


Abstract
Spliced alignment plays a central role in the precise identification of eukaryotic gene structures. Even though many spliced alignment programs have been developed, recent rapid progress in DNA sequencing technologies demands further improvements in software tools. Benchmarking algorithms under various conditions is an indispensable task for the development of better software; however, there is a dire lack of appropriate datasets usable for benchmarking spliced alignment programs. In this study, we have constructed two types of datasets: simulated sequence datasets and actual cross-species datasets. The datasets are designed to correspond to various real situations, i.e. divergent eukaryotic species, different types of reference sequences, and the wide divergence between query and target sequences. In addition, we have developed an extended version of our program Spaln, which incorporates two additional features to the scoring scheme of the original version, and examined this extended version, Spaln2, together with the original Spaln and other representative aligners based on our benchmark datasets. Although the effects of the modifications are not individually striking, Spaln2 is consistently most accurate and reasonably fast in most practical cases, especially for plants and fungi and for increasingly divergent pairs of target and query sequences.
INTRODUCTION
The central task in the annotation of eukaryotic genomes is to locate protein-coding and non-coding genes on the genomic sequence. For this purpose, several approaches are employed, including ab initio gene prediction methods, comparative genomic methods and evidence-based methods (1). Of these, the most accurate are the evidence-based methods that rely on known sequences of transcripts [complementary DNAs (cDNAs), expressed sequence tags (ESTs), or proteins] used as ‘reference’. This approach involves the alignment between the genomic sequence and cognate or homologous transcript sequences. In the alignment process, we ought to consider the possibility that introns can intervene between the exonic regions on the genome and hence such an alignment is often called ‘spliced alignment’ (2). Many spliced alignment programs have been developed so far, including EXALIN (3), Exonerate (4), GeneSeqer (5), GeneWise (6), GMAP (7), sim4 (8), Splign (9) and XAT (10), and new software continues to be developed, such as Pairagon (11), sim4cc (12) and genBlastG (13). Thus, we can now use a wide variety of spliced alignment programs besides those specialized for short reads (14,15), which are not generally applicable to long transcript sequences considered here and outside the scope of this investigation. However, it is often difficult for a non-specialist to choose the most appropriate ones for his/her specific problem. One solution to this problem is to objectively evaluate existing programs under various conditions with quality-controlled test datasets. Today, such benchmark tests prevail to compare the pros and cons of alternative algorithms and have greatly contributed to bioinformatics software development (16). For benchmark tests, we need sizable datasets used as ‘golden standards’. However, there are very few datasets publicly available for benchmarking spliced alignment programs under a variety of conditions, such as divergent species, different kinds of transcripts and various degrees of divergence between genomic and reference sequences. It is a prerequisite to develop high-quality reference datasets covering such a variety of conditions in order to not only evaluate existing programs, but also find the right direction for improving existing programs or designing new software.
Early spliced alignment algorithms (17,18) were formulated as an extension of the classical pairwise sequence alignment algorithm with long gaps (19) supplemented with the canonical GT-AG rule of splice junctions. To achieve more accurate gene recognition, more recent programs (5,6,20–22) tend to incorporate various lines of information that form the backbone of ab initio gene finding algorithms (23). Along this line, we have developed our own spliced alignment programs Aln (24) and Spaln (25,26). Spaln is more space-efficient and faster than Aln, but the target function to be optimized by the two programs is essentially the same. Meanwhile, the sequence signals involved in splice site recognition have been studied for many years (27). Whereas the basic process of splicing is conserved throughout eukaryotic species, most properties related to splice site recognition, such as nucleotide frequencies around splice junctions, length distribution of introns, strength of branch point (BP) signal, oligomer compositions within introns, etc. are species-specific (28–30). In a previous study (31), we have found that some properties that were not considered in Aln/Spaln have made significant contributions to splice site recognition in some species. For example, oligomer compositions within introns plus the BP signal may account for 10–20% of the total signals of short intron recognition in almost all species other than vertebrates. Hence, we are curious to know whether the fidelity of Spaln could be improved by the incorporation of these features into the scoring system.
In this article, we report the construction of a series of datasets used for benchmarking various spliced alignment algorithms under a variety of conditions. Based on the datasets named SPAliBASE (spliced alignment benchmark database), we compared the performance of the extended version of Spaln (Spaln2) with those of the original Spaln and other representative aligners. We found that although the effects of the modifications are not striking, they work well for increasingly divergent pairs of query and target sequences of plants and fungi, rendering Spaln2 the most accurate program in most practical situations.
