


Abstract
Eukaryotic genomes are repetitively wrapped into nucleosomes that then regulate access of transcription and DNA repair complexes to DNA. The mechanisms that regulate extrinsic protein interactions within nucleosomes are unresolved. We demonstrate that modulation of the nucleosome unwrapping rate regulates protein binding within nucleosomes. Histone H3 acetyl-lysine 56 [H3(K56ac)] and DNA sequence within the nucleosome entry-exit region additively influence nucleosomal DNA accessibility by increasing the unwrapping rate without impacting rewrapping. These combined epigenetic and genetic factors influence transcription factor (TF) occupancy within the nucleosome by at least one order of magnitude and enhance nucleosome disassembly by the DNA mismatch repair complex, hMSH2–hMSH6. Our results combined with the observation that ∼30% of Saccharomyces cerevisiae TF-binding sites reside in the nucleosome entry–exit region suggest that modulation of nucleosome unwrapping is a mechanism for regulating transcription and DNA repair.
INTRODUCTION
Eukaryotic genomes are organized into repeats of nucleosomes, which contain 147 bp of DNA wrapped ∼1.65 times around an octamer of H2A, H2B, H3 and H4 histone proteins (1). Nucleosomes function to regulate DNA processing by sterically occluding transcription (2) and repair (3) complexes from nucleosomal DNA. External factors such as histone post-translational modifications (PTMs) (4), chromatin remodeling (5) and histone chaperones (6) appear to provide access to nucleosomal DNA. In addition, the inherent property of nucleosomes to spontaneously partially unwrap directly exposes buried DNA sites within the nucleosome (7–9). The equilibrium between the fully wrapped and partially wrapped nucleosome states is termed nucleosome site exposure, and conversion into a partially unwrapped nucleosome occurs many times per second (10,11). Site exposure provides DNA access for transcription factors (TFs) (8,12–15) and DNA repair complexes (16–18). However, the mechanisms that regulate nucleosome site exposure remain undetermined.
Histone PTMs (14,15,19,20) and the DNA sequence bound by the core histones (21–23) influence the nucleosome site exposure equilibrium. Histone H3 lysine 56 acetylation [H3(K56ac)], which helps regulate eukaryotic transcription, replication and repair (24–30), is located in the globular core of H3 near the nucleosome entry–exit region. Structural studies of the acetylation mimic H3(K56Q) observe minimal changes in the fully wrapped nucleosome structure (31), while replacing histone H3 with its variant, CENP-A, does significantly alter the DNA in the crystal structure (32). H3(K56ac) shifts the site exposure equilibrium toward partially unwrapped nucleosome states (20). This enhances DNA accessibility for TF binding within the nucleosome (14), while the acetyl-lysine mimic H3(K56Q) enhances nucleosome disassembly by the mismatch repair (MMR) recognition complex, hMSH2–hMSH6 (17). These results suggest that H3(K56ac) reduces DNA–histone interactions that enhance site exposure without altering the nucleosome structure in the fully wrapped state. Determining the mechanisms by which H3(K56ac) and other factors such as DNA sequence influence site exposure is central to understanding how transcription and DNA repair complexes gain access to nucleosomal DNA.
Here we examine the influence of H3(K56ac) and DNA sequence on the nucleosome unwrapping and rewrapping rates in order to determine the changes in nucleosome dynamics associated with changes in site exposure equilibrium. We find that H3(K56ac) and DNA sequence within the nucleosome entry–exit region separately influence the nucleosome unwrapping rate without altering the rewrapping rate. H3(K56ac) and DNA sequence additively influence the nucleosome unwrapping rate by at least an order of magnitude and result in an equivalent change in TF occupancy within the nucleosome. Furthermore, the H3(K56ac)-enhanced DNA unwrapping rate causes a parallel increase in the hMSH2–hMSH6 induced nucleosome disassembly rate. These results are consistent with the conclusion that modulation of nucleosome unwrapping by PTMs and DNA sequence is a general mechanism for regulating DNA accessibility for transcription and DNA repair.
EXPERIMENTAL PROCEDURES
DNA constructs
The 601L (8), 5SL, 5SL-dyad, 5S(1–7), 5S(28–47) and 5S(1–47) molecules for fluorescence studies were prepared by PCR from plasmid containing the 601 nucleosome positioning sequence (NPS) or the Xenopus laevis 5S rDNA NPS with a LexA-binding site (TACTGTATGAGCATACAGTA) cloned into bases 8–27. Oligonucleotides for PCR (Supplementary Table S1) were conjugated to a 5′ or internal amine with Cy3-NHS (GE Healthcare) and purified by RP-HPLC on a 218TPTM C18 (Grace/Vydac) column. The 5SX-G/C and 5SX-G/T molecules were prepared and P32 labeled as previously described (17)
Preparation of histone octamers and LexA protein
Xenopus laevis recombinant histones were expressed and purified as previously described (33). Plasmids encoding histones H2A, H2A(K119C), H2B, H3 and H4 were generous gifts from Dr Karolin Luger (Colorado State University) and Dr Jonathan Widom (Northwestern University). Mutations H3(C110A) and H3(K56Q) were introduced by site-directed mutagenesis (Stratagene). Histone H3(K56Ac) was prepared as previously described (14). Each of the four histones were combined at equal molar ratios, refolded and purified as previously described (33). H2A(K119C) containing histone octamer (HO) was labeled with Cy5-maleamide (GE Healthcare) as previously described (14). LexA protein was expressed and purified from pJWL288 plasmid as previously described (34).
Nucleosome reconstitutions
Nucleosomes were reconstituted from DNA and purified HO by salt double dialysis and purified by sucrose gradient (14). Nucleosomes containing Cy3 labeled DNA for fluorescence studies were reconstituted with HO containing Cy5-labeled H2A(K119C). Nucleosomes containing 5SX-G/T or 5SX-G/C were reconstituted with HO containing unlabeled H2A. Nucleosomes reconstituted with 5S-Lv, 5S and 5SL(147) resulted in two nucleosome positions as previously reported (35). The central positioned 5SL(147) nucleosomes used in FRET measures were purified from the depositioned nucleosomes by sucrose gradient purification.
TF binding and site accessibility equilibrium measurements
TF binding and nucleosome site accessibility equilibrium constants were measured with LexA binding to its target site buried within the nucleosome as previously described (8,14). TF binding and DNA unwrapping were detected by a reduction in FRET. LexA binding to its target site traps the nucleosome into a partially unwrapped state (8). FRET efficiency measurements were determine by the (ratio)A method (36). Fluorescence emission spectra were measured as previously described (14). We previously determined that non-specific DNA binding of LexA does not reduce the FRET efficiency and that binding of LexA to its target sequence within the nucleosome does not induce dissociation of H2A–H2B heterodimers (14).
Stopped flow nucleosome kinetics measurements
Stopped flow experiments were performed on a KinTek 2004-SF instrument at room temperature as previously described (10). Samples were excited by a XeHG arclamp with a 525±22 nm excitation filter (Omega); simultaneous Cy3 and Cy5 emission was followed using a 570±5 nm bandpass filter (Newport) and 680±15 nm bandpass filter (Chroma), respectively. After rapid mixing, samples contained 7 nM Cy3/Cy5-labeled nucleosomes in 0.5× TE with 1, 75 or 130 mM NaCl and [LexA] varying from 0 to 50 μM. Data were smoothed by 10 point forward averaging and fit to a single exponential decay, except for unmodified nucleosomes in 0.5× TE, 1 mM NaCl, which was fit to a double exponential due to an additional slower process, which can be attributed to non-specific LexA binding (10).
