


Abstract
Translation of a gene is assumed to be efficient if the supply of the tRNAs that translate it is high. Yet high-abundance tRNAs are often also at high demand since they correspond to preferred codons in genomes. Thus to fully model translational efficiency one must gauge the supply-to-demand ratio of the tRNAs that are required by the transcriptome at a given time. The tRNAs’ supply is often approximated by their gene copy number in the genome. Yet neither the demand for each tRNA nor the extent to which its concentration changes across environmental conditions has been extensively examined. Here we compute changes in the codon usage of the transcriptome across different conditions in several organisms by inspecting conventional mRNA expression data. We find recurring dynamics of codon usage in the transcriptome in multiple stressful conditions. In particular, codons that are translated by rare tRNAs become over-represented in the transcriptome in response to stresses. These results raise the possibility that the tRNA pool might dynamically change upon stress to support efficient translation of stress-transcribed genes. Alternatively, stress genes may be typically translated with low efficiency, presumably due to lack of sufficient evolutionary optimization pressure on their codon usage.
INTRODUCTION
Organisms have evolved means to tune the translational efficiency of their genes to different desired levels. Facilitating this mode of regulation is the redundancy of the genetic code—synonymous codons are translated to the same amino acid, but their corresponding tRNAs might differ by their amounts in cells. Common measures of translational efficiency assess genes by either measuring the correlation between their codon usage pattern to that of selected ‘elite’ highly expressed genes (1,2), or by an explicit weight of the availability of tRNAs that translate them (3). One Such measure is the tRNA adaptation index, tAI (4), which deduces the abundance of the various tRNAs from their gene copy number (GCN) in the genome. The tAI measure predicts with reasonable accuracy both mRNA and protein levels (5,6).
Yet, recent studies suggest that models of translational efficiency should be more comprehensive [reviewed in (7); (8,9)]. In particular, the concentration of the various tRNAs can vary to different extents between conditions and tissues (9–13) and so is their base modification and amino acid loading (14–17). In addition codon usage was shown to vary between tissues (18). Toward a comprehensive model of translational efficiency we focus here on an unexplored important consideration—the supply-to-demand ratio of the different tRNAs that translate mRNAs.
Specifically, we explore the potential changes in the demand for each tRNA, namely the abundance of the corresponding codon(s) in the transcriptome at various biological conditions. We suggest a simple means to mine mRNA expression data in order to explore the dynamic codon usage across conditions and species. We find that under stress conditions the demand for tRNAs by certain codons increases, whereas the extent of representation of other codons in the transcriptome decreases. Interestingly the codons whose representation increases the most in the transcriptome upon stress correspond to tRNAs that are represented by the lowest GCNs in the genome in each of the examined species. This situation suggests two possible explanations: the supply for these codons increases too in stress, e.g. by increased production of the corresponding tRNAs, or that stress-related genes remain relatively poorly translated. We constructed a simulated evolution model that uses two minimalist assumptions: that stress genes are expressed infrequently during evolution, and that the supply of tRNAs in the cell is limited. Results from the simulation are consistent with the hypothesis that limited codon adaptiveness of the stress genes results from lack of evolutionary constraint to optimize them and due to the limitation in the supply of tRNAs. Reassuringly the simulation reproduces codon usage differences observed in genomes. We thus suggest that the stress transcriptome remains poorly translated due to compromised translational efficiency of the stress genes.
MATERIALS AND METHODS
Data sources
The affymetrix platform of micro-arrays was used to allow absolute, i.e. non-relative, measurements of mRNA abundance. Normalized mRNA abundances for Saccharomyces cerevisiae following oxidative stress and DNA damage by methyl methanesulfonate (MMS) were downloaded from (19) and in heat shock, oxidative stress and osmotic stress from (20). Normalized mRNA abundances of S. cerevisiae during a 15 days process of wine fermentation (21) were downloaded from GEO (Gene Expression Omnibus, www.ncbi.nlm.nih.gov/geo), GEO accession: GSE8536. Normalized mRNA abundances of Schizosaccharomyces pombe during a short-term response to nitrogen starvation (22) were downloaded from ArrayExpress (http://www.ebi.ac.uk/microarray-as/ae/) under accession E-TABM-784. Transcriptome profiling data for Caenorhabditis elegans in response to oxidative stress (23) were downloaded from GEO under accession number GSE9301. The tRNA GCNs of species were downloaded from the Genomic tRNA Database (http://lowelab.ucsc.edu/GtRNAdb/), (24).
