


Abstract
Genomic experiments (e.g. differential gene expression, single-nucleotide polymorphism association) typically produce ranked list of genes. We present a simple but powerful approach which uses protein–protein interaction data to detect sub-networks within such ranked lists of genes or proteins. We performed an exhaustive study of network parameters that allowed us concluding that the average number of components and the average number of nodes per component are the parameters that best discriminate between real and random networks. A novel aspect that increases the efficiency of this strategy in finding sub-networks is that, in addition to direct connections, also connections mediated by intermediate nodes are considered to build up the sub-networks. The possibility of using of such intermediate nodes makes this approach more robust to noise. It also overcomes some limitations intrinsic to experimental designs based on differential expression, in which some nodes are invariant across conditions. The proposed approach can also be used for candidate disease-gene prioritization. Here, we demonstrate the usefulness of the approach by means of several case examples that include a differential expression analysis in Fanconi Anemia, a genome-wide association study of bipolar disorder and a genome-scale study of essentiality in cancer genes. An efficient and easy-to-use web interface (available at http://www.babelomics.org) based on HTML5 technologies is also provided to run the algorithm and represent the network.
INTRODUCTION
There is now a wide consensus on the fact that most of the biological functionality of the cell arises from complex interactions between their molecular components (1). Such interacting components define operational entities or modules to which different elementary functions can be attributed. Understanding the organization and the dynamics of the complex intracellular network of interactions that contribute to the structure and function of a living cell is one of the main challenges in functional genomics (2) and constitutes the objective of systems biology (3).
Simple, unstructured module definitions, such as Gene Ontology (GO) (4), account only for the common functionality of their components. Despite its simplicity, they have been extensively used for the development of functional enrichment methods (5–11). Such methods have proven its usefulness in helping researchers to understand the relationships between the genes activated (or deactivated), mutated or affected in some way, found in a genomic experiment and the corresponding functional consequences. Functional enrichment methods aim at finding overrepresentations of genes belonging to some of these modules among a predefined list of genes. However, this approach was soon criticized because of its dependence on the initial selection of the set of genes to be analyzed (12). Then, a family of methods known under the generic name of Gene-Set Enrichment Analysis (GSEA) emerged that studied the distribution of modules across a list of genes ranked according to a parameter representative of the experiment, such as differential expression (13), association to a disease (14) and others (15–17).
Despite the success of methods based on GO (or other unstructured) modules for the biological interpretation of different types of genomic experiments (gene expression microarrays, large-scale genotyping), conceptualizing a function simply as a label shared by a set of genes resulted in a poor description of the cellular complexity. Actually, information on relationships among gene products is available and can be used to define other types of modules. In particular, protein–protein interactions (PPIs) provide a useful and extensively used representation of such relationships beyond categorical definitions such as GO (18). The use of the interactome as a theoretical scaffold that relates proteins among them allows depicting sub-networks of interacting proteins associated to features in genomic experiments (19), which can be considered functional modules (20). It is known that disease gene products exhibit an increased tendency to interact among them, tend to co-express and display coherent functions according to GO annotations (19). Actually, the relationship between common functionality, co-expression and interaction has also been reported in numerous studies (21–23). In fact, these properties are so tightly related that protein function has been successfully predicted from gene co-expression (24,25) and PPI (26,27) data. This relationship has also been observed for genotyping data, where gene interactions (28) or even single-nucleotide polymorphism (SNP) associations can be related to PPI networks (29,30). An additional advantage of PPI networks is that their topology and properties (e.g. connectivity, betweenness) provide a deal of information on the modules besides the own functional annotations of the components. Therefore, sub-networks, (sub sets of the interactome comprising proteins that directly interact among them) can be considered a higher level, structured description of functional modules operating in the cell.
Since it is increasingly clear that phenotypes and, more specifically, diseases are the consequence of the interactions between gene products, different methods have been proposed for finding disease-related sub-networks (31,32) or to predict disease-causing genes (33–36). Most of these methods have been designed to deal with gene expression data and use a scoring function based on the values of differential expression (20,37,38) (node-based methods) or co-expression (39–41) (edge-based methods). Such scoring function is applied within different search strategies to evaluate the highest scored sub-network (the largest possible number of proteins connected among them according to the interactome map) compatible with the gene expression experiment. However, the complexity of the interactome generates a search space of an enormous size, which makes of the task of finding sub-networks a NP-hard problem (37). This fact implies large runtimes and constitutes a drawback for the application of these methods. Other simpler methods rely on the pre-selection of gene sets (42–46), which constitutes a drawback, as mentioned above for the case of functional enrichment methods.
Here, we propose a simpler but powerful approach able to find the sub-network component associated to extreme values of a list of genes (or proteins) ranked by any criterion (differential gene expression, disease association in a genome-wide association study (GWAS), etc.). Contrarily to other methods, which need to optimize a number of parameters in a way that is sensitive to initial conditions, our proposal has only one parameter which is the rank. Moreover, in the proposed method not only direct connections among the genes (or proteins) studied are considered but also intermediate nodes not present in the dataset studied that link highly-ranked nodes of the dataset are considered. These intermediate links increase enormously the sensitivity of the search procedure, being thus more robust against false negatives (still abundant in genome-wide experiments).
The method is implemented in a publicly available and free web tool, Network Miner, designed to find, within a list of ranked genes, the largest network components among the best scored genes along with the corresponding statistical significance (i.e. the probability of finding such structured sub-networks just by chance) and represent the network found in an advanced visualization system. Network Miner can be found within the Babelomics package (47).
MATERIALS AND METHODS
Datasets used and data preprocessing
Gene expression data in Fanconi Anemia
Gene expression datasets for Fanconi Anemia (FA) were obtained from GEO (GSE16334) (Affymetrix Human Genome U133A Array). The original GEO normalization of each dataset was used. Differential gene expression control versus case samples were carried out using the Limma package (48) from Bioconductor (49), implemented into Babelomics web platform (47). Probes were ranked according to decreasing values of the statistic. When several probes mapped onto to the same Ensembl gene, the highest ranked probe was selected. Finally, probe identifiers were converted to the corresponding Ensembl Gene identifiers. Genes for which protein interactions have not yet been described in the literature were also discarded. The same process was repeated using the Robust Multichip Average (RMA) (50) normalization method to discard the influence of the normalization method in the results obtained.