Nucleosome competitive reconstitutions
Competitive reconstitutions were performed as previously described (37). Briefly, 0.6 μg of unmodified HO were combined with 0.5 μg high-affinity DNA and 2 μg low-affinity competitor DNA in 2 M NaCl, 0.5× TE, 1 mM benzamidine (BZA). Samples were placed in a 50 -μl engineered dialysis chamber which was then placed in a reservoir with 2 M NaCl, 0.5× TE, 1 mM BZA. The reservoir [NaCl] was then slowly lowered to ∼1 mM NaCl by pumping with 0.5× TE, 1 mM BZA over 36 h. Competitive reconstitutions were resolved by electrophoretic mobility gel shift analysis (EMSA) on a native 5% polyacrylamide gel and quantified as previously described (37).
Analysis of nucleosome position and TF-binding site databases
The fraction of TF-binding sites within the nucleosome dyad, entry exit and linker regions were determined with the reported consensus map of nucleosome positions (38) (http://refnucl.atlas.bx.psu.edu/) and TF-binding sites (39) (http://fraenkel.mit.edu/improved_map/) in S. cerevisiae. Nucleosomes with occupancy >10 and TF-binding sites with binding P-values <0.005 and no limitation on conservation were used. For each TF-binding site we determined the number of bases between the nearest nucleosome dyad and the center of the TF-binding site rounded down to the nearest base. The random distribution simply treats every base pair in the yeast genome as the center of a potential TF-binding site while retaining the same consensus map of nucleosome positions and is therefore the probability density for the distance to the nearest nucleosome from any position. The number of sites exactly on top of a nucleosome dyad was doubled to account for the two possible ways to be any distance >0. Distributions of TF-binding sites relative to nucleosome positions were also determined with different criteria on nucleosome occupancy and TF-binding sites.
hMSH2–hMSH6 nucleosome remodeling assay
Nucleosome disassembly reactions were carried out at 37°C as previously described (17) with 0.25 nM of unmodified H3 or H3(K56ac) containing nucleosomes. The fraction of disassembled nucleosomes were analyzed by gel shifts on polyacrylamide gels as previously described (17) A small fraction of free DNA appears at the zero time point because we do not completely purify naked DNA away from the nucleosome and a fraction of nucleosomes fall apart during rapid mixing with hMSH2–hMSH6. We control for this by not including the zero time point in the exponential decay fit.
RESULTS
H3(K56ac) enhances site accessibility by increasing the nucleosome unwrapping rate
We previously reported that H3(K56ac) increases the site exposure equilibrium for LexA binding by 3-fold (14). To investigate the kinetic mechanism of H3(K56ac) on nucleosome site exposure, we used stopped flow fluorescence resonance energy transfer (FRET). FRET was used to detect the binding of a model TF, LexA, to its target sequence within the nucleosome as previously described (10). The nucleosomes contained the 147-bp high-affinity 601 NPS (40), with the LexA target sequence inserted between base pairs 8–27 (601L, Figure 1A). The NPS DNA was labeled with Cy3 at the 5′-end nearest the LexA target sequence, while Cy5 was linked to the HO at H2A(K119C) (Figure 1B). The distance between Cy3 and Cy5 within the nucleosome is significantly less than the Cy3–Cy5 Förster radius (Figure 1C) and results in efficient FRET. Sucrose gradient–purified nucleosomes (Supplementary Figure S1A) were reconstituted with and without H3(K56ac), which was introduced into H3 by sequential native chemical ligation (14) (Figure 1B). In the presence of LexA, the nucleosome can be trapped in a partially unwrapped state (Figure 1C), which results in a reduction in the FRET efficiency.
Figure 1.

H3(K56ac) increases the rate of nucleosome unwrapping. (A) DNA constructs for FRET measurements of nucleosome unwrapping kinetics. Both higher affinity 601 NPS or lower affinity X. borealis contain a LexA protein binding site from bases 8–27 and a Cy3 molecule on the 5′-end. (B) Structure of FRET-labeled nucleosome (41) containing 601L or 5SL DNAs; the LexA-binding site in red, Cy3 in green, Cy5 on H2A(K119C) in magenta and H3(K56Ac) in orange. (C) A three-state model for LexA binding to its target site within a nucleosome. (D and E) Stopped Flow Cy5 emission versus time of 601L nucleosomes containing unmodified H3 or H3(K56Ac) nucleosomes, respectively, at 1 mM NaCl mixed with 0–900 nM LexA. (F) Normalized stopped flow Cy5 emission versus time at 900 nM LexA for nucleosomes containing unmodified H3 (blue circles), H3(K56Q) (red circles) and H3(K56ac) (orange circles) at 1 mM NaCl. (G) Relative unwrapping and calculated rewrapping rates of nucleosomes containing H3(K56Q) and H3(K56ac) versus unmodified at 1 mM NaCl. (H) Normalized stopped flow Cy5 emission versus time at 30 μM LexA for nucleosomes containing unmodified H3 (blue circles), H3(K56Q) (red circles) and H3(K56ac) (orange circles) at 130 mM NaCl. (I) Relative unwrapping and calculated rewrapping rates of nucleosomes containing H3(K56Q) and H3(K56ac) versus unmodified H3 at 130 mM NaCl (see also Supplementary Figure S1).
To measure the nucleosome unwrapping rate, k12 (Figure 1C), the LexA TF was rapidly mixed with Cy3–Cy5-labeled nucleosomes, and LexA binding to the nucleosome was detected by the measured reduction in Cy5-stimulated emission (Figure 1D and E). LexA concentrations (300–900 nM) were used to confirm that the rate of FRET reduction was independent of LexA concentration (Supplementary Figure S1D). Under these conditions the rate-limiting step of LexA binding is the nucleosome unwrapping rate, k12 (Figure 1C), and the observed rate of reduction in Cy5 fluorescence is equal to k12 (10). We determined at low ionic strength (0.5× TE, 1 mM NaCl) the unwrapping rates for nucleosomes containing unmodified H3 (8±1 s−1), H3(K56Q) (15±2 s−1) and H3(K56ac) (15±1 s−1; Figure 1F). The unwrapping rate for unmodified nucleosome is similar to previous measurements (10), while H3(K56ac) and H3(K56Q) increase the unwrapping rate by 1.9±0.3- and 2.0±0.3-fold, respectively (Figure 1G). The changes in unwrapping rates are identical to the changes in site exposure equilibrium, Keq (14), which implies that H3(K56ac) and H3(K56Q) do not alter the nucleosome rewrapping rate (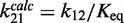 ).
).
We next investigated the influence of H3(K56ac) on the nucleosome unwrapping rate at physiological ionic strength (0.5× TE, 130 mM NaCl). We rapidly mixed Cy3/Cy5-labeled nucleosomes in the presence of 130 mM NaCl with LexA ranging between 20 and 50 µM. At these TF concentrations the decay rate of Cy5 fluorescence is independent of LexA concentration (Supplementary Figure S1E) and therefore equal to the nucleosome unwrapping rate. We determined the unwrapping rates for nucleosomes containing unmodified H3 (16±5 s−1), H3(K56Q) (43±11 s−1) and H3(K56ac) (50±11 s−1) (Figure 1H; Supplementary Figure S1B and S1C). H3(K56ac) and H3(K56Q) increased the nucleosome unwrapping rate by 3.0±1.1- and 2.6±1.0-fold, respectively (Figure 1I). This change in unwrapping rates is nearly identical to the increase in the site exposure equilibrium induced by H3(K56ac) and H3(K56Q) (14), providing additional support for the conclusion that the nucleosome rewrapping rate is unchanged by H3(K56ac) and H3(K56Q) (Figure 1I). Taken together, these results suggest that H3(K56ac) enhances TF binding within the nucleosome by increasing the nucleosome unwrapping rate without altering the rewrapping rate.