Generation of condition-dependent demand matrices
We created condition-dependent ‘Codon-Expression’ matrices for various environmental conditions. A ‘Codon-Expression’ matrix, is a 61 × m matrix, which denotes the representation of the 61 codons in the transcriptome over m conditions. This matrix thus depicts the demand for each tRNA at each condition. The representation of codon i in the transcriptome at a given time/condition k is defined by
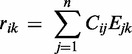 |
where j is a gene, Cij depicts the number of appearances of codon i in gene j, and Ejk indicates the mRNA abundance of gene j at condition or time point k. In the same manner, we generated ‘Amino acid Expression’ and ‘Nucleotide Expression’ matrices, which similarly depict the representation of the 20 amino acid in the translated transcriptome or the 4 nucleotides in the transcriptome at a given time/condition k.
Calculating translational efficiency by the tAI value
We calculated translation efficiencies of genes using the tAI (4). Throughout this article, we distinguish between translational efficiency of a gene, which corresponds to the original tAI measure, and translational efficiency of individual codons (originally defined by dos Reis et al. as the ‘absolute adaptiveness value’, Wi). Briefly, Wi defines the adaptiveness of an individual codon by the availability of the tRNAs that serve in translating it, incorporating both the fully matched tRNA, as well as tRNAs that contribute to translation through wobble rules (25–27). Formally, the ‘absolute adaptiveness value’ for the i-th codon is
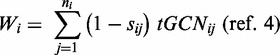 |
where n is the number of tRNA isoacceptors that recognize the i-th codon, 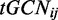 denotes the GCN of the j-th tRNA that recognizes the i-th codon, and
denotes the GCN of the j-th tRNA that recognizes the i-th codon, and 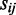 correspond to the wobble interaction, or selective constraint on the efficiency of the pairing between codon i and anticodon j. As done in the original tAI formalism by dos Reis et al. the absolute adaptiveness value of codon i is further divided by the maximum Wi (termed Wmax), obtaining the codon's relative adaptiveness value:
correspond to the wobble interaction, or selective constraint on the efficiency of the pairing between codon i and anticodon j. As done in the original tAI formalism by dos Reis et al. the absolute adaptiveness value of codon i is further divided by the maximum Wi (termed Wmax), obtaining the codon's relative adaptiveness value:
The tAI value of a gene with L codons is then simply calculated as the geometric mean of the wi's of its codons
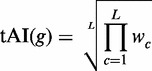 |
Based on (9) and (28) we interpret the codon adaptiveness values as representatives of translation speed of each codon.
Exploring the balance between drift and selection on codon usage by a computational simulation
We developed a computer evolutionary simulation of unicellular population of 1 000 000 haploid cells that cope with occasional stress periods. The genome of each cell consists of six archetypal genes, each of which is required during one or more of the simulated growth conditions (see Supplementary Material for details). We envisage a mapping function, such as the tAI score, that maps between sequence and expression level. During the simulation genes are mutated and as a consequence their expression level changes. At the beginning of the simulation, the six genes are equally scored with an initial value of expression level. The population then evolves at a fixed mutation rate of 0.001 mutations per genome per generation. Sequences are not represented explicitly in the simulation; instead genes are characterized by an expression level that implicitly corresponds to a genotype. Thus, ‘mutated’ expression levels at a given time step are computed by the previous step’s expression levels multiplied by a random number drawn from an exponential probability distribution of changes in expression [as estimated before (29)].
We also ran the simulation with a mode in which the tRNA supply is not unlimited. This mode of the run sets a constant maximal total expression level from the ‘genome’. In this mode of limited supply of tRNAs, the expression of the i-th gene in each generation, 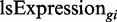 , (‘ls’ stands for limited supply) is defined by
, (‘ls’ stands for limited supply) is defined by
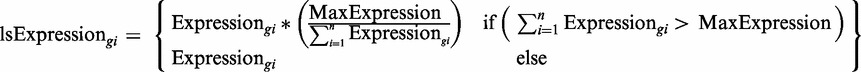 |
where n is the number of genes in the cell, Expressiongi is the expression of the i-th gene in the mutated population, and MaxExpression is a constant maximal total expression level from the whole ‘genome’.