Bipolar disorder genotyping
Anonymous genotypic data from the Wellcome Trust Case Control Consortium (WTCCC) (51) were downloaded in plink transposed format (52). A total of 2000 Caucasian UK patients of bipolar disorders and 1500 controls genotyped on the Affymetrix 500 K mapping array were studied. GWAS was performed using the basic association test of Plink toolset based on comparing allele frequencies between cases and control. Following a similar strategy than in pathway-based analysis (PBA) (14,29), we filter this list to retain the subset of SNPs mapping within genes or in the neighborhood (up to 500 bp up- and downstream of the gene limits). Then, we filter the list again leaving only one SNP per gene. The SNP retained is the one with the smaller P value, which is finally converted to the ID of the corresponding gene. Thus, we have a list of genes ranked by the P value of their most associated marker. Genes previously associated with bipolar disorder obtained from Uniprot database (53) were used as seed genes.
Essential genes in cancer cell lines datasets
In a recent study, gene essentiality for growth in different cancer cells was tested using a powerful genome-scale pooled short hairpin RNAs (shRNAs) screen (54). Cancer cell lines evaluated represented different cancer types: small-cell lung cancer (H187 and H82), non-small-cell lung cancer (A549, H1650, H1975 and HCC827), lymphocytic leukemia (Jurkat, REH and SUPT1), chronic myelogenous leukemia (K562) and glioblastoma (LN229 and U251). A statistic test called shRNA gene enrichment ranking (RIGER), which outputs a gene essentiality score, was used in the study. Genes were ranked according to the RIGER score (gene essentiality) for network analysis.
PPI data and curation
So far, there is not a common source for PPI data. In contrast, there are several primary interaction databases that vary in the way they store the interactions, their scope, annotation quality and public availability. Given the low overlap observed among the main general interaction databases (55), we collected and merged the data from the following databases: IntAct (2011-01-19 version) (56), MINT (2011-01-19 version) (57) and BioGRID (version 3.1.72) (58).
In order to integrate and unify PPIs coming from different databases, three steps of curation were applied. First, only proteins whose identifier could unequivocally be mapped to a reference protein in UniProt Swiss-Prot (59) were used. Next, only interactions whose type was ‘physical association’ were taken. This filtering step prevents from including other interactions types that do not necessarily imply physical contact between gene products, such as genetic interactions and other. Finally, potential artifactual PPIs, frequent in interactions data, especially those derived from high-throughput technologies, were also filtered out by considering only those PPIs detected with at least two different detection methods (60). To avoid selecting PPIs determined through experiments with a similar basis (e.g. ‘two hybrid array’ and ‘two hybrid gal4 vp16 complementation’), the six lower levels of depth in the Molecular Interaction (MI) ontology ‘interaction detection method’ (61) were used. Interactions reached at this step constitute the called curated interactome. The categories ‘physical association’ and ‘detection method’ are components of the xml format PSI-MI 2.5 (62) offered by the PPI databases used.
Interactomes were generated for the following species: Arabidopsis thaliana, Drosophila melanogaster, Escherichia coli (strain K12), Homo sapiens, Mus musculus and Saccharomyces cerevisiae.
False Discovery rate (FDR) and power tests
With the purpose of finding the feature that best characterizes a real PPI network, different parameters were evaluated in terms of power (true-positive rates) and false-positive rates. Power is defined as the probability of declaring a network as significantly different from a random network when it has been obtained from a list of actual functionally-related proteins. On the other hand, the false-positive rate is defined as the probability of declaring a network as significant when it is obtained from lists of randomly selected proteins.
Networks are defined here as the shortest network that connects all the physically interacting proteins within a set of proteins, also known as Minimum Connected Network (MCN). The shortest paths among these interacting proteins are calculated using Dijkstra algorithm (63).
MCNs can be characterized by topological parameters related to each node in the network, such as connection degree (defined as the number of connections of a node), relative betweenness (a measure of a node's centrality, which is the number of shortest paths from all nodes to all others that pass through that node (64))—and clustering coefficient (a measure of degree to which nodes in the MCN tend to cluster together (65)) or by parameters related to the whole network (number of nodes, number of connections and number of components). Furthermore, we can also combine the whole network features and test the average number of nodes or connections per component. The different nature of parameters requires a distinct strategy of network evaluation:
Testing node-level parameters. The comparison of two networks through their node-level parameters can be performed by testing whether the corresponding distributions of values are significantly different or not. Different tests were checked in order to determine which one provided more power for the discrimination. The tests checked were as follows: (i) two sample Wilcoxson (ii) Kolmogorov–Smirnov and (iii) the common area under both distributions (66). For a given node-level parameter, the distribution of reference for a random network of size N is derived from 2000 networks made of N components randomly selected among all the possible proteins.
Testing whole network parameters. Whole network parameters are described with a single number and are, consequently, easier to test. Given a distribution of values generated from 2000 random networks derived as above, the P value can be estimated by simply ranking the value of the parameter of interest on such distribution.
The tests were performed using the S. cerevisiae interactome, because it is the most comprehensive description of a whole protein interaction network in any organism. All statistical tests were performed using R software environment.
Generation of MCNs experimentally verified
Several networks described in the literature in (67–69) were used as bona fide real networks that should be detected by their peculiar network parameters’ values. We also used KEGG pathways (70) and GO (4) terms, which are also known to be rich in network component (18,21). Specifically, GO-defined modules among levels 6 and 12 were selected to avoid general and highly specific GO terms. Since we may work among a range of list sizes, lists containing 20, 50 and 100 proteins were collected from all these sources. A total of 156 modules were analyzed.
Generation of random MCNs
In order to calibrate the rate of false positives that the combination of a parameter with a particular test (as described above) produced, it was necessary to build up a collection of networks connecting sets of randomly chosen proteins. An extensive range of conditions has been tested that include random sets from 10 to 200 proteins, for both the curated and the non-curated interactome and for both direct connections and allowing one intermediate connection by means of a node external to the dataset analyzed. In order to obtain a distribution of values for any of these conditions, 2000 random samples have been obtained. That amounts a total of 1 520 000 MCNs to obtain an estimation of the expectation of the network parameters just by chance for an equivalent number of proteins not linked a priori by a network. These values can be used as a pre-calculated confidence interval when a MCN found in a new dataset is tested (see next section).