The influence of H3(K56ac) on the nucleosome unwrapping equilibrium is independent of DNA sequence
To test the possibility that the nucleosomal DNA sequence impacts the influence of H3(K56ac) on the site exposure equilibrium, we prepared Cy3–Cy5 nucleosomes with the Xenopus borealis 5S rDNA positioning sequence (42). This is a naturally occurring sequence containing part of the somatic 5S rDNA gene and has been used as a model NPS in numerous biophysical studies (43–45).
As with the 601 sequence, we inserted the LexA target sequence between base pairs 8–27 of the 5S NPS (5SL, Figure 1A). We then reconstituted nucleosomes with the 5SL DNA labeled with Cy3 at the 5′-end nearest the LexA target sequence and HO labeled with Cy5 at H2A(K119C) (Figure 1B). H3(K56ac) and H3(K56Q) induced a slight shift in 5SL nucleosome electrophoretic mobility relative to unmodified nucleosomes (Figure 2A), as previously reported for the 601 NPS (14). To determine if the change in electrophoretic mobility was due to a position change, we mapped the nucleosome positions on 5SL by hydroxyl radical mapping (35) using the FeBABE label as previously reported (14) and found that the H3(K56Q) did not impact the cleavage pattern (Supplementary Figure S2). Furthermore, the cleavage pattern with 5SL nucleosomes was indistinguishable from nucleosomes containing 601L (14). This indicates that the nucleosomes were well positioned within the 5S NPS and that nucleosome position was not altered by modifying H3(K56).
Figure 2.

DNA sequence does not influence H3(K56ac) enhancement of TF binding. (A) Cy3 fluorescence image of native PAGE analysis of purified FRET-labeled nucleosomes containing the 5SL NPS and unmodified H3 (lane 2), H3(K56Q) (lane 3) or H3(K56ac) (lane 4). (B and C) Fluoresence emission spectra of 5SL FRET-labeled nucleosomes containing unmodified H3 or H3(K56ac), respectively, excited at 510 nm (donor excitation) in the presence of 0.5× TE, 1 mM NaCl and LexA at 0 nM (black), 30 nM (red) or 1000 nM (blue), (D) Steady-state FRET efficiency, as determined by the (ratio)A method (36), versus LexA concentration for nucleosomes containing unmodified H3 (blue), H3(C110) (purple), H3(K56Q) (red), and H3(K56a) (orange) at 1 mM NaCl. Plots are the average of three LexA titrations and the error bars were determined from the SD of the three measurements. The data were fit to a non-cooperative binding curve, which determines S0.5-nuc, the LexA concentration at which 50% of the nucleosomes are bound by LexA. (E) Relative change in Keq for nucleosomes with the 601L (14) or 5SL NPS containing H3(K56Q) or H3(K56ac) versus unmodified H3 at 1 mM NaCl. This is inversely related to the relative change in S0.5.
To determine the influence of H3(K56ac) on the 5SL site exposure equilibrium, we carried out LexA titrations with unmodified, H3(K56ac) and H3(K56Q) nucleosomes containing the 5SL sequence (Figure 2B–D). We determined the FRET efficiency of unmodified and modified nucleosomes at each LexA concentration in triplicate and then fit the FRET efficiency at increasing LexA concentrations to a non-cooperative binding isotherm. This analysis determines the LexA concentration at which half of the nucleosomes are bound by LexA, S0.5_nuc. From this we use, Keq_modified/Keq_unmodified = S0.5_unmodified/S0.5_modified, to infer the relative nucleosome site exposure equilibrium (8,14). The resulting increase in 5SL site accessibility matches that observed for the 601L NPS [Keq_5SL-K56ac/Keq_5SL-unmod = 1.8±0.3 and Keq_5SL-K56Q/Keq_5SL-unmod = 2.0±0.4; Figure 2E and Table 1].
Table 1.
Summary of the TF-binding equilibrium and nucleosome unwrapping measurements
| DNA | Histone | Na+ (mM) | Eo | S0.5 (nM) | 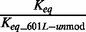 |
k12 (s−1) |  |
 |
|---|---|---|---|---|---|---|---|---|
| 601L | unmod | 1 | 0.68 ± 0.01 | 58 ± 6 | – | 7.8 ± 0.9 | – | – |
| 601L | H3(K56Q) | 1 | 0.45 ± 0.01 | 32 ± 3 | 1.8 ± 0.4 | 15 ± 2 | 2.0 ± 0.3 | 1.1 ± 0.2 |
| 601L | H3(K56Ac) | 1 | 0.43 ± 0.01 | 32 ± 3 | 1.8 ± 0.4 | 15 ± 1 | 1.9 ± 0.2 | 1.1 ± 0.2 |
| 601L | unmod | 130 | 0.74 ± 0.01 | 13 100 ± 500 | – | 17 ± 5 | – | – |
| 601L | H3(K56Q) | 130 | 0.52 ± 0.01 | 5200 ± 500 | 2.5 ± 0.5 | 40 ± 10 | 3 ± 1 | 1.0 ± 0.4 |
| 601L | H3(K56Ac) | 130 | 0.50 ± 0.01 | 4000 ±500 | 3.3 ± 0.7 | 50 ± 10 | 3 ± 1 | 0.9 ± 0.3 |
| 5SL | unmod | 1 | 0.40 ± 0.01 | 90 ± 10 | – | |||
| 5SL | H3(K56Q) | 1 | 0.28 ± 0.01 | 42 ± 8 | 2.0 ± 0.5 | |||
| 5SL | H3(K56Ac) | 1 | 0.31 ± 0.01 | 48 ± 6 | 1.8 ± 0.3 | |||
| 5SL | H3(C110) | 1 | 0.42 ± 0.01 | 90 ± 10 | 1.0 ± 0.2 | |||
| 601L | unmod | 75 | 0.67 ± 0.02 | 2500 ± 100 | – | 17 ± 3 | – | – |
| 5SL(1–147) | unmod | 75 | 0.47 ± 0.02 | 6700 ± 300 | 0.36 ± 0.03 | 5.4 ± 0.7 | 0.33 ± 0.07 | 1.1 ± 0.3 |
| 5SL(1–7) | unmod | 75 | 0.48 ± 0.02 | 4600 ± 300 | 0.53 ± 0.05 | 7 ± 2 | 0.40 ± 0.1 | 1.3 ± 0.4 |
| 5SL(28–27) | unmod | 75 | 0.54 ± 0.02 | 2600 ± 200 | 0.96 ± 0.08 | 17 ± 3 | 1.0 ± 0.2 | 0.9 ± 0.2 |
| 5SL(1–47) | unmod | 75 | 0.48 ± 0.02 | 5300 ± 400 | 0.46 ± 0.05 | 7 ± 1 | 0.40 ± 0.09 | 1.1 ± 0.3 |
| 601L | H3(K56Q) | 75 | 0.52 ± 0.01 | 870 ± 100 | 2.8 ± 0.4 | 50 ± 10 | 3.1 ± 0.8 | 0.9 ± 0.3 |
| 5SL(1–7) | H3(K56Q) | 75 | 0.45 ± 0.01 | 1400 ± 100 | 1.2 ± 0.2 | 20 ± 3 | 1.3 ± 0.3 | 1.3 ± 0.3 |
Column 1 is the type of the DNA construct. Column 2 is the type of histone octamer. Column 3 is the concentration of sodium. Column 4 is the average FRET efficiency without LexA. Column 5 is the half saturation concentration of LexA. Column 6 is the site exposure equilibrium relative to the measurement with nucleosomes containing 601L and unmodified histone octamer. Column 7 is the nucleosome unwrapping rate to exposure the LexA target sequence for binding. Column 8 is the nucleosome unwrapping rate relative to the unwrapping rate of unmodified nucleosomes containing 601L. Column 9 is the calculated rewrapping rate relative to unmodified nucleosomes containing 601L.