The fitness of a given cell is determined by the arithmetic mean of expression of the m genes which are required in a given environmental condition
where 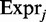 corresponds to either
corresponds to either 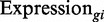 or
or 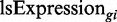 , depending on the mode of simulation, i.e. if supply is limited or not.
, depending on the mode of simulation, i.e. if supply is limited or not.
Our model consists of drift and selection, thus we propagate individuals between consecutive generations (t−1) to (t) in two stages. First, a population (whose size can be different from that of the population at generation t−1) is formed in which the i-th genotype population size is given by its size in the previous generation and its fitness by
Then, to keep a constant population size stochastic rescaling is applied that implements a Kimura-governed (30) allele sampling as the random drift step of the simulation.
RESULTS
The supply-to-demand balance in translational efficiency
The speed at which a codon is translated is expected to increase with the availability of its supply—amino acid-loaded tRNAs. Yet, if the codon is highly represented in the genome, and even more importantly, in transcriptome at a given condition, i.e. if the demand for the tRNA is high, then the codon’s translational efficiency might be compromised. Translational efficiency should thus be modeled as a supply-to-demand process, i.e. the ratio between the availability of the tRNAs that translate a codon—the ‘supply’, to the extent of representation of that codon in the transcriptome at a given moment—the ‘demand’. The supply component is effectively captured by the tAI of a codon (4), originally defined as Wi and termed codon ‘adaptiveness value’ (see ‘Materials and Methods’ section). The demand component is simply modeled as the representation of the i-th codon in the transcriptome, 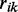 (see ‘Materials and Methods’ section).
(see ‘Materials and Methods’ section).
Translational efficiency of the i-th codon is the supply-to-demand ratio:
In the following sections we will focus on measuring the dynamics of codon usage across conditions in different species. Note that although in the original tAI model the supply is constant (modeled by copy number of the tRNA genes in the genome) it could in future be represented as a dynamic entity too, accounting for the possibility that tRNA levels may change across conditions and cell types (9–12,14–17).
mRNA expression data can be mined to deduce dynamics of cellular codon usage
Conventional mRNA expression micro-arrays are typically used to study transcription and more recently mRNA decay (19,31–37). Here we realized that micro-arrays may contain data pertinent to translational efficiency. In particular we mine expression micro-array data to compute changes in the representation of the various codons in the transcriptome in various growth conditions in several model organisms. Figure 1 shows as an example the change in usage of the six codons coding for arginine during response to a DNA damaging agent in the yeast S. cerevisiae (see ‘Materials and Methods’ section). The representation of some of the codons increases by as much as 25% in stress, whereas others decrease in the stressed transcriptome. These changes in codon representation might indicate a change in the demand for the various tRNAs, and potentially also a change in translational efficiency of some genes in stress. Interestingly the summed usage of all six arginine codons hardly changes, and as so is the usage of the four nucleotides.
Figure 1.

Variations in representation of amino acids, codons and their constituent nucleotides in the transcriptome. Illustration of the case of amino acid arginine: shown on the y-axis is the (log2) change in the representation of the six codons of this amino acid in the transcriptome during response to the chemical MMS. Four of the codons are increased in their representation following the stress compared with a reference condition, and two are diminished. This change is not accompanied by an appreciable change in the representation of arginine in the translated transcriptome, nor can it be reduced to a putative change in the amount of the constituent nucleotides which are shown to hardly vary during the process.
We systemize our inspection of fluctuations of codons representation in the transcriptome. By multiplying the number of occurrences of each codon in each gene by the expression level of each gene in each condition we obtain a ‘Codon-Expression’ matrix which depicts the representation of each codon in the transcriptome in each condition (see ‘Materials and Methods’ section).
Similarly, for further control purposes, we create ‘Amino acid Expression’ and ‘Nucleotide-Expression’ matrices, which correspond, respectively, to the sum of codons for each amino acid or the representation of each nucleotide in the transcriptome at various conditions in a given species. The amino acid expression matrix allows us to ask whether changes at the codon expression matrix simply reflect changes in the relative appearance of the different amino acids at the translated transcriptome [as shown in some cases, c.f. (38)] whereas the nucleotide expression matrix weighs the putative changes in the nucleotide composition of the transcriptome (39).