The algorithm for network enrichment
Instead to take a sub-selection of the genes, the algorithm starts with the complete list of gene or protein identifiers involved in a genomic experiment, ranked by a given criteria. In principle, ranking values are supposed to be derived from a genomic experiment and must have, consequently, a biological meaning. For example, it can be the value of a t-test statistics derived from a differential expression experiment, thus accounting for the higher level of expression in one of the conditions compared; it can also be a P value in a genotyping association experiment, thus accounting for the association of each of the genes with a disease, etc. Obviously, this methodology is not restricted to genotyping or differential gene expression and other ranking values representing the results of other types of experiments are also possible. Then, the interpretation must be done accordingly to the biological property that this particular ranking value is representing. The ranking parameter is, therefore, used as a guide to scan for sub-network enrichment through the entire ranked list of molecules. This strategy, similar to the GSEA strategy, avoids the imposition of a gene-based threshold to pre-select a limited number of genes for further network enrichment analysis. The algorithm seeks for sets of genes connected among them, moderately but coordinately associated to high (or low) values of the ranking parameter. Since we look for a set-based property, there is no point in pre-selecting a fixed number of genes based on a conventional gene-based test. The algorithm proposed follows the steps listed below:
The ranked list S = (g1,…, gn) of n molecules is subdivided into a sequence of additive partitions Sk = (gi ϵ S: i = 1,…, k; k ≤ n) of size k.
The proteins corresponding to any of the partitions are mapped onto the interactome scaffold and the MCN, that is, the minimal network that connects the maximum number of nodes in the partition is found. The shortest paths among all the pairs of nodes in the list are calculated using Dijkstra algorithm (63). Then, the parameter of interest (zk, defined as the average nodes per component of the MCN) is calculated.
- Then we seek for the most relevant partition (the sub-list Sbest) as follows:
- First, ordering the parameter of interest zk according to the ranked list, all relative maxima are identified. The partitions so selected (
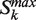 ) represent situations where a new protein capable of connecting to the previous ones is added to the previous partitions.
) represent situations where a new protein capable of connecting to the previous ones is added to the previous partitions. - Second, the score Lk is computed as Lk = (zk − 1)/(k−1) for all the selected partitions
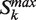 . The score can be seen as a balance between the increase in connected nodes and the distance to the top of the ranked list (k = 1).
. The score can be seen as a balance between the increase in connected nodes and the distance to the top of the ranked list (k = 1). - Third, we choose the partition Sbest and index kbest corresponding to the highest Lk computed in b) form the
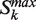 chosen in (a).
chosen in (a).
Finally, an empirical P value is calculated as the proportion of 2000 random sub-lists of kbest molecules (which corrects the size effect) with a value for the network parameter greater than zkbest.
In the step two, only proteins contained in the partition are considered to find the MCN. That is, only direct PPIs are considered. However, we can also consider another scenario in which proteins not included in the partition are used to connect proteins contained in the partition. Thus, MCNs can be found that connect proteins in the partition using some connections external to the partition. Given the potential density of connections of the interactome, only one-step connections are used. Allowing external nodes to participate in the MCN if they directly connect two proteins in the partition is a quite realistic assumption in several genomic experimental designs. Thus, false-negative occurrences would remove connecting proteins from the partitions analyzed and would consequently have a negative effect on the ability of finding MCNs. Allowing these proteins to participate in the connections would overcome this effect. In other cases, the own measurement can remove interesting proteins from the analyzed dataset. For example, in the case of differential expression experiments, it might happen that proteins participating in the networks are always expressed but not differentially expressed. In this case, these would not appear among the differentially expressed proteins despite they participate in the differentially expressed network.
Optionally, a list of seed molecules may be incorporated. In this case, a seed list Sseed = (g1,…, gm) of m molecules is forced to be part of the whole list, SK, defined as:
The selection procedure is the same than described above but keeping always the Sseed molecules within the list.
Software implementation details
The NetworkMiner web interface was implemented using HTML, CSS, JavaScript and Java. The queries to the server are implemented in Java connecting to a MySQL Server and a plain text file with the interactomes information. The visualization tool was implemented using HTML, JavaScript and SVG from HTML5. This new standard allows implementing advanced SVG graphics directly on the web browser without the necessity of installing any plug-in or Adobe Flash or using any Java applet. Moreover, HTLM5 standard is extensively supported by modern web browsers such as Google Chrome 14+, Microsoft Internet Explorer 9, Mozilla Firefox 6+ or Apple Safari 5+. CSS and JavaScript are used at the client side together with HTML5, while Java is used at the server side of the NetworkMiner application to query a MySQL Server and connect it to a plain text file with the interactomes information.
The program inputs a list of ordered genes (in a simple column). An extra column can be added, which will be taken as the value of the ranking parameter. Furthermore, an extra list of seed data can be provided. Typically, this list represents the already known gene diseases, and the program will try to include them in the list. The user can also chose between using all the described interactions or a more curated version of the interactome containing only those interactions reported for more than one detection method. The order in which the ranked list will be explored can be ascendant or descendent and the threshold for the P value can also be defined.
The interactomes of the following species are supported: A. thaliana, D. melanogaster, E. coli (strain K12), H. sapiens, M. musculus and S. cerevisiae.
Once the calculations have been done, the result is presented in a network viewer box (see Figure 1 as an example). The HTML5 technology allows a straightforward dynamic representation of the networks found, on which many operations can be performed. Different layouts are possible that implement different algorithms for distributing the net components in different ways. Furthermore, different backgrounds, including different cell views over which the network can be represented, can be seen in Figure 1.
Figure 1.

Screen snapshot of the network viewer box of NetworkMiner represented over a cell image background.
Thus, nodes, intermediate nodes (not present in the partition analyzed, but connecting nodes in the partition), edges and edges between intermediate nodes can individually or collectively selected and deselected for different manipulations such as moving them, collapsing or expanding them. In addition, node and edge properties including name, size, opacity adjustment, color, stroke, edge shape, etc., can be customized.
The complete view can be zoomed in and out, and the labels can also be customized in size. Different filters based on specific attributes of the network can be applied. These attributes include gene/protein ID, GO, KEGG, Reactome, Interpro, Jaspar and Ensembl terms. Finally, all the information relative to nodes can be extracted and displayed in a pop-up window, including functional and regulatory information, protein interaction information, etc.
NetworkMiner is available within the Babelomics environment (http://www.babelomics.org) within the section of functional analysis.