MATERIALS AND METHODS
Preparation of simulated benchmark dataset
The complete genomic sequences of human (Build #36.3), Arabidopsis thaliana (accession nos. NC_003070, NC_003071, NC_003074, NC_003075 and NC_003076) and Neurospora crassa (accession nos. NC_001570, NW_001091935, … , NW_001092755) were downloaded from NCBI (http://www.ncbi.nlm.nih.gov/). Those species were chosen as representatives of the three major kingdoms of eukaryotes: animals, plants and fungi, respectively, as their genome sequences are of the highest quality in each kingdom or the largest number of transcript data is available. We developed three types of benchmark datasets for each species, i.e. cDNA sequence (cDNA dataset), protein coding sequence (CDS dataset) and protein sequence (protein dataset). Full length or partial cDNA sequences were downloaded from the ‘unique’ sets of UniGene database of NCBI (32), whereas CDS data were obtained from GenBank. We used both GMAP (7) and Spaln (25) to map and align those two types of nucleotide sequences to the genomic sequence of the corresponding species. Then, we collected only those sequences that exactly matched the genomic sequence (100% identities at the nucleotide level after splicing) with both GMAP and Spaln. As GMAP and Spaln employ substantially different strategies, the exon–intron structures identically inferred by the two programs will be highly reliable; hence, we regard them as the true gene organization corresponding to the transcript. For the evaluation of various programs and/or conditions, we used the genomic segment that covered the corresponding transcript with a margin of 10 kb before and after the ends of the exact alignment (Supplementary Figure S1). We defined the genomic segment and the transcript as ‘target’ and ‘query’, respectively. The protein datasets were obtained from CDS datasets by conceptual translation.
We randomly selected 1000 samples from the above collection for each species and query type. Then, we mutated the query sequences to various degrees by using a detailed sequence evolution simulator, indel-Seq-Gen version 2.1 (iSGv2.1) (33). The use of a simulator made it possible to finely control the sequence divergence between the query and the target. We considered that the mutation of query sequences while keeping the genomic sequence intact is more realistic than the opposite operation, as random mutations in the genomic sequence can destroy the intrinsic gene properties within the genomic sequence. In particular, the real outcome of a change in critical sites for splicing would be generally unexpected. iSGv2.1 can simulate the evolution of multi-partitioned nucleotide or codon sequences through the processes of insertion, deletion and substitution in continuous time. We used ‘nucleotide substitution models’ for cDNA dataset and ‘codon substitution models’ for CDS dataset. In this experiment, we mutated each original query sequence to six degrees of evolutionary changes (denoted by D0 to D5), resulting in final nucleotide identities of 100% to ∼70% (Table 1).
Table 1.
Details of simulated datasets
| Species and transcript type | Human |
Arabidopsis thaliana |
Neurospora crassa |
||||
|---|---|---|---|---|---|---|---|
| cDNA | CDS | cDNA | CDS | cDNA | CDS | ||
| Downloaded sequences | 32 661 | 37 337 | 30 576 | 33 200 | 17 096 | 10 038 | |
| GMAP: aligned sequences (100% identity) | 25 280 | 28 285 | 21 852 | 31 422 | 4376 | 9784 | |
| Spaln: aligned sequences (100% identity) | 24 518 | 29 030 | 20 714 | 31 703 | 4131 | 9663 | |
| Aligned sequences (100% identity) | 17 870 | 19 369 | 15 442 | 28 074 | 1994 | 8802 | |
| Percentage identity between transcript sequence and genomic sequence (%) | D0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
| D1 | 92.9 | 92.6 | 92.9 | 92.6 | 93.0 | 92.6 | |
| D2 | 86.8 | 86.4 | 86.7 | 86.4 | 86.7 | 86.4 | |
| D3 | 81.0 | 81.2 | 81.1 | 81.2 | 81.1 | 81.1 | |
| D4 | 76.1 | 76.8 | 76.0 | 76.9 | 76.0 | 76.8 | |
| D5 | 71.6 | 73.1 | 71.8 | 73.2 | 71.7 | 73.1 | |
We also prepared another dataset (RefSeq human cDNA dataset) independent of the above procedure that relies on particular aligners (GMAP and Spaln), i.e. we randomly chose 1000 human RefSeq entries (34), obtained cDNA/genomic segment pairs according to their annotations and sequences, and then generated a series of mutated cDNA datasets in the same way as described above.
Preparation of cross-species benchmark dataset
For the cross-species benchmark test, we regarded the above three species, human, A. thaliana and N. crassa, as the main species, where the reference datasets are the same as the CDS datasets described above. To collect test sequences, we downloaded CDSs of a few related species from GenBank and aligned them to their respective genome sequences by Spaln. We retained only the CDSs that aligned to respective genome sequences without any ordinary (non-intronic) gap to ensure the quality of the CDSs. Next, we searched for CDS pairs that are putatively orthologous between the reference and the test species by means of the reciprocal-best-BLAST-hits approach (34). We then examined if each putative orthologous pair represents true orthologs or different isoforms derived from orthologous genes, on the basis of their global alignment generated by Aln (19) with a double-affine gap penalty function. In practice, we retained CDS pairs that fulfilled the following two conditions. (i) The alignment fully ranges from the start codon to the stop codon of both transcripts; (ii) the alignment contains no gap longer than 15 bp. The target sequence is the genomic segment of the main species and the query sequence is the CDS of the other species, thus selected. The protein sequence was obtained by the conceptual translation of the CDS. Details of the cross-species data are shown in Table 2.