The codon usage of the transcriptome varies upon stress
We analyzed mRNA expression data for the yeast S. cerevisiae in diversity of stressful environmental conditions, and also during recovery from stress (19–21). For each time point in each condition we computed the ‘Codon-Expression’ matrix (Figure 2). Qualitatively, the pattern of variation in codon usage is highly correlated between the various stresses and anti-correlated between the stresses and the stress-recovery experiments. The codons that are changed the most in stress show an increased representation of up to 40% relative to the reference condition (Figure 2). In contrast, changes in amino acid representation are very minor (Supplementary Figure S1), and it is often the case that codons for the same amino acid change in opposite directions. The representation of the four nucleotides is also relatively constant throughout conditions—fold-change values vary between 0.99–1.01 and 0.98–1.03 for codon position-independent and codon position-dependent usage of nucleotides, respectively (Supplementary Figure S1). Thus codon representation changes are not explained by a need to change the amino acid composition of the proteome and cannot be reduced to potential change in nucleotide availability. In turn this dynamics likely reflects changes in translational efficiency.
Figure 2.

The fold-changes in codons representation in S. cerevisiae's transcriptome across environmental changes. Left panel—fold-changes in the representation of the 61 codons in response to heat shock, osmotic and MMS stresses, and when potassium chloride (KCL) and heat-shock stresses were removed (time is denoted in minutes). Second panel from left—fold-changes in representation of the 61 codons in yeast's transcriptome throughout a 15-day wine fermentation. Columns marked with ‘stress’ denote the fold-changes at a given stress compared with time point zero, before the stress was applied, whereas ‘recovery’ marks columns which show the fold-changes after the stress, as cells were transferred to non-stressful conditions, compared with the stress conditions (refers to time point 45 of the respective stress). Columns marked with 'WF' represent the various time points (in hours) during the wine fermentation process. The matrices were normalized by dividing the fold-change values of individual codons at each time point to the total fold-change values across all codons. In addition the right most two columns depict for each codon the gene copy number of its fully matched tRNAs (tRNA's GCN), as well as its codon adaptiveness value. The codons in the matrices are sorted according to their fold-change increase in the stresses. As seen, codons that are increased in representation during either stress or fermentation have low tRNA GCN and low adaptiveness values.
Low-efficiency codons are over-represented in the stress transcriptome
We were next interested to check whether changes in codon representation in stress, or during recovery from stress, are correlated with the abundance of the corresponding tRNAs. A common simplified proxy for tRNA availability is the copy number of the tRNA genes in the genome. tRNA GCNs correlate with tRNA concentrations, at least in non-stressful conditions (9,11,40,41). This correlation was recently corroborated in a study that examined in several mammals RNA Pol III occupancy in the vicinity of tRNA genes (13). We thus plotted for each codon its representation in the transcriptome at various conditions along with the GCN of the corresponding tRNA, and in addition a more refined measure of tRNA availability (called codon adaptiveness value, 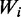 , see ‘Materials and Methods’ section) that also incorporates contributions to the translation of a codon through wobble interaction (25) from non-perfectly matching tRNAs (Figure 2). Interestingly the codon change pattern during recovery from stress correlates positively with these two measures of tRNA availability, while the changes during stress showed negative correlation with the tRNA availability (Figure 3a, Supplementary Figure S2 and legends for numerical details). This result means that during stress the transcriptome’s codon usage shifts from codons that correspond to the tRNAs that are represented by high gene counts toward codons that correspond to tRNAs that are typically encoded by few gene copies.
, see ‘Materials and Methods’ section) that also incorporates contributions to the translation of a codon through wobble interaction (25) from non-perfectly matching tRNAs (Figure 2). Interestingly the codon change pattern during recovery from stress correlates positively with these two measures of tRNA availability, while the changes during stress showed negative correlation with the tRNA availability (Figure 3a, Supplementary Figure S2 and legends for numerical details). This result means that during stress the transcriptome’s codon usage shifts from codons that correspond to the tRNAs that are represented by high gene counts toward codons that correspond to tRNAs that are typically encoded by few gene copies.
Figure 3.