All the examples presented in the next section are also available as pre-loaded case studies in the program.
RESULTS AND DISCUSSION
Study of network parameters characteristic of real networks
A previous step in this study involves determining if a real network can be distinguished from a random network and, if so, what network parameter has the better discriminatory power. With this purpose, we have collected a number of validated biological networks as a test set on which the efficiency of the combination of the most common local and global network parameters with different statistical tests has been tried. Reference networks covering three sizes (20, 50 and 100 nodes), which include KEGG pathways, sub-networks described in the literature and some GO modules with the proteins highly interconnected, were used.
We have checked two scenarios (i) networks found within sets of proteins with direct connections among them and (ii) networks found within sets of proteins with either direct connections or connected through one intermediate protein not present in the set. This second scenario represents a common situation in large-scale genomic analysis. In many cases, in proteomic analyses, some of the proteins activated in an experiment are simply not detected, because of the sensitivity of the technique. In the case of transcriptomics experiments, it is quite common that the noise affecting to individual probes representative of the genes (and the corresponding gene products) makes some of them present different values of the statistic. In an ideal situation, a group of proteins that co-express and conform, for example, a complex should appear together in a differential expression experiment and should easily be detected by a conventional test that look for network enrichment. In a real situation, it is quite common that as a consequence of noise or experimental errors some proteins of the sub-network are missing in the experiment (in spite of being actually involved in the network structure). It can also happen that some proteins (key in the definition of the network) do not change their expression across the compared conditions, thus a differential expression experiment did not report them in the result. Thus, looking for networks within a set of proteins, allowing for some connections provided by proteins not in the set, increases enormously the sensitivity of the network detection method and makes it more robust against noise. It also allows overcoming some intrinsic limitations of experimental designs based on differential expression, such as the difficulty of detecting networks in which some of the nodes do not differentially express across the conditions compared.
In order to derive the expected values for the topological network parameters for random networks different approaches can be used. For a given number of genes N, an empirical simulated distribution can be derived by repeatedly selecting N genes randomly from the genome, then looking for the MCN that connects them and measuring the parameter of interest. Repeating this procedure 2000 times allows deriving the distribution sought.
Figure 2 summarizes the results obtained in the combined study of network parameters and tests. In the case of local network parameters, the connection degree in combination with the Kolmogorov–Smirnov test provided the highest sensitivity in distinguishing real networks from random networks. However, sensitivity decreases if intermediate nodes (not present in the partition analyzed but connecting nodes from the partition) are included in the MCN. On the other hand, Figure 2 shows that the most sensitive among the global network parameters is the average number of nodes per component. This feature also demonstrates to be robust to the inclusion of intermediate nodes and will be used throughout the case studies illustrating the usefulness of the approach proposed.
Figure 2.

Comparative analysis of the discriminatory power of different network parameters to distinguish a true biological network from a random network. The x-axis accounts for the MCN size. Arrangement of charts in rows and columns corresponds to intermediate node inclusion and false/true-positive rates (FDR: false-positive rate; power: true-positive rate). Color corresponds to the feature tested.
All the network calculations have been carried out using the server-side NetworkMiner application implemented behind the web interface.
A case study with differential gene expression: sub-networks activated or deactivated in FA
FA is a rare inherited disease complicated by aplastic anemia. There is evidence that hematopoietic stem cells have lost self-replicative capacity and undergo apoptosis when exposed to inhibitory cytokines including interferon gamma and tumor necrosis factor-alpha (71). Moreover, there is a known susceptibility to leukemia in FA patients (72). A recent study uses gene expression microarrays to identify differences at the transcription level between bone marrow cells from normal volunteers and from children and adults with FA (73). FA patients were identified using mitomycin C and/or diepoxybutane chromosomal breakage analysis. Eleven normal volunteers and 21 patients were studied.
Gene expression datasets for FA were obtained from the GEO database (see Materials and Methods for details). In both the cases, the most differentially expressed genes in FA when compared with cases and vice versa were analyzed, respectively. Since the statistic accounts for differential expression high and low values of the rank list account for gene more expressed in cases and in controls, respectively. Thus, the analysis has been done twice, in both extremes of the list.
In order to discard any possible influence of the normalization method in the results, we have used both the default settings obtained from GEO and the RMA normalization method. The results obtained were the same (data not shown). The network analysis allowed the introduction of one intermediate node connecting any two nodes in the network to increase the capability for discovery of whole functional modules with nodes not differentially expressed.
Figure 3A shows the sub-network found among the genes activated in FA. A total of 44 highly expressed genes are densely connected to a big network component, comprising 560 proteins (P = 0.0015), many of them involved in the spliceosome and the ribosome. A simpler network is obtained (P = 0.041) in the case of genes downregulated in FA (Figure 3B), and the 30 genes belonged to up to 54 different KEGG pathways (see Supplementary Table 1) that include, among other cancer pathways, Chronic myeloid leukemia, which is particularly relevant given the known link between FA and several cancers (74) and specifically with leukemia (72).
Figure 3.

Sub-network found among the genes in a differential expression experiment that compares FA patients to controls. (A) Genes up-regulated in FA. A total of 44 of 55 highly expressed genes are densely connected to a big network component (P = 0.0015), comprising 560 proteins. These genes are related to ribosome and spliceosome processes. (B) A total of 24 of 30 genes significantly down-regulated in FA (P = 0.041), connected by 70 intermediate proteins conforms the network which is differentially more expressed in controls than in FA. As mentioned in the text, some of these genes are related to the mTOR signalling pathway (in green) and to the Chronic myeloid leukaemia pathway (in red).
Functional modules, when composed by physically connected proteins, are easily detected by network analysis. However, differential gene expression data reveal only that part of the module which shows a different behavior among the conditions studied. In this way, experiments of differential gene expression provide only an incomplete description of the functionalities operating in the compared conditions. The advantage of the approach proposed here that allows the introduction of extra nodes not differentially expressed (in many cases because these are expressed in both conditions) reveals a picture much closer to the real functional modules operating in the conditions compared.