Table 2.
Details of cross-species datasets
| Reference transcript species | Human |
Arabidopsis thaliana |
Neurospora crassa |
||||
|---|---|---|---|---|---|---|---|
| Mouse | Chicken | Arabidopsis lyrata | Poplar | Rice | Gibberella zeae | Magnaporthe grisea | |
| Downloaded sequences | 34 950 | 16 754 | 8226 | 2281 | 23 313 | 11 558 | 1166 |
| Sequences mapped on genome | 11 905 | 8095 | 7685 | 2228 | 22 087 | 4957 | 1041 |
| Putative orthologous pairs | 5046 | 2895 | 5909 | 552 | 4648 | 1013 | 192 |
| Orthologs without long gaps | 3003 | 1527 | 5089 | 415 | 3036 | 503 | 74 |
| Average identity (%) | 85.5 | 74.3 | 94.8 | 70.4 | 63.6 | 66.6 | 64.6 |
Extension of Spaln scoring scheme
We tried to incorporate two additional signals into the scoring scheme of Spaln (26). The first one is the oligomer composition preference within intron that is added to the objective function by modifying the intron penalty function. The second signal is BP score obtained with a simple 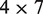 weight matrix (31). This score is added to the nearest downstream splicing acceptor signal as BP is the target for acceptor site recognition (35). Details of these extensions are described in Supplementary Methods. Use of these additional signals will be denoted by ‘TBZ’ option hereafter.
weight matrix (31). This score is added to the nearest downstream splicing acceptor signal as BP is the target for acceptor site recognition (35). Details of these extensions are described in Supplementary Methods. Use of these additional signals will be denoted by ‘TBZ’ option hereafter.
Other modifications
Besides the above-mentioned extension, Spaln2 has undergone a few modifications compared with the original version. First, a heuristic routine is added between the high scoring segment pair (HSP) search and the restricted dynamic programming (DP) routine called ‘Sandwich’ or ‘attack by both sides’ algorithm, when the projections to the query axis of two adjacent HSPs overlap. In such a case, juxtaposing 5′ and 3′ canonical splice site pairs are looked for within the overlapped region of the HSPs allowing no indel. If more than one candidate is found, the one with the largest 5′ + 3′ splicing signals is chosen. The DP routine is invoked only when this heuristic step fails. This type of heuristics is already used for the rapid identification of splice sites in sim4 (8), and XAT (10). Second, the code has been rewritten in C++ and is now compatible with multi-thread operation, ensuring a considerable increase in speed under a multi-core system. Concomitantly, several minor revisions are made concerning the recursive HSP search routines. Other modifications not directly relevant to the present study will be described in the document attached to the Spaln2 distribution.
Programs and parameters used for evaluation
We compared the performance of Spaln2 under various conditions and also to that of other aligners. The programs and the options used are summarized in Supplementary Table S1. In the present study, we focused on the alignment phase as the genome mapping phase is supported by only a few programs. Generally, the default parameters were used, but the ‘global’ option was set for GeneWise as our previous experiment (26) indicated that this option improved the overall performance of GeneWise. Moreover, if there was a specific flag for cross-species comparison, that parameter set was tested separately. The results of such a cross-species setting were distinguished from those of the default setting by the suffix ‘X’ (e.g. PairagonX versus Pairagon). Note that the default setting of Spaln2 is slightly different depending on the query type: both cross-species (–yX) and splice signal (–yS) switches are off for DNA, whereas both are on for protein. However, throughout the examination except for the data collection process through genome mapping, we always set the ‘–yS’ option even for DNA queries indicating full use of the species-specific splice signals around splice junctions. As the cross-species switch is set by default, the suffix ‘X’ was occasionally omitted for protein queries.
Throughout this article, we referred to only exon-level accuracy implying the harmonic mean of sensitivity and specificity, or ‘F-measure’, at the exon level. Other statistics, such as nucleotide-level, junction-level and gene-level sensitivities and specificities were generally well correlated with the exon-level accuracy. Non-parametric statistical tests were performed with the R package (http://www.R-project.org).
Run times were measured on a Linux machine with 3.47 GHz Intel® Xeon® (64bit CPU) with 24 GB memory. Although some programs including Spaln2 could be accelerated under a multi-core environment, all tests were performed with a single core mode.