To verify that the increase in the site exposure equilibrium was due to unwrapping and not repositioning, we monitored LexA-induced nucleosome sliding by placing the Cy3 fluorophore on the 80th bp of the DNA (Supplementary Figure S3A) (14). This results in juxtaposition of the Cy3–Cy5 FRET pair near the nucleosome dyad (Supplementary Figure S3B). Repositioning induced by LexA binding would result in a FRET change, while unwrapping will not. We do not observe any change in FRET, even at saturating concentrations of LexA (Supplementary Figure S3C). As an independent verification, we conducted hydroxyl radical mapping (35) using the FeBABE label (14) of 5SL nucleosomes pre-incubated with saturating concentrations of LexA (1 μM). There was no observable difference between the cleavage of 5SL and 601L nucleosomes in the presence and absence of LexA (Supplementary Figure S2). The combination of the site exposure equilibrium measurements for TF binding within 5SL and 601L containing nucleosomes as well as the nucleosome position mapping strongly suggests that H3(K56ac) increases the nucleosome site exposure equilibrium independent of the underlying nucleosomal DNA sequence.
DNA sequence within the nucleosome entry–exit region influences accessibility by modulating the DNA unwrapping rate
We investigated the influence of DNA sequence changes within the 601 NPS on nucleosome unwrapping and rewrapping rates. First, we determined the LexA concentrations at which half of the nucleosomes are bound by LexA, S0.5, for nucleosomes containing 5SL and 601L. We find at low ionic strength (0.5× TE with 1 mM NaCl) that S0.5 of unmodified nucleosomes containing 5SL is increased relative to unmodified nucleosomes with 601L, indicating lower site exposure, Keq, in the entry–exit region [Keq_5SL-unmod/Keq_601L-unmod = 0.6±0.1]. Changing the DNA sequence from 601L to 5SL had a similar influence on the Keq for LexA binding within nucleosomes containing either H3(K56Q) [Keq_5SL-K56Q/Keq_601L-K56Q = 0.8±0.2] or H3(K56ac) [Keq_5SL-K56ac/Keq_601L-K56ac = 0.7±0.1].
We also investigated the influence of DNA sequence on Keq closer to physiological ionic strength. While we were unable to fully saturate LexA binding to nucleosomes containing 5SL in 130 mM NaCl, LexA binding to nucleosomes did saturate in 75 mM NaCl (Supplementary Figure S4B). We used these conditions (0.5× TE with 75 mM NaCl) to further examine the DNA sequence dependence of Keq. Under these conditions, changing the NPS from 601L to 5SL significantly increased S0.5, which implied that the site exposure equilibrium constant, Keq, was reduced by ∼3-fold [Keq_5SL/Keq_601L = 0.36±0.03].
The observation that the X. borealis 5S NPS reduced the site exposure equilibrium relative to the 601 NPS was unexpected because the sea urchin 5S NPS has a significantly higher free energy for nucleosome formation (lower HO affinity) relative to the 601 positioning sequence (46). However, these results are consistent with the recent report that H2A-H2B heterodimers have a higher affinity to sea urchin 5S NPS than the 601 NPS (47). To determine which region(s) of the DNA sequence is responsible for this 3-fold reduction in Keq, we created three DNA chimeras where segments of 601L were replaced with segments from 5SL (Figure 3A). These chimeric DNAs were 5′ labeled with Cy3 and reconstituted into nucleosomes with Cy5-labeled HO (Supplementary Figure S4A). We carried out LexA-binding analysis with nucleosomes containing the chimeric DNA sequences to determine the relative changes in S0.5 to detect alterations in the site exposure equilibrium, Keq (Figure 3B and C; Supplementary Figure S4B). We found that most of the increase in S0.5 and therefore reduction in Keq is induced by changing base pairs 1–7, located between the nucleosome entry–exit and the LexA target sequence [Keq_5SL(1–7)/Keq_601L = 0.53±0.04]. Changing base pairs 28–47, located on the opposite side of the LexA target sequence, does not reduce the Keq [Keq_5SL(28–47)/Keq_601L = 0.98±0.08]. Furthermore, the combined influence of changing base pairs 1–7 and 28–47 on Keq is similar to the influence of changing only base pairs 1–7 [Keq_5SL(1–47)/Keq_601L = 0.46±0.05]. This suggests that the DNA sequence located between the nucleosome entry–exit and the TF-binding site can significantly influence the site exposure equilibrium.
Figure 3.

DNA sequence between the nucleosome entry–exit and the TF-binding site impacts nucleosome unwrapping. (A) DNA constructs with the 601 sequence (blue) iteratively replaced by the 5S sequence (lavender), where the parenthetical numbers represents the bases in 601 replaced by 5S; lexA site in red. (B) Steady-state FRET efficiency, as determined by the (ratio)A method, versus lexA concentration for nucleosomes containing 601L and unmodified H3 (blue), 601L and H3(K56Q) (red), 5SL(1–7) and unmodified H3 (green), and 5SL(1–7) and H3(K56Q) (olive) at 75 mM NaCl. Plots are the average of three LexA titrations and the error bars were determined from the SD of the three measurements. The data were fit to a non-cooperative binding curve. (C) Relative Keq for the indicated nucleosome versus nucleosomes containing 601L and unmodified H3 at 75 mM NaCl. (D) Normalized stopped flow Cy5 emission versus time at 15 μM LexA for nucleosomes containing 601L and unmodified H3 (blue), 601L and H3(K56Q) (red), 5SL(1–7) and unmodified H3 (green), and 5SL(1–7) and H3(K56Q) (olive) at 75 mM NaCl. (E and F) Relative unwrapping and calculated rewrapping rates, respectively, for the indicated nucleosomes versus nucleosome containing 601L and unmodified H3 at 75 mM NaCl (see also Supplementary Figure S4).
We performed stopped flow experiments with nucleosomes containing each of the 601L-5SL chimeric DNA to examine the mechanism by which DNA sequence modulates site accessibility (Figure 3D and Supplementary Figure S4C). The change in Cy5 fluorescence was measured at LexA concentrations between 10 and 30 µM where the rate of change of Cy5 fluorescence was independent of LexA concentration (Supplementary Figure S4D) and equal to the nucleosome unwrapping rate. We determined the nucleosome unwrapping rate, k12, for each DNA chimera (Figure 3D and Table 1). Replacing base pairs 1–7 of 601L with 5SL significantly reduced the unwrapping rate [k12_5S(1–7)/k12_601L = 0.4±0.1], while replacing base pairs 28–47 of 601L with 5SL did not influence the unwrapping rate [k12_5S(28–47)/k12_601L = 1.0±0.2]. The combined change of base pairs 1–7 and 28–47 reduced the unwrapping rate [k12_5S(1–47)/k12_601L = 0.4±0.1], which appears indistinguishable to changing only base pairs 1–7. Importantly, the DNA sequence-induced changes in the unwrapping rates are nearly identical to the induced changes in site exposure equilibrium (Figure 3C and E), which implies that the changes in DNA sequence do not influence the calculated nucleosome rewrapping rates, 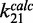 (Figure 3F). Taken together, these results demonstrate that alterations in DNA sequence influence TF binding within nucleosomes by changing the DNA unwrapping rate without influencing the rewrapping rate.
(Figure 3F). Taken together, these results demonstrate that alterations in DNA sequence influence TF binding within nucleosomes by changing the DNA unwrapping rate without influencing the rewrapping rate.