A case study with GWAS: sub-networks associated to bipolar disorder
Anonymous genotypic data from the WTCCC (51) were downloaded. A total of 2000 Caucasian UK patients of bipolar disorders and 1500 controls genotyped on the Affymetrix 500 K mapping array were studied. GWAS was performed using the basic association test of Plink toolset (52), based on comparing allele frequencies between cases and control. The association study rendered a list of SNPs ranked by P value. Following a strategy similar to PBA (14,29), we filter this list retaining SNPs mapping within or in the neighborhood of genes. Then, we filter the list again leaving only one SNP per gene. The SNP retained is the one with the smaller (most significant) P value, which is finally converted to the ID of the corresponding gene. This ID conversion process is identical to the performed by similar functional-based approaches such as PBA (29). Thus, a list of genes ranked by the best P value of one of their markers is obtained. This list is used by NetworkMiner, which looks for the significant sub-networks associated to the lowest P values of the association test, i.e. sub-networks associated to the bipolar disorder. The network analysis was performed allowing an intermediate node in the MCN. Genes previously known to be associated with bipolar disorder obtained from Uniprot database (53) were used as seed genes (see Materials and Methods).
Figure 4 shows the network significantly associated to bipolar disorder (P = 0.028), which includes 11 genes highly associated to the disease and 12 additional genes connected to them. One of the genes already known to be associated to the disease, FXYD6 (75), belongs to the network found. The network is enriched in genes belonging to four GO biological processes, one of them significant, learning (P = 0.0364), and the three others cognition, nervous system development and, specifically, nerve growth factor receptor signaling pathway marginally significant. All these processes are likely to be associated to the bipolar disorder.
Figure 4.

Sub-network found among the genes most associated to bipolar disorder in a GWAS. A total of 11 genes highly associated to the disease, in addition to 12 intermediate proteins, are significantly connected (P = 0.028). Subcellular location of the genes is displayed by the cell layout used by the NetworkMiner software, based on GO subcellular locations.
This example is the typical case of a GWAS of a common disease where clear associations are not found mainly because heritability of complex traits is due to multiple genes of small effect size (76). None of these small effect genes will obtain a significant value in a gene-based test, but all of them will have simultaneously a low P value and consequently will be closer to the top side of the ranked list. If these genes are part of an interacting network, then network analysis methodologies will discover them as collectively associated to the disease through their connections.
Sub-networks that characterize essential genes in cancer
Beyond the conventional genome-wide studies on gene expression or gene association, any other parameter than can be measured for lists of genes that account for any interesting biological property can be used to find gene networks associated to this parameter. A very interesting example is provided by a recent study of the essentiality of genes in different cancer cells (54). As commented in the methods section, the genes were ranked according to the RIGER score provided by the authors, which, basically, accounts for gene essentiality. As can be seen in Table 1, in almost all of the cell lines, the most essential genes configure significant sub-networks. These results demonstrate that essential genes in different cancer cell lines are close in the interactome (77). These sub-networks are probably depicting signaling pathways and molecular complexes that are essential for the phenotype of cancer lines (31).
Table 1.
MCNs found in the different cell lines with the corresponding size and P value
| Cell lines | Size of MCN | P value | Score |
|---|---|---|---|
| H82 | 181 | <0.001 | 0.064 |
| LN229 | 16 | <0.001 | 0.346 |
| REH | 25 | <0.001 | 0.225 |
| K562 | 19 | 0.003 | 0.173 |
| H1650 | 45 | 0.016 | 0.067 |
| H187 | 14 | 0.018 | 0.119 |
| H1975 | 144 | 0.02 | 0.042 |
| SUPT1 | 155 | 0.039 | 0.036 |
| JURKAT | 66 | 0.049 | 0.043 |
| A549 | 192 | 0.076 | 0.032 |
| HCC827 | 14 | 0.1 | 0.0615 |
| U251 | 110 | 0.241 | 0.028 |
A significant network (P < 0.05) could not be found for the three last cell lines (in italics).
As previously observed (78,79), the essential sub-networks are enriched in a number of molecular pathways that include mRNA processing and splicing, translation and cell cycle regulation (see Supplementary Table 2). Although some general functions are common for all the sub-networks identified here, each of them also shows enrichment in specific functionalities. Just to mention an example: while essential sub-network in Jurkat, a human leukemic helper T-lymphocyte line, is enriched in pathways related to cell death such as apoptosis and programmed cell death regulation, the K562 sub-network is enriched in mRNA processing and splicing process. This is in accordance with the fact that Jurkat is highly dependent on the CD95 (Fas/APO-1)-induced apoptosis pathway (80). In contrast, the characteristic sub-network of essential genes found in K562 sub-network bears a mutated gene for the p53 protein and an abnormal fusion gene BCR-ABL. As a result, the pro-apoptotic regulatory function is annulled and cells are resistant to drug-induced apoptosis, which may explain that apoptosis pathway elements are not essential for K562 survival (81,82). A recent study has demonstrated that the transcription regulatory machinery is highly active in K562 and that the alteration of such process causes a decrease in cell growth (83). Much more relevant information can be extracted from the detailed analysis of the essentiality networks but is beyond the aim of this work. Figure 5 shows the significant networks obtained for both cell lines, Jurkat and K562.
Figure 5.

Significant sub-networks (see also Table 1) found among the lists of most essential genes, ranked by the RIGER parameter, obtained for both cell lines: (A) Jurkat and (B) K562. Genes are colored according to their main functions (KEGG terms): pale blue: connecting protein; blue: MAPK signaling pathway; green: ribosome; yellow: proteasome; pink: cell cycle; magenta: apoptosis; orange: spliceosome and pale green: RIG-I-like receptor signaling pathway.
Supplementary Table 2 shows the number of genes corresponding to different KEGG pathways that appear connected within the essential gene networks in the different cell lines. Pathways like Ribosome and Spliceosome are quite common since the corresponding networks are significantly enriched in genes belonging to these. The same can be said of other pathways relevant in cancer such as MAPK signaling pathway, Toll-like receptor signaling pathway, NOD-like receptor signaling pathway and RIG-I-like receptor signaling pathway. Obviously, smaller networks result in less significant enrichments but, in general, most of the pathways in Supplementary Table 2 seem to be shared by the different essential networks in the different cell lines, despite the genes involved in the different networks are not the same. This fact suggests that in different cancer cell lines the same processes triggering cancer are acting in different ways, through different genes, to produce different flavors of the same cancer hallmarks (84). Supplementary Figure 1 shows the different networks found for the cell lines analyzed.