RESULTS AND DISCUSSION
Benchmark datasets
The initial and one of the most important steps in the construction of a benchmark dataset is to collect reliable reference data used as golden standards. To construct the reference datasets, we adopted an automatic and high-throughput, but rather conservative approach based on the coincidence of the results of two independent programs (Materials and Methods). GMAP (7) and Spaln (25) could align almost all the downloaded transcript sequences to the corresponding genomic sequences of all the three species. We retained only those transcripts that exactly (100% at the nucleotide level) matched the genome sequence after conceptual splicing by both programs. This procedure also eliminates all low-quality sequences, including most ESTs. For human, e.g. the numbers of transcripts that satisfied this condition were 25 280 cDNAs and 28 285 CDSs with GMAP and 24 518 cDNAs and 29 030 CDSs with Spaln. Of these, 17 870 cDNAs and 19 369 CDSs were identically mapped and aligned by GMAP and Spaln, and were pooled for subsequent analyses. We randomly chose 1000 sequences from each class of transcripts thus selected and mutated the sequences to various degrees using iSGv2.1 (33). Table 1 presents the details of primary and simulated datasets.
One potential drawback of this procedure is that the dataset thus constructed may be biased toward ‘easy’ samples, because those samples that harbor short exons, minor-type or non-canonical splice junctions or repetitive exon structures are less likely to be identically aligned by independent programs than ‘ordinary’ samples that are devoid of such an irregularity. We considered this might be really the case. However, ordinary samples are more suitable than peculiar ones for the initial examination of the basic performance of various methods. It would be a future task to construct specialized datasets to study the behavior of each method under individual specific conditions.
Details of the cross-species benchmark dataset are summarized in Table 2. We used the CDS datasets mentioned above as the golden standard and the orthologous CDSs from related species as the queries. Ideally, the gene pair from the two species should be orthologous and the transcripts should be of the same isoform type. Using our operational procedure (Materials and Methods), we found more than 1000 putatively orthologous gene pairs for the majority of species pairs examined, although the size of the fungal data was considerably small. The average sequence identities between the orthologs for the seven comparisons varied from 60% to 95%. Although it was difficult to estimate the fraction of these gene pairs that strictly satisfied the above mentioned orthologous conditions, our datasets seemed to be superior to those obtained with HomoloGene (36) or the datasets of Cui et al. (10) in terms of both wider coverage and methodically controlled quality. Although it is feasible to construct datasets similar to those described here for some other species, e.g. Drosophila melanogaster and Caenorhabditis elegans, the benefits gained from such datasets would be limited at least for the present purpose.
We did not prepare cross-species cDNA datasets because it was difficult to define the range within which two cDNA sequences could be regarded as orthologous at the nucleotide level. The inclusion of foreign sequences could invoke uncontrollable confusion in the test procedure. As a CDS is delineated by a start codon and a stop codon, this problem is largely avoided. Another merit of using CDS is that we can directly evaluate the effects of translation.
Improvement in performance of Spaln
Spaln2 had undergone a few revisions (Materials and Methods) compared with the original version (25,26). We evaluated separately the effects of the basic revisions and the effects of the additional features. An example is shown in Figure 1 that exhibits the exon-level error rates of Spaln and Spaln2 with or without the ‘cross-species’ option tested on the simulated datasets of A. thaliana. The effects of the additional features were examined only for Spaln2. The results of the other species are shown in Supplementary Figure S2. Comparison of the results of the default settings of Spaln and Spaln2 indicated that the basic revisions only slightly affected the overall accuracy for human genes, but significantly increased the accuracy for A. thaliana and N. crassa genes with low-identity test sets (D4 and D5), particularly with the protein datasets. We considered that this improvement could be largely ascribed to the revised HSP routines, the most influential of which was the refinement of the method to delimit the sequences transferred to the lower level recursion. Spaln with ‘–yS –yX’ options produced some errors even with D0 datasets, whereas Spaln2 yielded little error. This improvement could be ascribed to the newly employed heuristic routine that bypassed the formal procedure to optimize the objective function (Equation 1 in Supplementary Methods), favoring sequence matches over signal strengths. We initially intended to introduce this hasty splice-site identification routine to speed up Spaln (Supplementary Table S2). Thus, it is rather fortuitous that the heuristics effectively reduced the errors with high-identity test sets. It is also noteworthy that the full-DP mode of Spaln2 (‘–Q0’ option) is not necessarily more accurate than the heuristic modes in spite of the much longer computation time (data not shown).
Figure 1.

Exon-level error rates of different versions or settings of Spaln tested with Arabidopsis thaliana cDNA (a), CDS (b) and protein (c) datasets. Statistically significant difference (P < 0.05 with the Wilcoxon signed-rank tests) between adjacent methods is marked by two asterisks.