Note that while the Keq measured by LexA binding increased for nucleosomes containing 5SL relative to 601L, the average FRET efficiency without LexA was lower for nucleosomes containing 5SL (0.47±0.02) than 601L (0.67±0.02). This contradictory result could be due to increased nucleosome unwrapping of the first few base pairs of 5SL. However, the average FRET efficiency is influenced by numerous parameters (36). In particular, the structure of a nucleosome containing the 601 positioning sequence was recently reported to contain 145 bp as compared to 147 bp for an alpha satellite DNA sequence (48). This could significantly alter the Cy3–Cy5 distance. In contrast, determining changes in the site exposure equilibrium from the S0.5 measurements of LexA binding to nucleosomes does not rely on the absolute average FRET efficiency. Furthermore, the agreement between the DNA sequence-induced changes in nucleosome unwrapping and the changes to the equilibrium of LexA binding to a nucleosome confirms the reliability of the LexA-binding measurements. Therefore, we used the S0.5 measurements from LexA titrations to determine the relative change in site exposure of the LexA target sequence.
DNA sequence and modification of H3(K56) additively regulate TF binding within nucleosomes
Our observation that DNA sequence does not alter the influence of either H3(K56ac) or H3(K56Q) on nucleosome site exposure equilibrium suggests that the influence of DNA sequence is independent of the modification state of H3(K56). To test this hypothesis, we prepared Cy3–Cy5-labeled nucleosomes that contained the DNA chimera 5S(1–7) and the acetyl-lysine mimic H3(K56Q). We found that the influence of replacing the first 7 bp of 601L with 5SL did not alter the impact of H3(K56Q) on the LexA concentration to bind half of the nucleosomes, S0.5, and therefore the site exposure equilibrium, Keq [Keq_5S(1–7)-K56Q/Keq_601L-K56Q = 0.36±0.06 and Keq_5S(1–7)-unmod/Keq_601L-unmod = 0.36±0.03; Figure 3B and C]. We then determined that the influence of replacing the first 7 bp of 601L with 5SL on the unwrapping rate was unaffected by H3(K56Q) [k12_5S(1–7)-K56Q/k12_601L-K56Q = 0.4±0.1 and k12_5S(1–7)-unmod/k12_601L-unmod = 0.33±0.07; Figure 3D and E]. These results are consistent with the conclusion that H3(K56) and the DNA sequence influence nucleosome unwrapping rate and ultimately TF binding independently. This implies that the DNA sequence and the modification of H3(K56) may additively combine to significantly influence TF binding. In fact, the unwrapping rate of nucleosomes modified at H3(K56) within the 601L NPS is 10±2 times larger than unmodified nucleosomes within the 5SL NPS. This difference enhances TF binding by 8±2 times.
The 601 positioning sequence has a higher HO binding affinity but lower nucleosome unwrapping equilibrium than a 5S positioning sequence
The observation that substitution of the first 7 bp of the 601 NPS with the X. borealis 5S NPS reduces the site exposure equilibrium was unexpected. To further investigate this, we determined the difference in free energies for nucleosome formation between the 601-5S chimeras with competitive reconstitutions (49). This method determines the free energy difference, 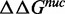 , for nucleosome formation between distinct DNA sequences. Nucleosomes are reconstituted with a fluorophore-labeled NPS in the presence of low-affinity competitor DNA and HO. The naked DNA and HO establish a dynamic equilibrium with nucleosomes during the reconstitution by gradual salt dialysis. The equilibrium constant, Keq, is then determined by an EMSA (Figure 4A) and the free energy for nucleosome formation relative to a reference DNA sequence is determined from:
, for nucleosome formation between distinct DNA sequences. Nucleosomes are reconstituted with a fluorophore-labeled NPS in the presence of low-affinity competitor DNA and HO. The naked DNA and HO establish a dynamic equilibrium with nucleosomes during the reconstitution by gradual salt dialysis. The equilibrium constant, Keq, is then determined by an EMSA (Figure 4A) and the free energy for nucleosome formation relative to a reference DNA sequence is determined from: 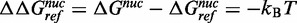 ln(Keq/Keq-ref). We then compared this to the difference in free energy for the nucleosome to remain fully wrapped (Figure 4), which we determined from site exposure equilibrium measurements
ln(Keq/Keq-ref). We then compared this to the difference in free energy for the nucleosome to remain fully wrapped (Figure 4), which we determined from site exposure equilibrium measurements  .
.
Figure 4.

Relation between DNA–histone binding and DNA unwrapping free energies. (A) Cy3 fluorescence image of native PAGE analysis of competitive reconstitutions performed in triplicate for unmodified HO with the DNA chimeras used for site exposure measures. (B) Change in free energy of nucleosome formation (ΔΔGnuc, light gray) relative to 601L as determined by competitive reconstitution from the gel in (A), and the change in nucleosome free energy for wrapping (ΔΔGwrap, dark gray) relative to 601L as determined from site accessibility measures (Figure 3B and C). The error bars for ΔΔGnuc are the SD of the three independent measurements for each nucleosome type. (C) Table of the change in free energy values in kcal/mol for nucleosome formation of each DNA species relative to Lumbriculus variegatus 5S NPS 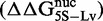 , for nucleosome formation of each DNA species relative to 601L DNA
, for nucleosome formation of each DNA species relative to 601L DNA 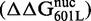 and for nucleosome wrapping for each DNA species relative to 601L DNA
and for nucleosome wrapping for each DNA species relative to 601L DNA 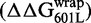 .
.
We find that substitution of the first 7 bp of the X. borealis 5SL sequence into the 601L sequence reduced the nucleosome formation free energy 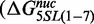
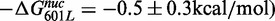 . This observation is in agreement with the reduced site exposure equilibrium induced by this chimeric NPS
. This observation is in agreement with the reduced site exposure equilibrium induced by this chimeric NPS 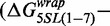
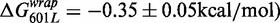 . The insertion of 5SL base pairs 28–47 into the 601L sequence increased the ΔΔG
. The insertion of 5SL base pairs 28–47 into the 601L sequence increased the ΔΔG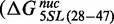
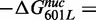
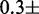
 , but did not impact the site exposure free energy
, but did not impact the site exposure free energy 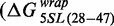
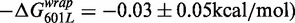 . The insertion of both 5SL base pairs 1–7 and 28–47 into the 60L sequence displayed a free energy change equal to the sum of the free energy changes of the separate substitutions
. The insertion of both 5SL base pairs 1–7 and 28–47 into the 60L sequence displayed a free energy change equal to the sum of the free energy changes of the separate substitutions 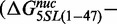
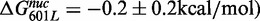 . In contrast, the
. In contrast, the 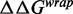 induced by changing both base pairs 1–7 and 28–47
induced by changing both base pairs 1–7 and 28–47 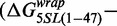
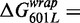
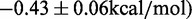 and between 601L and 5SL(1–147)
and between 601L and 5SL(1–147) 
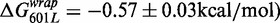 is similar to the free energy difference induced by base pairs 1–7 alone. These comparisons are consistent with the conclusion that the influence of base pairs 1–7, 28–47 and 48–147 on the nucleosome stability free energy is additive but that only base pairs 1–7 influence the exposure of the LexA TF-binding site. Taken as a whole these results suggest that changes in the nucleosomal DNA sequence can separately tune TF-binding site exposure as well as overall nucleosome stability.
is similar to the free energy difference induced by base pairs 1–7 alone. These comparisons are consistent with the conclusion that the influence of base pairs 1–7, 28–47 and 48–147 on the nucleosome stability free energy is additive but that only base pairs 1–7 influence the exposure of the LexA TF-binding site. Taken as a whole these results suggest that changes in the nucleosomal DNA sequence can separately tune TF-binding site exposure as well as overall nucleosome stability.