CONCLUSIONS
Here, we propose a simple but powerful approach able to find the sub-network component associated to extreme values of a list of genes (or proteins) ranked by any criterion (differential gene expression, disease association in a GWAS, etc.). Contrarily to other methods, which need to optimize a number of parameters in a way that is sensitive to initial conditions, our proposal has only one parameter which is the value used to rank the list of genes. This rank is not restricted to a particular biological property (e.g. differential gene expression, gene association to a disease or trait) and can consequently be applied in a large variety of experimental or theoretical scenarios. An exhaustive analysis for finding the combination of parameter and test that best distinguishes between a real network and a random network has been performed. The results point to the average number of nodes per component of the MCN as the most sensitive parameter to discover real networks and distinguish them from random networks. An advantage over almost all similar approaches is that this approach allows the inclusion of molecules originally not in the portion of the list analyzed that connect molecules in the subset analyzed. This provides a more realistic and efficient approach to real scenarios where not all the proteins involved in the network are in the portion of the list analyzed either because of experimental sensitivity or because the experiment does not allow to target them (e.g. a differential expression experiment will not point on genes relevant but expressed in both of the conditions compared).
As part of our efforts to provide the scientific community with user-friendly web tools, a web server to run this test, named NetworkMiner, is freely available within the Babelomics (47) environment. This web interface and visualization tool was implemented using the latest web standard HTML5. This new standard allows implementing advanced SVG graphics directly on the web browser without the necessity of installing any plug-in or Adobe Flash or using any Java applet. Moreover, HTLM5 standard is extensively supported by most of the modern web browsers.
The web server offers a large number of options for the graphical representation of the network, easily customizable, including different layouts for the network and backgrounds, within the context of the range of graphical possibilities of HTML5. The program is connected to other Babelomics tools such as FatiGO (5), which allows studying whether there is a functional enrichment analysis in the MCN based on GO terms, KEGG or Reactome pathways.
The approach has been applied in several case examples that illustrate the power of this methodology to uncover the network component contained in different groups of genes selected by different experiments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1 and 2 and Supplementary Figure 1.
FUNDING
The Spanish Ministry of Science and Innovation [BIO2011-27069]; the GVA-FEDER [PROMETEO/2010/001]; The CIBER de Enfermedades Raras is an initiative of the Instituto de Salud Carlos III (ISCIII), Ministry of Economy and Competitiveness (MINECO); the MINECO [PFIS FI10/00020 to L.G.-A.]. Funding for open access charge: The Spanish Ministry of Science and Innovation [BIO2011-27069].
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank the WTCCC for providing us with the bipolar disorder genotyping samples as well as the corresponding controls.
REFERENCES
- 1.Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402:C47–C52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- 2.Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 3.Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- 4.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Shahrour F, Diaz-Uriarte R, Dopazo J. FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics. 2004;20:578–580. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
- 6.Draghici S, Khatri P, Bhavsar P, Shah A, Krawetz SA, Tainsky MA. Onto-Tools, the toolkit of the modern biologist: Onto-Express, Onto-Compare, Onto-Design and Onto-Translate. Nucleic Acids Res. 2003;31:3775–3781. doi: 10.1093/nar/gkg624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khatri P, Draghici S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 2005;21:3587–3595. doi: 10.1093/bioinformatics/bti565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 10.Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, et al. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003; 4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dopazo J. Functional interpretation of microarray experiments. Omics. 2006;10:398–410. doi: 10.1089/omi.2006.10.398. [DOI] [PubMed] [Google Scholar]
- 12.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 13.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medina I, Montaner D, Bonifaci N, Pujana MA, Carbonell J, Tarraga J, Al-Shahrour F, Dopazo J. Gene set-based analysis of polymorphisms: finding pathways or biological processes associated to traits in genome-wide association studies. Nucleic Acids Res. 2009;37:W340–W344. doi: 10.1093/nar/gkp481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goeman JJ, Buhlmann P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics. 2007;23:980–987. doi: 10.1093/bioinformatics/btm051. [DOI] [PubMed] [Google Scholar]
- 16.Nam D, Kim SY. Gene-set approach for expression pattern analysis. Brief Bioinform. 2008;9:189–197. doi: 10.1093/bib/bbn001. [DOI] [PubMed] [Google Scholar]
- 17.Dopazo J. Formulating and testing hypotheses in functional genomics. Artif. Intell. Med. 2009;45:97–107. doi: 10.1016/j.artmed.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Minguez P, Dopazo J. Functional genomics and networks: new approaches in the extraction of complex gene modules. Expert Rev. Proteomics. 2010;7:55–63. doi: 10.1586/epr.09.103. [DOI] [PubMed] [Google Scholar]
- 19.Ideker T, Sharan R. Protein networks in disease. Genome Res. 2008;18:644–652. doi: 10.1101/gr.071852.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dittrich MT, Klau GW, Rosenwald A, Dandekar T, Muller T. Identifying functional modules in protein-protein interaction networks: an integrated exact approach. Bioinformatics. 2008;24:i223–i231. doi: 10.1093/bioinformatics/btn161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minguez P, Dopazo J. Assessing the biological significance of gene expression signatures and co-expression modules by studying their network properties. PLoS One. 2011;6:e17474. doi: 10.1371/journal.pone.0017474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jansen R, Greenbaum D, Gerstein M. Relating whole-genome expression data with protein-protein interactions. Genome Res. 2002;12:37–46. doi: 10.1101/gr.205602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ge H, Liu Z, Church GM, Vidal M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat. Genet. 2001;29:482–486. doi: 10.1038/ng776. [DOI] [PubMed] [Google Scholar]