The effects of the two additional features inferred from a comparison of the results of Spaln2 (or Spaln2X) with or without ‘TBZ’ options were rather limited. As mentioned earlier, our previous study (31) suggested that the oligomer composition in intron plus BP signal accounts for 10–20% of the total information contents of short-intron recognition in plants, fungi and protists, whereas they make negligible contributions to the intron recognition of vertebrate genes. Thus, it is not surprising that these features little affect the accuracy of Spaln2 tested with human genes. For plants and fungi, we were able to recognize the positive effects of these features in both low-identity simulated datasets (D4 and D5 in Supplementary Figures S1 and S2) and cross-species datasets (Figure 2), although they were not as remarkable as initially expected. This result is not necessarily disappointing because it suggests that we do not need to care much about the species specificity of parameter sets used by Spaln2. The relatively weak species dependence is an advantage of spliced alignment methods over ab initio or comparative genomic approaches for gene identification. From another point of view, our observation also indicates that we can expect maximal performance at little additional computational cost if we have once prepared such species-specific parameters. Such parameter sets are already available for 61 divergent eukaryotic species, although it is yet to be confirmed whether or not the number of supporting data in every species is sufficiently large to allow robust estimation of the parameters.
Figure 2.

Exon-level accuracies of spliced alignment programs tested on cross-species CDS (a) and protein (b) datasets. The genomic segments of human, Arabidopsis thaliana, and Neurospora crassa are used as target sequences of vertebrate, plant and fungal query sequences, respectively.
Performance evaluation of various aligners
We next used our benchmark datasets to evaluate the performance of Spaln2 with ‘TBZ’ options (denoted by Spaln2TBZ below) relative to that of the other aligners, EXALIN, Exonerate, GeneSeqer, GMAP, Pairagon, sim4, sim4cc, Splign and XAT with nucleotide datasets, and Exonerate, genBlastG, GeneSeqer, GeneWise and ProSplign with protein datasets. We also examined the performance of the default setting and the cross-species setting if the program supported such an option.
Results of simulated dataset
Figure 3 and Supplementary Figures S3 and S4, respectively present the results of the examinations with A. thaliana, human and N. crassa datasets for each of the three query types. The results clearly demonstrated that Spaln2TBZX outperformed the other aligners throughout all the species and query types for low-identity datasets (D3–D5). For high-identity datasets (D0–D2), the difference in performance of various aligners became smaller; nevertheless, a significant difference was occasionally recognized even at the D0 level as discussed later. The D0 level is somewhat special where GMAP is always 100% accurate because of our dataset construction procedure (‘Materials and Methods’). Closer inspections demonstrated that either Pairagon or Spaln2TBZ followed GMAP at the D0 level of cDNA or CDS datasets depending on the species and query type. At the D1 and D2 levels, Spaln2TBZ was the best performer in most cases, whereas Spaln2TBZX outperformed Spaln2TBZ at the D3–D5 levels. To confirm the fairness of our evaluation, we also performed an additional examination with another dataset (RefSeq human cDNA dataset) prepared independently of SPAliBASE (Materials and Methods). The results shown in Supplementary Figure S5 indicate that the above observations were highly reproducible regardless of the test datasets.
Figure 3.

Exon-level accuracies of various aligners tested with three kinds of Arabidopsis thaliana simulated datasets: (a) cDNA, (b) CDS and (c) protein. Panel (d) demonstrates the effects of translation on the performance of the same programs.
Spaln2TBZ performed best for all protein datasets except two; ProSplign was nearly 100% accurate for A. thaliana and N. crassa protein datasets at the D0 level where Spaln2TBZ made a few errors. Curiously, however, ProSplign worked poorly with human protein dataset at the D0 level (Supplementary Figure S4). In addition, the performance of ProSplign rapidly worsened with increasing divergence between the query and target sequences for all species.
Besides Spaln2, the programs that used full-blown DP or pair-hidden Markov model (HMM) (EXALIN, GeneSeqer and Pairagon) were generally more accurate than the others that adopted some heuristic acceleration procedures. However, none of the programs in the former group consistently outperformed others throughout the variety of situations. Of the latter group with a heuristic routine, sim4cc performed best in most situations, sometimes even outperforming the programs in the former group. Considering its high speed (see below), sim4cc was noticeably good in terms of cost performance for nucleotide queries. GeneSeqer that is specifically tuned for Arabidopsis genes worked quite well for Arabidopsis protein queries. However, its human counterpart was absurdly inaccurate when tested on human protein and CDS datasets. For protein queries, GeneWise showed the highest accuracy in most cases, whereas GeneSeqer and genBlastG performed similarly well for plants and fungi.
Exonerate, GeneSeqer and Spaln2 accept both nucleotide and protein sequences as queries. As our protein sequences were derived from the corresponding CDSs, we could directly compare the effects of translation. Panel (d) in Figure 3, Supplementary Figures S3 and S4 indicates that translation improved the accuracy in almost all situations except for the D0 level, where translation resulted in small decreases in accuracy with all programs.