TF-binding sites (30%) reside in the entry–exit regions of S. cerevisiae nucleosome positions
Our observation that H3(K56ac) and DNA sequence have a combined influence on TF binding within nucleosomes suggests that the regulation of DNA unwrapping could be a major regulator of TF occupancy. To investigate if TF-binding sites are poised for this regulatory mechanism in vivo, we determined the fraction of TF-binding sites that reside within S. cerevisiae nucleosome entry–exit regions. We examined the consensus maps of S. cerevisiae nucleosome occupancy (38) and the map of TF-binding sites (39) to determine the distance from the center of a TF-binding site to the dyad center of the nearest nucleosome (Figure 5A). The nucleosome dyad and entry–exit regions were defined to be 0–36 and 37–74 bp from the nucleosome dyad center, respectively. We found that 31 and 33% of TF-binding sites are within the dyad and entry–exit regions, respectively (Figure 5B). Approximately 36% of TF-binding sites are located in linker DNA between nucleosomes. Variation in the criteria for TF and nucleosome occupancy modestly altered these results (Supplementary Figure S5). We compared the observed distance distribution of TF-binding sites from the nearest nucleosome dyad to the distance distribution of every position in the genome to the nearest nucleosome dyad (Figure 5A, black line). The expected fractions of TF-binding sites within the dyad, entry–exit and linker DNA regions for randomly positioned nucleosome are 36, 37 and 22%, respectively (Figure 5B). These observations suggest that while TF-binding sites appear somewhat biased toward being in linker DNA between nucleosomes (50–52), there is rather little suppression of TF-binding sites within the nucleosome; thus a significant fraction of TF-binding sites are located within nucleosome entry–exit regions and hence poised to be regulated through nucleosome unwrapping. Furthermore, these observations are consistent with the report that within S. cerevisiae the positions of TF-binding sites at gene promoters are correlated with the nucleosome entry–exit region for nucleosomes containing the histone variant H2A.Z (53).
Figure 5.

TF-binding sites predominantly reside within nucleosomes. (A) Distribution of TF-binding site distances from the nearest nucleosome dyad (black circles), with distances of 0–36 bases considered within the dyad region, distances of 37–74 bases considered within the entry–exit region, and distance greater than 74 bases considered to be in naked DNA (see Experimental Procedures and Supplementary Figure S5 for details). The black line represents the expected TF-binding site distribution if they were distributed randomly throughout the genome. (B) Measured and expected percentages of TFs in each region calculated from the distribution in (A).
H3(K56ac) facilitates nucleosome removal by the DNA-mismatch recognition complex hMSH2–hMSH6
The modulation of nucleosome unwrapping is also a potential mechanism for regulating the interaction between DNA repair complexes and nucleosomal DNA. We investigated this mechanism in the context of DNA repair by determining the influence of H3(K56ac) on nucleosomal disassembly by the DNA-mismatch recognition complex, hMSH2–hMSH6 (17,18). MMR often occurs behind replication forks (54), where newly assembled nucleosomes are found with the histone PTM, H3(K56ac) (30). Previous analysis of nucleosome disassembly by hMSH2–hMSH6 suggested that regulation of site exposure qualitatively accounts for the impact of PTMs on nucleosome disassembly (18). To investigate this hypothesis, we reconstituted unmodified and H3(K56ac)-containing nucleosomes with DNA containing a mismatch 55 bp from the X. borealis 5S rDNA positioning sequence (17) (Figure 6A). Incubation with hMSH2–hMSH6 showed that these nucleosomes were disassembled by hMSH2–hMSH6 1.7±0.3 times faster (τH3(K56Ac) = 112±5 min) than unmodified nucleosomes [τH3(C110) = 189±33 min; Figure 6]. This difference is statistically significant with a P-value of 0.005. The preparation of H3(K56ac) required the mutation H3(C110A), while unmodified H3 contained the native cysteine. To control for this difference, we confirmed that the conversion of the cysteine at the 110th amino acid of H3 to an alanine did not alter the nucleosome site accessibility with the X. borealis 5S NPS (Figure 2D and Table 1). As a control we showed that nucleosomes without a mismatch were not disassembled significantly (Supplementary Figure S6) (17). Furthermore, H3(K56Q) with the native cysteine at the 110th amino acid, enhances hMSH2–hMSH6-induced nucleosome disassembly by 2-fold (17), similarly to H3(K56ac). The combination of these measures indicates that the increase in DNA unwrapping by H3(K56ac) enhances hMSH2–hMSH6-induced nucleosome disassembly adjacent to a DNA mismatch. Additional studies are required to determine if this 2-fold increase is biologically significant. However, 2-fold changes often have significant biological consequences as in dosage compensation and haploinsufficiency diseases.
Figure 6.

H3(K56ac) facilitates nucleosome dissociation by the mismatch recognition complex hMSH2–hMSH6. (A) DNA constructs containing the X. borealis 5S NPS with (5S-GT) or without (5S-GC) a DNA mismatch and a 3′-biotin. (B and C) Electrophoretic mobility shift analysis of H3(C110) nucleosomes adjacent to a GT mismatch, and H3(K56ac) nucleosomes adjacent to a GT mismatch disassembled by hMSH2–hMSH6, respectively. Lane 1: sucrose gradient–purified nucleosomes, Lane 2: nucleosomes bound by streptavidin, Lane 3: nucleosomes bound by streptavidin and hMSH2–hMSH6, Lanes 4–9: kinetic analysis of streptavidin-bound nucleosome disassembly by hMSH2–hMSH6 in the presence of 1 mM ATP. (D) The fraction of unmodified 5S-GC NPS nucleosomes (black square), H3(C110) 5S-GT NPS nucleosomes (lavender diamond) and H3(K56ac) 5S-GT 5S-GT NPS nucleosomes (orange triangle) versus time in the presence of hMSH2–hMSH6 (250 nM) and ATP (1 mM). The error bars were determined from the SD of at least three separate experiments. The fraction of nucleosomes versus time were fit excluding the zero time point to a single exponential decay, A × e−t/τ (see also Supplementary Figure S6).
DISCUSSION
Site exposure is an inherent property of the nucleosome that appears to provide transcription (8,12–15) and DNA repair (16–18) complexes direct access to DNA. Here, we demonstrate that H3(K56ac) and DNA sequence influence TF occupancy by altering the nucleosome unwrapping rate, but not the rewrapping rate. We find that DNA sequence and H3(K56ac) additively influence the nucleosome unwrapping rate, which allows these two factors to function together to enhance or counteract their influence on TF occupancy (Figure 7). We observe 3-fold changes in TF occupancy induced by either H3(K56ac) or DNA sequence alone and a 10-fold effect in combination. These results, in combination with the observation that ∼30% of S. cerevesia TF-binding sites are located within nucleosome entry–exit regions, suggests that the modulation of the nucleosome unwrapping rate could be a mechanism for regulating TF occupancy in vivo.
Figure 7.

Kinetic model of H3(K56Ac) and DNA sequence modulation of nucleosome unwrapping rate. (A and B) Nucleosomes containing TF-binding sites at two different loci within the genome (nucleosome 1 with entry–exit in blue, nucleosome 2 with entry–exit in magenta). The DNA sequence between the entry–exit and TF-binding site influences the inherent nucleosome unwrapping rate to regulate TF binding within each nucleosome. Acetylation/de-acetylation of H3 lysine 56 at nucleosome 1 enhances/suppresses the DNA unwrapping rate to adjust TF occupancy. This influences both the TF occupancy within nucleosome 1 but also TF occupancy relative to nucleosome 2.
The observation that the H3(K56ac) and DNA sequence influence the nucleosome unwrapping rates but not the rewrapping rates suggests that they influence the free energy of the fully wrapped state while not affecting the free energies of the unwrapped and the transition states. This could be caused by a reduction in DNA–histone binding. The influence of H3(K56ac) removes a negative charge, which could disrupt water-mediated hydrogen bonding near K56 as is observed for H3(K56Q) (31). The first 7 bp of the 601 DNA sequence contain a GG dinucleotide that is in phase with the GC/GG dinucleotides, which are important for the strong positioning. However, this predicts that the first 7 bp of the 601 sequence should be more tightly wrapped than the 5S sequence, which is not what we observe. Instead, the DNA sequence may alter the precise DNA structure near the entry–exit region as is observed for nucleosomes containing the 601 and alpha satellite DNA molecules (48). This could alter direct and water-mediated hydrogen bonding in the entry–exit region and influence the free energy of the fully wrapped nucleosome state. In contrast, it appears the DNA bending involved in nucleosome rewrapping is not significantly different between the 601 and 5S sequences since the rewrapping rate is unaffected.