- 24.Brown MP, Grundy WN, Lin D, Cristianini N, Sugnet CW, Furey TS, Ares M, Jr, Haussler D. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc. Natl. Acad. Sci. U. S. A. 2000;97:262–267. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mateos A, Dopazo J, Jansen R, Tu Y, Gerstein M, Stolovitzky G. Systematic learning of gene functional classes from DNA array expression data by using multilayer perceptrons. Genome Res. 2002;12:1703–1715. doi: 10.1101/gr.192502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vazquez A, Flammini A, Maritan A, Vespignani A. Global protein function prediction from protein-protein interaction networks. Nat. Biotechnol. 2003;21:697–700. doi: 10.1038/nbt825. [DOI] [PubMed] [Google Scholar]
- 27.Deng M, Tu Z, Sun F, Chen T. Mapping Gene Ontology to proteins based on protein-protein interaction data. Bioinformatics. 2004;20:895–902. doi: 10.1093/bioinformatics/btg500. [DOI] [PubMed] [Google Scholar]
- 28.Hannum G, Srivas R, Guenole A, van Attikum H, Krogan NJ, Karp RM, Ideker T. Genome-wide association data reveal a global map of genetic interactions among protein complexes. PLoS Genet. 2009;5:e1000782. doi: 10.1371/journal.pgen.1000782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang K, Li M, Hakonarson H. Analysing biological pathways in genome-wide association studies. Nat. Rev. Genet. 2010;11:843–854. doi: 10.1038/nrg2884. [DOI] [PubMed] [Google Scholar]
- 30.Jia P, Zheng S, Long J, Zheng W, Zhao Z. dmGWAS: dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics. 2011;27:95–102. doi: 10.1093/bioinformatics/btq615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pujana MA, Han JD, Starita LM, Stevens KN, Tewari M, Ahn JS, Rennert G, Moreno V, Kirchhoff T, Gold B, et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat. Genet. 2007;39:1338–1349. doi: 10.1038/ng.2007.2. [DOI] [PubMed] [Google Scholar]
- 32.Nibbe RK, Koyuturk M, Chance MR. An integrative -omics approach to identify functional sub-networks in human colorectal cancer. PLoS Comput. Biol. 2010;6:e1000639. doi: 10.1371/journal.pcbi.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oti M, Snel B, Huynen MA, Brunner HG. Predicting disease genes using protein-protein interactions. J. Med. Genet. 2006;43:691–698. doi: 10.1136/jmg.2006.041376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franke L, van Bakel H, Fokkens L, de Jong ED, Egmont-Petersen M, Wijmenga C. Reconstruction of a functional human gene network, with an application for prioritizing positional candidate genes. Am. J. Hum. Genet. 2006;78:1011–1025. doi: 10.1086/504300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lage K, Karlberg EO, Storling ZM, Olason PI, Pedersen AG, Rigina O, Hinsby AM, Tumer Z, Pociot F, Tommerup N, et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007;25:309–316. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- 36.Vidal M, Cusick ME, Barabasi AL. Interactome networks and human disease. Cell. 2011;144:986–998. doi: 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ideker T, Ozier O, Schwikowski B, Siegel AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18(Suppl. 1):S233–S240. doi: 10.1093/bioinformatics/18.suppl_1.s233. [DOI] [PubMed] [Google Scholar]
- 38.Ulitsky I, Krishnamurthy A, Karp RM, Shamir R. DEGAS: de novo discovery of dysregulated pathways in human diseases. PLoS One. 2010;5:e13367. doi: 10.1371/journal.pone.0013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nacu S, Critchley-Thorne R, Lee P, Holmes S. Gene expression network analysis and applications to immunology. Bioinformatics. 2007;23:850–858. doi: 10.1093/bioinformatics/btm019. [DOI] [PubMed] [Google Scholar]
- 40.Guo Z, Li Y, Gong X, Yao C, Ma W, Wang D, Li Y, Zhu J, Zhang M, Yang D, et al. Edge-based scoring and searching method for identifying condition-responsive protein protein interaction sub-network. Bioinformatics. 2007;23:2121–2128. doi: 10.1093/bioinformatics/btm294. [DOI] [PubMed] [Google Scholar]
- 41.Ma H, Schadt EE, Kaplan LM, Zhao H. COSINE: COndition-SpecIfic sub-NEtwork identification using a global optimization method. Bioinformatics. 2011;27:1290–1298. doi: 10.1093/bioinformatics/btr136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minguez P, Gotz S, Montaner D, Al-Shahrour F, Dopazo J. SNOW, a web-based tool for the statistical analysis of protein-protein interaction networks. Nucleic Acids Res. 2009;37:W109–W114. doi: 10.1093/nar/gkp402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu J, Vallenius T, Ovaska K, Westermarck J, Makela TP, Hautaniemi S. Integrated network analysis platform for protein-protein interactions. Nat. Methods. 2009;6:75–77. doi: 10.1038/nmeth.1282. [DOI] [PubMed] [Google Scholar]
- 44.Brohee S, Faust K, Lima-Mendez G, Sand O, Janky R, Vanderstocken G, Deville Y, van Helden J. NeAT: a toolbox for the analysis of biological networks, clusters, classes and pathways. Nucleic Acids Res. 2008;36:W444–W451. doi: 10.1093/nar/gkn336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sama IE, Huynen MA. Measuring the physical cohesiveness of proteins using physical interaction enrichment. Bioinformatics. 2010;26:2737–2743. doi: 10.1093/bioinformatics/btq474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rho K, Kim B, Jang Y, Lee S, Bae T, Seo J, Seo C, Lee J, Kang H, Yu U, et al. GARNET—gene set analysis with exploration of annotation relations. BMC Bioinformatics. 2011;12(Suppl. 1):S25. doi: 10.1186/1471-2105-12-S1-S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Medina I, Carbonell J, Pulido L, Madeira SC, Goetz S, Conesa A, Tarraga J, Pascual-Montano A, Nogales-Cadenas R, Santoyo J, et al. Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res. 2010;38:W210–W213. doi: 10.1093/nar/gkq388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smyth G. Linear Models for Microarray Data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 49.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 51.WTCCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.UNIPROT_Consortium. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38:D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, Hinkle G, Boehm JS, Beroukhim R, Weir BA, et al. Highly parallel identification of essential genes in cancer cells. Proc Natl. Acad. Sci. U. S. A. 2008;105:20380–20385. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ooi HS, Schneider G, Chan YL, Lim TT, Eisenhaber B, Eisenhaber F. Databases of protein-protein interactions and complexes. Methods Mol. Biol. 2010;609:145–159. doi: 10.1007/978-1-60327-241-4_9. [DOI] [PubMed] [Google Scholar]