Lu et al. (20) have performed the most comprehensive evaluation so far of various spliced alignment programs. In that study, they used two sets of test data: D. melanogaster CDSs and corresponding artificially mutated genomic sequences, and mammalian cross-species CDS-genome pairs. Their observations indicated that Pairagon performed best among a total of 12 aligners examined, including the previous version of Spaln and SpalnX. Using the same D. melanogaster test data as those used by Lu et al., we examined the performance of Spaln2TBZ and confirmed that Pairagon works slightly better than Spaln2TBZ except for the two lowest-identity levels where Spaln2TBZ or Spaln2TBZX were more accurate than Pairagon. We consider that this observation may be interpreted from three points of view. First, random mutations in the genomic sequence as done to create the D. melanogaster test data might render realistic performance evaluation difficult, because they can destroy the intrinsic gene properties within the genomic sequence. Second, the pair-HMM of Pairagon is trained with CDSs so that it will capture CDS-specific features, whereas the objective function of Spaln2 for DNA queries (Equation 1 in Supplementary Methods) accepts no such training. Finally, the D. melanogaster test data are richer than our datasets in short coding exons, particularly short terminal coding exons. As Spaln2 for DNA queries is ignorant about the codon architecture within CDS, it would be intrinsically weak in discerning short terminal coding exons.
Result of cross-species dataset
Figure 2 shows the results obtained from the cross-species CDS and protein datasets. Here, we compared the performances of Spaln2TBZX, SpalnX (older version) and eight other programs for CDS dataset, and five other programs for protein dataset. In this test, we also applied the ‘double affine’ option to both Spaln and Spaln2, as our previous studies (24,26) indicated that a double-affine gap penalty function worked better than the default single-affine gap penalty function for distant genome-transcript pairs.
The general trends of the results were the same as those observed with the simulated datasets for the two query types. Comparison of the results of the two versions of Spaln indicated that the revisions exerted only marginal effects on vertebrate and close target-query pairs, i.e. human–mouse (Wilcoxon signed-rank tests CDS: 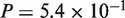 , protein:
, protein: 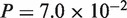 ), human–chicken (CDS:
), human–chicken (CDS: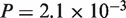 , protein:
, protein: 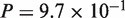 ), and A. thaliana–A. lyrata (CDS:
), and A. thaliana–A. lyrata (CDS: 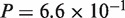 , protein:
, protein: 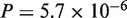 ), and an excessively large number of ties prohibited proper evaluation of P-value for the N. crassa–Magnaporthe grisea pair. In contrast, significant improvement in accuracy was observed for the other pairs, A. thaliana–poplar (CDS:
), and an excessively large number of ties prohibited proper evaluation of P-value for the N. crassa–Magnaporthe grisea pair. In contrast, significant improvement in accuracy was observed for the other pairs, A. thaliana–poplar (CDS: 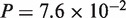 , protein:
, protein: 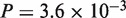 ), A. thaliana–rice (CDS:
), A. thaliana–rice (CDS: 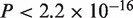 , protein:
, protein: 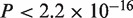 ), and N. crassa–Gibberella zeae (CDS:
), and N. crassa–Gibberella zeae (CDS: 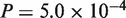 , protein:
, protein: 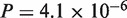 ). Thus, the revisions were confirmed to make significant contributions to accuracy improvement with the real datasets, as well as with the simulated datasets.
). Thus, the revisions were confirmed to make significant contributions to accuracy improvement with the real datasets, as well as with the simulated datasets.
Throughout all the cross-species datasets examined, Spaln2TBZX performed best followed by SpalnX, whereas the third best-performing program varied depending on the query type and the species pair. It also became clear that translation was effective for accurate exon recognition. Specifically, Spaln2TBZX yielded >80% average accuracies for all the cross-species pairs when examined with protein datasets, implying a maximal increase in accuracy of ∼40% compared with the CDS counterpart.
Computational time
Supplmentary Table S2 shows the computational times taken by the programs to analyze D0 level datasets and cross-species datasets. Sim4 and sim4cc are the fastest of all programs for DNA. Spaln2 belongs to the next fastest group together with Exonerate, GMAP, Splign and XAT. EXALIN, GeneSeqer and Pairagon are much slower than the second group. For protein queries, genBlastG and Spaln2TBZ are one to two orders of magnitude faster than Exonerate, ProSplign and GeneSeqer, whereas GeneWise is the slowest of all programs.
