foundation (fellowship to C.D.S.). Funding for open access charge: Defense Advanced Research Projects Agency.
Conflict of interest statement. The authors declare competing financial interests in the form of a pending patent application.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank C. Crumpton and M. Bigos of the Stanford Shared FACS facility. J.C.L. designed and performed the research and wrote the article; A.L.C. designed and performed the research associated with the two-color FACS-based screen and wrote the article; A.B.K. designed and performed the research associated with the in vitro characterization of ribozyme cleavage rates and the actuator screen and wrote the article; and C.D.S. designed the research and wrote the article.
REFERENCES
- 1.Chen YY, Jensen MC, Smolke CD. Genetic control of mammalian T-cell proliferation with synthetic RNA regulatory systems. Proc. Natl. Acad. Sci. USA. 2010;107:8531–8536. doi: 10.1073/pnas.1001721107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Culler SJ, Hoff KG, Smolke CD. Reprogramming cellular behavior with RNA controllers responsive to endogenous proteins. Science. 2010;330:1251–1255. doi: 10.1126/science.1192128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ro D-K, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440:940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- 4.Sinha J, Reyes SJ, Gallivan JP. Reprogramming bacteria to seek and destroy an herbicide. Nat. Chem. Biol. 2010;6:464–470. doi: 10.1038/nchembio.369. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Joyce GF. Forty years of in vitro evolution. Angew. Chem. Int. Ed. Engl. 2007;46:6420–6436. doi: 10.1002/anie.200701369. [DOI] [PubMed] [Google Scholar]
- 6.Seeman NC. From genes to machines: DNA nanomechanical devices. Trends Biochem. Sci. 2005;30:119–125. doi: 10.1016/j.tibs.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Win MN, Liang JC, Smolke CD. Frameworks for programming biological function through RNA parts and devices. Chem. Biol. 2009;16:298–310. doi: 10.1016/j.chembiol.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fedor MJ, Williamson JR. The catalytic diversity of RNAs. Nat. Rev. Mol. Cell Biol. 2005;6:399–412. doi: 10.1038/nrm1647. [DOI] [PubMed] [Google Scholar]
- 9.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Win MN, Smolke CD. A modular and extensible RNA-based gene-regulatory platform for engineering cellular function. Proc. Natl. Acad. Sci. USA. 2007;104:14283–14288. doi: 10.1073/pnas.0703961104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Link KH, Guo L, Ames TD, Yen L, Mulligan RC, Breaker RR. Engineering high-speed allosteric hammerhead ribozymes. Biol. Chem. 2007;388:779–786. doi: 10.1515/BC.2007.105. [DOI] [PubMed] [Google Scholar]
- 12.Wittmann A, Suess B. Selection of tetracycline inducible self-cleaving ribozymes as synthetic devices for gene regulation in yeast. Mol. Biosyst. 2011;7:2419–2427. doi: 10.1039/c1mb05070b. [DOI] [PubMed] [Google Scholar]
- 13.Chen X, Denison L, Levy M, Ellington AD. Direct selection for ribozyme cleavage activity in cells. RNA. 2009;15:2035–2045. doi: 10.1261/rna.1635209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lynch SA, Gallivan JP. A flow cytometry-based screen for synthetic riboswitches. Nucleic Acids Res. 2009;37:184–192. doi: 10.1093/nar/gkn924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fowler CC, Brown ED, Li Y. A FACS-based approach to engineering artificial riboswitches. Chembiochem: Eur. J. Chem. Biol. 2008;9:1906–1911. doi: 10.1002/cbic.200700713. [DOI] [PubMed] [Google Scholar]
- 16.Desai SK, Gallivan JP. Genetic screens and selections for small molecules based on a synthetic riboswitch that activates protein translation. J. Am. Chem. Soc. 2004;126:13247–13254. doi: 10.1021/ja048634j. [DOI] [PubMed] [Google Scholar]
- 17.Topp S, Gallivan JP. Random walks to synthetic riboswitches—a high-throughput selection based on cell motility. Chembiochem: Eur. J. Chem. Biol. 2008;9:210–213. doi: 10.1002/cbic.200700546. [DOI] [PubMed] [Google Scholar]
- 18.Nomura Y, Yokobayashi Y. Dual selection of a genetic switch by a single selection marker. Biosystems. 2007;90:115–120. doi: 10.1016/j.biosystems.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 19.Sambrook JRD. Molecular Cloning: A Laboratory Manual. 3rd edn. Cold Spring Harbor, NY: Cold Spring Harbor Lab Press; 2001. [Google Scholar]