Our conclusion that the nucleosome rewrapping rate is not influenced by DNA sequence and H3(K56ac) relies on our observation that the relative changes in the rate of unwrapping and the LexA-binding equilibrium are identical. This observation implies that the combined influence of k21, k23 and k32 (Figure 1C) does not alter the LexA-binding equilibrium to nucleosomes. If the rewrapping rate, k21, were to change, k23 and k32 would need to change to exactly compensate. Instead, the simplest explanation is that these rates are not influenced by H3(K56ac) and DNA sequence. However, further studies that directly measure k21, k23 and k32 are required to confirm this interpretation.
Our studies provide additional insight into the influence of DNA sequence on site exposure equilibrium. A previous study showed introduction of a polyA track placed at the first 16 bp within a 601-like sequence increased nucleosome site accessibility ∼1.5-fold (22). Here we find that changing only the first 7 bases of the 601 DNA sequence to the X. borealis 5S sequence decreases the rate of DNA unwrapping by 2.5-fold. In contrast, the central 80 bp of the 601 NPS are largely responsible for the enhanced binding free energy relative to the 5S sequence (55). These results, in combination with ΔΔG measurements, indicate that while 601 was selected for optimal nucleosome stability (40), it is not optimized to suppress partial DNA unwrapping at the entry–exit region.
In vivo, nucleosomes are embedded within chromatin where higher order compaction could impact the influence of nucleosome unwrapping/rewrapping kinetics on TF occupancy. Recent studies of nucleosome unwrapping/rewrapping fluctuations indicate that higher order chromatin compaction does not significantly impact TF occupancy within the nucleosome but does impact occupancy in linker DNA (56,57). While this suggests that the influence of H3(K56ac) and DNA sequence on TF occupancy within the nucleosome will occur in the context of chromatin, additional studies are required to investigate the influence of chromatin higher order structure.
Other histone PTMs in the nucleosome entry–exit region could function like H3(K56ac) to enhance site accessibility by altering the nucleosome unwrapping rate. H4(K77ac) and H4(K79ac), which are located at the DNA–histone interface 40–45 bp within the nucleosome, enhanced TF binding 2-fold to their site between base pairs 8–27 within the nucleosome (15). Furthermore, histone H3 PTMs at P38 (58), Y41 (59) and T45 (60,61) reside at the nucleosome entry–exit. Further studies are required to determine if additional histone PTMs in the nucleosome entry–exit regulate the DNA unwrapping rate to control protein binding within nucleosomes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–6.
FUNDING
American Heart Association Predoctoral Fellowship [0815460D to J.A.N.]; [10PRE3150036 to A.M.M.]; OSUCCC and the James Pelotonia Fellowship (to J.A.N.); National Institutes of Health (NIH) [CA067007 and GM080176 to R.F.]; [GM083055 to M.G.P. and J.J.O.]; Career Award in the Basic Biomedical Sciences from the Burroughs Wellcome Fund (to M.G.P.) and National Science Foundation [MCB0845695 to J.J.O.]; [DMR1105458 to R.B.]. Funding for open access charge: NIH [GM083055].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Jonathan Widom and Karolin Luger for the X. laevis histone and LexA expression vectors and Karin Musier-Forsyth for access to a Typhoon Trio fluorescence scanner and a fluorescence plate reader.
REFERENCES
- 1.Luger K, Rechsteiner TJ, Flaus AJ, Waye MM, Richmond TJ. Characterization of nucleosome core particles containing histone proteins made in bacteria. J. Mol. Biol. 1997;272:301–311. doi: 10.1006/jmbi.1997.1235. [DOI] [PubMed] [Google Scholar]
- 2.Cairns BR. The logic of chromatin architecture and remodelling at promoters. Nature. 2009;461:193–198. doi: 10.1038/nature08450. [DOI] [PubMed] [Google Scholar]
- 3.Ryan DP, Owen-Hughes T. Snf2-family proteins: chromatin remodellers for any occasion. Curr. Opin. Chem. Biol. 2011;15:649–656. doi: 10.1016/j.cbpa.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annu. Rev. Biochem. 2011;80:473–499. doi: 10.1146/annurev-biochem-061809-175347. [DOI] [PubMed] [Google Scholar]
- 5.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 6.Park YJ, Luger K. Histone chaperones in nucleosome eviction and histone exchange. Curr. Opin. Struct. Biol. 2008;18:282–289. doi: 10.1016/j.sbi.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polach KJ, Widom J. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J. Mol. Biol. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 8.Li G, Widom J. Nucleosomes facilitate their own invasion. Nat. Struct. Mol. Biol. 2004;11:763–769. doi: 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- 9.Koopmans WJ, Buning R, Schmidt T, van Noort J. spFRET using alternating excitation and FCS reveals progressive DNA unwrapping in nucleosomes. Biophys. J. 2009;97:195–204. doi: 10.1016/j.bpj.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li G, Levitus M, Bustamante C, Widom J. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 2005;12:46–53. doi: 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- 11.Tims HS, Gurunathan K, Levitus M, Widom J. Dynamics of nucleosome invasion by DNA binding proteins. J. Mol. Biol. 2011;411:430–448. doi: 10.1016/j.jmb.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adams CC, Workman JL. Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol. Cell. Biol. 1995;15:1405–1421. doi: 10.1128/mcb.15.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Polach KJ, Widom J. A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J. Mol. Biol. 1996;258:800–812. doi: 10.1006/jmbi.1996.0288. [DOI] [PubMed] [Google Scholar]
- 14.Shimko JC, North JA, Bruns AN, Poirier MG, Ottesen JJ. Preparation of fully synthetic histone H3 reveals that acetyl-lysine 56 facilitates protein binding within nucleosomes. J. Mol. Biol. 2011;408:187–204. doi: 10.1016/j.jmb.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simon M, North JA, Shimko JC, Forties RA, Ferdinand MB, Manohar M, Zhang M, Fishel R, Ottesen JJ, Poirier MG. Histone fold modifications control nucleosome unwrapping and disassembly. Proc. Natl Acad. Sci. USA. 2011;108:12711–12716. doi: 10.1073/pnas.1106264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bucceri A, Kapitza K, Thoma F. Rapid accessibility of nucleosomal DNA in yeast on a second time scale. EMBO J. 2006;25:3123–3132. doi: 10.1038/sj.emboj.7601196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Javaid S, Manohar M, Punja N, Mooney AM, Ottesen JJ, Poirier MG, Fishel R. Nucleosome Remodeling by hMSH2-hMSH6. Mol. cell. 2009;36:1086–1094. doi: 10.1016/j.molcel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forties RA, North JA, Javaid S, Tabbaa OP, Fishel R, Poirier MG, Bundschuh R. A quantitative model of nucleosome dynamics. Nucleic Acids Res. 2011;39:8306–8313. doi: 10.1093/nar/gkr422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson JD, Lowary PT, Widom J. Effects of histone acetylation on the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol. 2001;307:977–985. doi: 10.1006/jmbi.2001.4528. [DOI] [PubMed] [Google Scholar]
- 20.Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell. 2009;36:153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson JD, Widom J. Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol. 2000;296:979–987. doi: 10.1006/jmbi.2000.3531. [DOI] [PubMed] [Google Scholar]