SUMMARY
To summarize our evaluation study, we combined the results of the three species for each of the six identity levels of simulated datasets or seven pairs of cross-species datasets. According to the accuracies averaged over the accumulated data, we ranked the 12 aligners including the older version of Spaln for cDNA and CDS or the seven aligners for protein, and then evaluated the significance of the difference in performance between succeeding aligners by means of Wilcoxon signed-rank tests (37). The results shown in Figure 4 and Supplementary Table S3 indicate that Spaln2 performs, at least equivalently, best among currently popular aligners in nearly all situations. A single exception is observed at the D2 level, where the older version of Spaln significantly outperformed Spaln2. The cross-species option is preferable in most cases, except for D0 and D1 levels. Because of the intrinsic nature of our datasets, GMAP ranked highest at the D0 level, but its performance abruptly worsened at the D1 level. Similar abrupt declines were observed in the rankings of Splign and GMAP tested on the RefSeq dataset (Supplementary Figure S5) that may rely on these two programs (34). These observations suggest that the bias toward the programs that were used for data construction rapidly damps out with increasing sequence divergence. Thus, Spaln or Splan2 with the default setting are most preferable at the D0 and D1 levels. A majority of the remaining errors originate from the very short coding exons, some of which may be recognized by time-consuming probability-based aligners, such as Pairagon and PALMA (22), or with the help of an external routine (38). Sim4cc might be preferred to Spaln2 for DNA queries if speed is essential. However, sim4cc significantly falls behind Spaln2 and some other aligners in terms of accuracy even at the D0 level.
Figure 4.

Ranking diagrams of various aligners. The 12 aligners for cDNA and CDS (a) or the seven aligners for protein (b) are ranked according to exon-level accuracies averaged over the three species for each of the six identity levels of simulated datasets or seven pairs of cross-species datasets. Succeeding aligners are connected by a shaded box if the difference in their performance is not significant (P ≥ 0.01) or by a shaded triangle if the difference is weakly significant (0.001 ≤ P < 0.01).
Spaln2 is particularly advantageous for cross-species genome–transcript pairs. By using protein queries, we can expect >97% exon-level accuracy of the coding part of a mammalian gene with a bird template and vice versa, and >95% exon-level accuracy for a monocot–dicot pair. Spaln2 is 1300-fold and 460-fold faster than the next best-performing competitors, Pairagon and GeneWise, for DNA and protein queries, respectively. In addition to the default output format that provides ample information about the predicted exons in a compact form, several other formats, such as GFF3 (http://www.sequenceontology.org/gff3.shtml) and BED (http://genome.ucsc.edu/FAQ/FAQformat.html#format1) are supported for convenience of subsequent processing. Spaln2 is one of the few existing programs that can perform mapping and alignment phases seamlessly with a single command, and the sole one that can do such a task for protein queries. Spaln2 is also quite memory efficient even if the mapping phase is included, and now supports parallel operations to deal with a large dataset. The benchmark datasets constructed here, the species-specific parameter sets for 61 divergent eukaryotic species, as well as the source codes of Spaln2 can be downloaded for free from http://www.genome.ist.i.kyoto-u.ac.jp/∼aln_user/.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–4, Supplementary Figures 1–7 and Supplementary Methods.
FUNDING
Kakenhi (Grant-in-Aid for Scientific Research) B [22310124]; Ministry of Education, Culture, Sports, Science and Technology of Japan. Funding for open access charge: National Institute of Advanced Industrial Science and Technology (AIST).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We would like to thank Dr D. Lu for providing the test data and the detailed results of the Pairagon project. We also thank Drs T. Yada, N. Ichinose and M. Suyama for valuable discussions.
REFERENCES
- 1.Brent MR. Steady progress and recent breakthroughs in the accuracy of automated genome annotation. Nat. Rev. Genet. 2008;9:62–73. doi: 10.1038/nrg2220. [DOI] [PubMed] [Google Scholar]
- 2.Gelfand MS, Mironov AA, Pevzner PA. Gene recognition via spliced sequence alignment. Proc. Natl Acad. Sci. USA. 1996;93:9061–9066. doi: 10.1073/pnas.93.17.9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang M, Gish W. Improved spliced alignment from an information theoretic approach. Bioinformatics. 2006;22:13–20. doi: 10.1093/bioinformatics/bti748. [DOI] [PubMed] [Google Scholar]
- 4.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Usuka J, Zhu W, Brendel V. Optimal spliced alignment of homologous cDNA to a genomic DNA template. Bioinformatics. 2000;16:203–211. doi: 10.1093/bioinformatics/16.3.203. [DOI] [PubMed] [Google Scholar]