- 20.Uppaluri R, Towle HC. Genetic dissection of thyroid hormone receptor beta: identification of mutations that separate hormone binding and transcriptional activation. Mol. Cell. Biol. 1995;15:1499–1512. doi: 10.1128/mcb.15.3.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 22.Gari E, Piedrafita L, Aldea M, Herrero E. A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast. 1997;13:837–848. doi: 10.1002/(SICI)1097-0061(199707)13:9<837::AID-YEA145>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 23.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- 25.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 2006;1:755–768. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 26.Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 27.Raser JM, O’Shea EK. Control of stochasticity in eukaryotic gene expression. Science. 2004;304:1811–1814. doi: 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jenison RD, Gill SC, Pardi A, Polisky B. High-resolution molecular discrimination by RNA. Science. 1994;263:1425–1429. doi: 10.1126/science.7510417. [DOI] [PubMed] [Google Scholar]
- 29.Zimmermann GR, Jenison RD, Wick CL, Simorre JP, Pardi A. Interlocking structural motifs mediate molecular discrimination by a theophylline-binding RNA. Nat. Struct. Biol. 1997;4:644–649. doi: 10.1038/nsb0897-644. [DOI] [PubMed] [Google Scholar]
- 30.Khvorova A, Lescoute A, Westhof E, Jayasena SD. Sequence elements outside the hammerhead ribozyme catalytic core enable intracellular activity. Nat. Struct. Biol. 2003;10:708–712. doi: 10.1038/nsb959. [DOI] [PubMed] [Google Scholar]
- 31.London RE. Methods for measurement of intracellular magnesium: NMR and fluorescence. Annu. Rev. Physiol. 1991;53:241–258. doi: 10.1146/annurev.ph.53.030191.001325. [DOI] [PubMed] [Google Scholar]
- 32.Canny MD, Jucker FM, Kellogg E, Khvorova A, Jayasena SD, Pardi A. Fast cleavage kinetics of a natural hammerhead ribozyme. J. Am. Chem. Soc. 2004;126:10848–10849. doi: 10.1021/ja046848v. [DOI] [PubMed] [Google Scholar]
- 33.Koch AL, Lamont WA. The metabolism of methylpurines by Escherichia coli. II. Enzymatic studies. J. Biol. Chem. 1956;219:189–201. [PubMed] [Google Scholar]
- 34.Win MN, Smolke CD. Higher-order cellular information processing with synthetic RNA devices. Science. 2008;322:456–460. doi: 10.1126/science.1160311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dulude D, Theberge-Julien G, Brakier-Gingras L, Heveker N. Selection of peptides interfering with a ribosomal frameshift in the human immunodeficiency virus type 1. RNA. 2008;14:981–991. doi: 10.1261/rna.887008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dufour D, de la Pena M, Gago S, Flores R, Gallego J. Structure-function analysis of the ribozymes of chrysanthemum chlorotic mottle viroid: a loop-loop interaction motif conserved in most natural hammerheads. Nucleic Acids Res. 2009;37:368–381. doi: 10.1093/nar/gkn918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perreault J, Weinberg Z, Roth A, Popescu O, Chartrand P, Ferbeyre G, Breaker RR. Identification of hammerhead ribozymes in all domains of life reveals novel structural variations. PLoS Comput. Biol. 2011;7:e1002031. doi: 10.1371/journal.pcbi.1002031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seehafer C, Kalweit A, Steger G, Graf S, Hammann C. From alpaca to zebrafish: hammerhead ribozymes wherever you look. RNA. 2011;17:21–26. doi: 10.1261/rna.2429911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beisel CL, Smolke CD. Design principles for riboswitch function. PLoS Comput. Biol. 2009;5:e1000363. doi: 10.1371/journal.pcbi.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Tuning genetic control through promoter engineering. Proc. Natl. Acad. Sci. USA. 2005;102:12678–12683. doi: 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nevoigt E, Kohnke J, Fischer CR, Alper H, Stahl U, Stephanopoulos G. Engineering of promoter replacement cassettes for fine-tuning of gene expression in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2006;72:5266–5273. doi: 10.1128/AEM.00530-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Babiskin AH, Smolke CD. A synthetic library of RNA control modules for predictable tuning of gene expression in yeast. Mol. Syst. Biol. 2011;7:471. doi: 10.1038/msb.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Darty K, Denise A, Ponty Y. VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hertel KJ, Pardi A, Uhlenbeck OC, Koizumi M, Ohtsuka E, Uesugi S, Cedergren R, Eckstein F, Gerlach WL, Hodgson R, et al. Numbering system for the hammerhead. Nucleic Acids Res. 1992;20:3252. doi: 10.1093/nar/20.12.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.