Correlation between variation in codons' usage and their translational efficiency across environmental changes. Each panel denotes the correlation between the adaptiveness values (Wi) of the 61 codons and the fold-changes in their representation in the transcriptome (a) Left panel—response to osmotic stress in S. cerevisiae; Pearson correlation = −0.66, P-value = 5.37 × 10−9. Right panel—recovery from osmotic stress; Pearson correlation = 0.69, P-value = 1.01 × 10−9. (b) Left panel—a short-term response to nitrogen starvation in S. pombe. Pearson correlation = −0.8, P-values = 1.02 × 10−14; Right panel—response to oxidative stress in C. elegans, Pearson correlation = −0.59, P-value = 5.52 × 10−7.
Stressful conditions induce a similar pattern of changes in codon usage in additional species
We next wanted to check whether the tendency to increase the representation of codons that correspond to low gene copy tRNAs in stressful conditions is shared in other species too. We analyzed the fission yeast S. pombe during a short-term response to nitrogen starvation (22) and the worm C. elegans in response to oxidative stress (23). The results clearly show a consistent trend—increased representation in the transcriptome upon stress of codons that correspond to tRNAs whose GCN is low—the Pearson correlation between the change in codon representation in the transcriptome in stress and the availability of the corresponding tRNA is −0.59 (P-value = 5.52 × 10−7) and −0.8 (P-value = 1.02 × 10−14), for C. elegans and S. pombe, respectively (Figure 3b).
In some cases a given codon may have high tRNA availability in one species and a low availability in another. We found out that such codons show increased representation in the stressed transcriptome only in the species where their corresponding tRNAs are at low level. For instance, codon GGA (Gly) has the highest 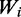 (codon adaptiveness value) in C. elegans, and relatively low value in S. cerevisiae, and reassuringly it shows decreased representation in the transcriptome of worm upon oxidative stress, whereas in yeast it is among the ten most elevated codons in stress. Thus the similarity of our trend across species does not simply result from a similar behavior of the same codon across species, but may rather reflect a commonality in which the codons that correspond to the rare tRNAs in each species respond similarly.
(codon adaptiveness value) in C. elegans, and relatively low value in S. cerevisiae, and reassuringly it shows decreased representation in the transcriptome of worm upon oxidative stress, whereas in yeast it is among the ten most elevated codons in stress. Thus the similarity of our trend across species does not simply result from a similar behavior of the same codon across species, but may rather reflect a commonality in which the codons that correspond to the rare tRNAs in each species respond similarly.
Distinct stress-specific genes induce a similar signature of changes in codon usage in various stresses
Our results in S. cerevisiae show a similar signature of codon usage change in stress across multiple stress types. This similarity could simply arise from a common set of general-stress genes that respond similarly to all stresses (32). In contrast we wanted to examine the possibility that distinct genes sets, that are each specific to each of the stresses, may also present the same trend. To examine this possibility we created distinct gene sets, each shows induction or repression of at least 2-fold in only one of the stress conditions. In addition we created a ‘general stress gene set’ that consists of genes that were either induced or repressed by at least 2-fold in all examined stresses. We characterized each of these gene sets by the change in the representation of each of the 61 codons in the various stress types (considering both the codon usage and the mRNAs levels see Materials and Methods for details). Then, we compared between the variations in codon representation of the gene sets across environmental changes (Figure 4).
Figure 4.

Clustering of codon usage profiles of general stress genes and stress-specific genes. This analysis compared between the codon usage profiles of various sets of genes that are characterized by their mRNA expression pattern across environmental changes. The fold-changes in the codon usage at each time point of a given stress were calculated separately for the stress specific and the general stress genes. In addition the codon-tAI values (Wi) are shown. Each column and row corresponds to one time point in a particular stress. The name of each codon set consists of the condition, followed by time point and a ‘S’ or ‘G’ designation indicating whether it was derived from the genes that were specifically changed at that stress or the general stress responsive genes, respectively. Hierarchical clustering was performed with 1-Pearson correlation as a distance metric and ‘average linkage’.
Interestingly we found that all stress-specific gene sets show highly correlated pattern of change, and that each of them correlated negatively with tRNA availability in terms of codons' tAI values (Pearson correlations between −0.62 and −0.73; P-values << 0.05). In contrast, the general stress genes have a different signature that does not correlate significantly with tRNA availability (Pearson correlations between −0.22 and 0.13). This analysis indicates that in each stress type distinct genes converge upon the same pattern of increased representation of codons that correspond to low GCN tRNAs. On the other hand, the general stress genes are actually better adapted to the high gene copy tRNAs.