- 22.Anderson JD, Widom J. Poly(dA-dT) promoter elements increase the equilibrium accessibility of nucleosomal DNA target sites. Mol. Cell. Biol. 2001;21:3830–3839. doi: 10.1128/MCB.21.11.3830-3839.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelbauskas L, Chan N, Bash R, Yodh J, Woodbury N, Lohr D. Sequence-dependent nucleosome structure and stability variations detected by Forster resonance energy transfer. Biochemistry. 2007;46:2239–2248. doi: 10.1021/bi061289l. [DOI] [PubMed] [Google Scholar]
- 24.Han J, Zhou H, Horazdovsky B, Zhang K, Xu RM, Zhang Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science. 2007;315:653–655. doi: 10.1126/science.1133234. [DOI] [PubMed] [Google Scholar]
- 25.Xu F, Zhang Q, Zhang K, Xie W, Grunstein M. Sir2 deacetylates histone H3 lysine 56 to regulate telomeric heterochromatin structure in yeast. Mol. Cell. 2007;27:890–900. doi: 10.1016/j.molcel.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Celic I, Verreault A, Boeke JD. Histone H3 K56 hyperacetylation perturbs replisomes and causes DNA damage. Genetics. 2008;179:1769–1784. doi: 10.1534/genetics.108.088914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008;134:231–243. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113–117. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie W, Song C, Young NL, Sperling AS, Xu F, Sridharan R, Conway AE, Garcia BA, Plath K, Clark AT, et al. Histone h3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol. Cell. 2009;33:417–427. doi: 10.1016/j.molcel.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ransom M, Dennehey BK, Tyler JK. Chaperoning histones during DNA replication and repair. Cell. 2010;140:183–195. doi: 10.1016/j.cell.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwasaki W, Tachiwana H, Kawaguchi K, Shibata T, Kagawa W, Kurumizaka H. Comprehensive structural analysis of mutant nucleosomes containing lysine to glutamine (KQ) substitutions in the H3 and H4 histone-fold domains. Biochemistry. 2011;50:7822–7832. doi: 10.1021/bi201021h. [DOI] [PubMed] [Google Scholar]
- 32.Tachiwana H, Kagawa W, Shiga T, Osakabe A, Miya Y, Saito K, Hayashi-Takanaka Y, Oda T, Sato M, Park SY, et al. Crystal structure of the human centromeric nucleosome containing CENP-A. Nature. 2011;476:232–235. doi: 10.1038/nature10258. [DOI] [PubMed] [Google Scholar]
- 33.Luger K, Rechsteiner TJ, Richmond TJ. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 34.Little JW, Kim B, Roland KL, Smith MH, Lin LL, Slilaty SN. Cleavage of LexA repressor. Methods Enzymol. 1994;244:266–284. doi: 10.1016/0076-6879(94)44022-0. [DOI] [PubMed] [Google Scholar]
- 35.Flaus A, Luger K, Tan S, Richmond TJ. Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proc. Natl Acad. Sci. USA. 1996;93:1370–1375. doi: 10.1073/pnas.93.4.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clegg RM. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 37.Manohar M, Mooney AM, North JA, Nakkula RJ, Picking JW, Edon A, Fishel R, Poirier MG, Ottesen JJ. Acetylation of histone H3 at the nucleosome dyad alters DNA-histone binding. J. Biol. Chem. 2009;284:23312–23321. doi: 10.1074/jbc.M109.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang C, Pugh BF. A compiled and systematic reference map of nucleosome positions across the Saccharomyces cerevisiae genome. Genome Biol. 2009;10:R109. doi: 10.1186/gb-2009-10-10-r109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 41.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 42.Rhodes D. Structural analysis of a triple complex between the histone octamer, a Xenopus gene for 5S RNA and transcription factor IIIA. EMBO J. 1985;4:3473–3482. doi: 10.1002/j.1460-2075.1985.tb04106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nightingale KP, Pruss D, Wolffe AP. A single high affinity binding site for histone H1 in a nucleosome containing the Xenopus borealis 5 S ribosomal RNA gene. J. Biol. chem. 1996;271:7090–7094. doi: 10.1074/jbc.271.12.7090. [DOI] [PubMed] [Google Scholar]
- 44.Pruss D, Wolffe AP. Histone-DNA contacts in a nucleosome core containing a Xenopus 5S rRNA gene. Biochemistry. 1993;32:6810–6814. doi: 10.1021/bi00078a002. [DOI] [PubMed] [Google Scholar]
- 45.Kurumizaka H, Wolffe AP. Sin mutations of histone H3: influence on nucleosome core structure and function. Mol. Cell. Biol. 1997;17:6953–6969. doi: 10.1128/mcb.17.12.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thastrom A, Lowary PT, Widlund HR, Cao H, Kubista M, Widom J. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J. Mol. Biol. 1999;288:213–229. doi: 10.1006/jmbi.1999.2686. [DOI] [PubMed] [Google Scholar]
- 47.Andrews AJ, Chen X, Zevin A, Stargell LA, Luger K. The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions. Molecular cell. 2010;37:834–842. doi: 10.1016/j.molcel.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makde RD, England JR, Yennawar HP, Tan S. Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature. 2010;467:562–566. doi: 10.1038/nature09321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thastrom A, Lowary PT, Widom J. Measurement of histone-DNA interaction free energy in nucleosomes. Methods. 2004;33:33–44. doi: 10.1016/j.ymeth.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 50.Ganapathi M, Palumbo MJ, Ansari SA, He Q, Tsui K, Nislow C, Morse RH. Extensive role of the general regulatory factors, Abf1 and Rap1, in determining genome-wide chromatin structure in budding yeast. Nucleic Acids Res. 2011;39:2032–2044. doi: 10.1093/nar/gkq1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou X, O’Shea EK. Integrated approaches reveal determinants of genome-wide binding and function of the transcription factor Pho4. Mol. Cell. 2011;42:826–836. doi: 10.1016/j.molcel.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rhee HS, Pugh BF. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 2011;147:1408–1419. doi: 10.1016/j.cell.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446:572–576. doi: 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- 54.Fishel R. Mismatch repair, molecular switches, and signal transduction. Genes Dev. 1998;12:2096–2101. doi: 10.1101/gad.12.14.2096. [DOI] [PubMed] [Google Scholar]
- 55.Thastrom A, Bingham LM, Widom J. Nucleosomal locations of dominant DNA sequence motifs for histone-DNA interactions and nucleosome positioning. J. Mol. Biol. 2004;338:695–709. doi: 10.1016/j.jmb.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 56.Poirier MG, Oh E, Tims HS, Widom J. Dynamics and function of compact nucleosome arrays. Nat. Struct. Mol. Biol. 2009;16:938–944. doi: 10.1038/nsmb.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Poirier MG, Bussiek M, Langowski J, Widom J. Spontaneous access to DNA target sites in folded chromatin fibers. J. Mol. Biol. 2008;379:772–786. doi: 10.1016/j.jmb.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nelson CJ, Santos-Rosa H, Kouzarides T. Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell. 2006;126:905–916. doi: 10.1016/j.cell.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 59.Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, Kouzarides T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461:819–822. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baker SP, Phillips J, Anderson S, Qiu Q, Shabanowitz J, Smith MM, Yates JR, III, Hunt DF, Grant PA. Histone H3 Thr 45 phosphorylation is a replication-associated post-translational modification in S. cerevisiae. Nat. Cell. Biol. 2010;12:294–298. doi: 10.1038/ncb2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hurd PJ, Bannister AJ, Halls K, Dawson MA, Vermeulen M, Olsen JV, Ismail H, Somers J, Mann M, Owen-Hughes T, et al. Phosphorylation of histone H3 Thr-45 is linked to apoptosis. J. Biol. Chem. 2009;284:16575–16583. doi: 10.1074/jbc.M109.005421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.