- 56.Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, Derow C, Feuermann M, Ghanbarian AT, Kerrien S, Khadake J, et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010;38:D525–D531. doi: 10.1093/nar/gkp878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ceol A, Chatr Aryamontri A, Licata L, Peluso D, Briganti L, Perfetto L, Castagnoli L, Cesareni G. MINT, the molecular interaction database: 2009 update. Nucleic Acids Res. 2010;38:D532–D539. doi: 10.1093/nar/gkp983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stark C, Breitkreutz BJ, Chatr-Aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Van Auken K, Wang X, Shi X, et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011;39:D698–D704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.UniProt_Consortium. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2011;39:D214–D219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.von Mering C, Krause R, Snel B, Cornell M, Oliver SG, Fields S, Bork P. Comparative assessment of large-scale data sets of protein-protein interactions. Nature. 2002;417:399–403. doi: 10.1038/nature750. [DOI] [PubMed] [Google Scholar]
- 61.Minguez P, Dopazo J. Protein Interactions for Functional Genomics. In: Li X-L, Ng S-K, editors. Biological Data Mining in Protein Interaction Networks. Hershey, PA: IGI Global; 2009. 17033, EE.UU, pp. 223–238. [Google Scholar]
- 62.Kerrien S, Orchard S, Montecchi-Palazzi L, Aranda B, Quinn AF, Vinod N, Bader GD, Xenarios I, Wojcik J, Sherman D, et al. Broadening the horizon—level 2.5 of the HUPO-PSI format for molecular interactions. BMC Biol. 2007;5:44. doi: 10.1186/1741-7007-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dijkstra E. A note on two problems in connexion with graphs. Numerische Mathematik. 1959;1:269–271. [Google Scholar]
- 64.Wilkinson DM, Huberman BA. A method for finding communities of related genes. Proc. Natl. Acad. Sci. USA. 2004;101(Suppl. 1):5241–5248. doi: 10.1073/pnas.0307740100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Watts DJ, Strogatz SH. Collective dynamics of ‘small-world' networks. Nature. 1998;393:440–442. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- 66.Martinez-Camblor P, De Una-Alvarez J, Corral N. k-Sample test based on the common area of kernel density estimators. J. Stat. Plann. Infer. 2008;138:4006–4020. [Google Scholar]
- 67.Han JD, Bertin N, Hao T, Goldberg DS, Berriz GF, Zhang LV, Dupuy D, Walhout AJ, Cusick ME, Roth FP, et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature. 2004;430:88–93. doi: 10.1038/nature02555. [DOI] [PubMed] [Google Scholar]
- 68.Shachar R, Ungar L, Kupiec M, Ruppin E, Sharan R. A systems-level approach to mapping the telomere length maintenance gene circuitry. Mol. Syst. Biol. 2008;4:172. doi: 10.1038/msb.2008.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roguev A, Bandyopadhyay S, Zofall M, Zhang K, Fischer T, Collins SR, Qu H, Shales M, Park HO, Hayles J, et al. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science. 2008;322:405–410. doi: 10.1126/science.1162609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Taniguchi T, D'Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 2006;107:4223–4233. doi: 10.1182/blood-2005-10-4240. [DOI] [PubMed] [Google Scholar]
- 72.Tischkowitz M, Dokal I. Fanconi anaemia and leukaemia—clinical and molecular aspects. Br. J. Haematol. 2004;126:176–191. doi: 10.1111/j.1365-2141.2004.05023.x. [DOI] [PubMed] [Google Scholar]
- 73.Vanderwerf SM, Svahn J, Olson S, Rathbun RK, Harrington C, Yates J, Keeble W, Anderson DC, Anur P, Pereira NF, et al. TLR8-dependent TNF-(alpha) overexpression in Fanconi anemia group C cells. Blood. 2009;114:5290–5298. doi: 10.1182/blood-2009-05-222414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood. 2003;101:822–826. doi: 10.1182/blood-2002-05-1498. [DOI] [PubMed] [Google Scholar]
- 75.Choudhury K, McQuillin A, Puri V, Pimm J, Datta S, Thirumalai S, Krasucki R, Lawrence J, Bass NJ, Quested D, et al. A genetic association study of chromosome 11q22-24 in two different samples implicates the FXYD6 gene, encoding phosphohippolin, in susceptibility to schizophrenia. Am. J. Hum. Genet. 2007;80:664–672. doi: 10.1086/513475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Plomin R, Haworth CM, Davis OS. Common disorders are quantitative traits. Nat. Rev. Genet. 2009;10:872–878. doi: 10.1038/nrg2670. [DOI] [PubMed] [Google Scholar]
- 77.Hernandez P, Huerta-Cepas J, Montaner D, Al-Shahrour F, Valls J, Gomez L, Capella G, Dopazo J, Pujana MA. Evidence for systems-level molecular mechanisms of tumorigenesis. BMC Genomics. 2007;8:185. doi: 10.1186/1471-2164-8-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zotenko E, Mestre J, O'Leary DP, Przytycka TM. Why do hubs in the yeast protein interaction network tend to be essential: reexamining the connection between the network topology and essentiality. PLoS Comput. Biol. 2008;4:e1000140. doi: 10.1371/journal.pcbi.1000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hart GT, Lee I, Marcotte ER. A high-accuracy consensus map of yeast protein complexes reveals modular nature of gene essentiality. BMC Bioinformatics. 2007;8:236. doi: 10.1186/1471-2105-8-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thiede B, Kretschmer A, Rudel T. Quantitative proteome analysis of CD95 (Fas/Apo-1)-induced apoptosis by stable isotope labeling with amino acids in cell culture, 2-DE and MALDI-MS. Proteomics. 2006;6:614–622. doi: 10.1002/pmic.200500120. [DOI] [PubMed] [Google Scholar]
- 81.Law JC, Ritke MK, Yalowich JC, Leder GH, Ferrell RE. Mutational inactivation of the p53 gene in the human erythroid leukemic K562 cell line. Leuk. Res. 1993;17:1045–1050. doi: 10.1016/0145-2126(93)90161-d. [DOI] [PubMed] [Google Scholar]
- 82.McGahon A, Bissonnette R, Schmitt M, Cotter KM, Green DR, Cotter TG. BCR-ABL maintains resistance of chronic myelogenous leukemia cells to apoptotic cell death. Blood. 1994;83:1179–1187. [PubMed] [Google Scholar]
- 83.Addya S, Keller MA, Delgrosso K, Ponte CM, Vadigepalli R, Gonye GE, Surrey S. Erythroid-induced commitment of K562 cells results in clusters of differentially expressed genes enriched for specific transcription regulatory elements. Physiol. Genomics. 2004;19:117–130. doi: 10.1152/physiolgenomics.00028.2004. [DOI] [PubMed] [Google Scholar]
- 84.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]