- 6.Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 8.Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kapustin Y, Souvorov A, Tatusova T, Lipman D. Splign: algorithms for computing spliced alignments with identification of paralogs. Biol. Direct. 2008;3:20. doi: 10.1186/1745-6150-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui X, Vinar T, Brejova B, Shasha D, Li M. Homology search for genes. Bioinformatics. 2007;23:i97–i103. doi: 10.1093/bioinformatics/btm225. [DOI] [PubMed] [Google Scholar]
- 11.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou L, Pertea M, Delcher AL, Florea L. Sim4cc: a cross-species spliced alignment program. Nucleic Acids Res. 2009;37:e80. doi: 10.1093/nar/gkp319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.She R, Chu JS, Uyar B, Wang J, Wang K, Chen N. genBlastG: using BLAST searches to build homologous gene models. Bioinformatics. 2011;27:2141–2143. doi: 10.1093/bioinformatics/btr342. [DOI] [PubMed] [Google Scholar]
- 14.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Homer N. A survey of sequence alignment algorithms for next-generation sequencing. Brief. Bioinform. 2010;11:473–483. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aniba MR, Poch O, Thompson JD. Issues in bioinformatics benchmarking: the case study of multiple sequence alignment. Nucleic Acids Res. 2010;38:7353–7363. doi: 10.1093/nar/gkq625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang X, Zhang J. Methods for comparing a DNA sequence with a protein sequence. Comput. Appl. Biosci. 1996;12:497–506. doi: 10.1093/bioinformatics/12.6.497. [DOI] [PubMed] [Google Scholar]
- 18.Mott R. EST_GENOME: a program to align spliced DNA sequences to unspliced genomic DNA. Comput. Appl. Biosci. 1997;13:477–478. doi: 10.1093/bioinformatics/13.4.477. [DOI] [PubMed] [Google Scholar]
- 19.Gotoh O. Optimal sequence alignment allowing for long gaps. Bull. Math. Biol. 1990;52:359–373. doi: 10.1007/BF02458577. [DOI] [PubMed] [Google Scholar]
- 20.Lu DV, Brown RH, Arumugam M, Brent MR. Pairagon: a highly accurate, HMM-based cDNA-to-genome aligner. Bioinformatics. 2009;25:1587–1593. doi: 10.1093/bioinformatics/btp273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Nimwegen E, Paul N, Sheridan R, Zavolan M. SPA: a probabilistic algorithm for spliced alignment. PLoS Genet. 2006;2:e24. doi: 10.1371/journal.pgen.0020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulze U, Hepp B, Ong CS, Ratsch G. PALMA: mRNA to genome alignments using large margin algorithms. Bioinformatics. 2007;23:1892–1900. doi: 10.1093/bioinformatics/btm275. [DOI] [PubMed] [Google Scholar]
- 23.Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 24.Gotoh O. Homology-based gene structure prediction: simplified matching algorithm using a translated codon (tron) and improved accuracy by allowing for long gaps. Bioinformatics. 2000;16:190–202. doi: 10.1093/bioinformatics/16.3.190. [DOI] [PubMed] [Google Scholar]
- 25.Gotoh O. A space-efficient and accurate method for mapping and aligning cDNA sequences onto genomic sequence. Nucleic Acids Res. 2008a;36:2630–2638. doi: 10.1093/nar/gkn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gotoh O. Direct mapping and alignment of protein sequences onto genomic sequence. Bioinformatics. 2008b;24:2438–2444. doi: 10.1093/bioinformatics/btn460. [DOI] [PubMed] [Google Scholar]
- 27.Lim LP, Burge CB. A computational analysis of sequence features involved in recognition of short introns. Proc. Natl Acad. Sci. USA. 2001;98:11193–11198. doi: 10.1073/pnas.201407298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Senapathy P, Shapiro MB, Harris NL. Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods Enzymol. 1990;183:252–278. doi: 10.1016/0076-6879(90)83018-5. [DOI] [PubMed] [Google Scholar]
- 29.Sheth N, Roca X, Hastings ML, Roeder T, Krainer AR, Sachidanandam R. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res. 2006;34:3955–3967. doi: 10.1093/nar/gkl556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartz SH, Silva J, Burstein D, Pupko T, Eyras E, Ast G. Large-scale comparative analysis of splicing signals and their corresponding splicing factors in eukaryotes. Genome Res. 2008;18:88–103. doi: 10.1101/gr.6818908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwata H, Gotoh O. Comparative analysis of information contents relevant to recognition of introns in many species. BMC Genomics. 2011;12:45. doi: 10.1186/1471-2164-12-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pontius JU, Wagner L, Schuler GD. UniGene: A Unified View of the Transcriptome. Bethesda, MD: National Center for Biotechnology Information; 2003. [Google Scholar]
- 33.Strope CL, Abel K, Scott SD, Moriyama EN. Biological sequence simulation for testing complex evolutionary hypotheses: indel-Seq-Gen version 2.0. Mol. Biol. Evol. 2009;26:2581–2593. doi: 10.1093/molbev/msp174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 35.Patel AA, Steitz JA. Splicing double: insights from the second spliceosome. Nat. Rev. Mol. Cell Biol. 2003;4:960–970. doi: 10.1038/nrm1259. [DOI] [PubMed] [Google Scholar]
- 36.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2007;35:D5–D12. doi: 10.1093/nar/gkl1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehmann EL, D'abrera H. Nonparametrics: Statistical Methods Based on Ranks. San Francisco: Holden-Day; 1975. [Google Scholar]
- 38.Volfovsky N, Haas BJ, Salzberg SL. Computational discovery of internal micro-exons. Genome Res. 2003;13:1216–1221. doi: 10.1101/gr.677503. [DOI] [PMC free article] [PubMed] [Google Scholar]