Why are not all genes codon-optimized? A potential effect of genetic drift and a limited tRNA supply
An intriguing question is why did not evolution optimize the codon usage of all genes in a genome? We hypothesize that at least three factors may account for this situation: (i) Drift versus selection balance: drift may erode codon optimization of genes, thus counteracting the force of purifying selection and codon optimization in particular. Intuitively, while drift should constantly act on all genes at all times, selection should mainly act on a gene during evolutionary time periods and conditions in which it is needed. Thus genes that are expressed rarely during the life-history would experience compromised selection-to-drift ratio. (ii) Limitation in the supply: if the pool of amino acid-loaded tRNAs is not in great excess (see Discussion) and all genes were biased toward codons that correspond to abundant tRNAs then the demand for such tRNAs might be too high and translation would not be efficient even in genes in which high translational efficiency is most needed and thus expressed. Hence some genes may need to ‘give way’ to others. (iii) In addition, it is entirely possible that some genes may actually be needed in low level of expression and are thus deliberately not codon-optimized (42,43).
Focusing on testing the first two scenarios we constructed an evolutionary dynamics computer simulation to test the effect of drift-to-selection ratio and of demand-to-supply ratio on codon optimization levels of genes. Our simulated model consists of a constant-size population of 1 000 000 cells, each possess a genotype of 6 archetypal genes—a house-keeping gene which is expressed in every environment and growth condition, a ‘good-life' gene which is expressed only at favorable growth conditions, three ‘stress-specific' genes, each uniquely associated with one out of three different stress types, and a ‘general-stress' gene, which is expressed in any of the stress types, but not during the favorable growth condition. The population experiences changes in the environmental conditions that could be either stressful or optimal. Stresses come in three different types. The different genes are either expressed or not depending on the prevailing conditions at a given moment. During cell doublings mutations are randomly seeded in the genome. These mutations are modeled here as changing codon usage and we refer to them by their effect on expression levels. The fitness of each cell in a given condition is determined by the expression level of all the genes that are needed at that condition, and growth rate is proportional to fitness (see ‘Materials and Methods’ section for further details). For example, under a particular stress the fitness would be affected by the expression level of the gene that is needed specifically at that condition, and also by the expression level of the ‘house-keeping’ and general-stress genes. Thus each gene is subject to the effect of drift throughout the simulation, but selection is acting upon it only during times in which the environment is presenting the conditions that require it. We ran the simulation for 10 000 generations (see ‘Materials and Methods’ section) and followed the extent of expression level of the various genes. We ran the simulation in two modes. In the first mode, there was no bound on the total expression level from all the genes in the genome. This mode simulates a situation in which the tRNA supply was not limiting and only drift limits expression of genes. In the second mode of the simulation the supply of the tRNA is limited so that not all genes can be optimized simultaneously. Hence, whereas in the first mode only drift can compromise expression level of genes, in the second one it was a combination of drift and limited tRNA supply that could act together in limiting expression levels of genes. For each mode, we applied three different environmental regimes, in which the total duration of stressful conditions constitutes 20, 50 or 80% of the total evolutionary time.
The results of the simulation, under the two modes (Figure 5a) show that on average, the ‘house-keeping’ gene has the highest expression level, followed by either the good-life gene or general stress gene, depending on the fraction of evolutionary time that the population spent in stressful or non-stressful conditions. Regardless of the duration of stressful or non-stressful conditions, the stress-specific genes show the lowest expression level. The differences are observed when drift alone limits genes expression and it becomes even more pronounced when imposing a limitation on the tRNA supply.
Figure 5.

Translational efficiency of different gene sets: comparison of genome data and simulations. (a) Measurements of translational efficiency for environmental-dependent gene sets by computational simulation. We simulated population of cells, each possessing a genotype of six genes—a house-keeping gene (green) which is expressed constitutively, a ‘good-life' gene (purple), which is expressed only at favorable growth conditions, three stress-specific genes, each expressed in only one of the stress conditions (represented by their average, colored in gray), and, a ‘general stress’ gene (blue), which is expressed in all of the stress types. Shown for each case is the averaged expression level across the entire simulated evolutionary time, each average, in turn, represents a mean of 50 independent runs of the simulation. The upper panel displays a simulation mode in which only drift limits expression of genes, whereas in the lower panel the tRNA supply too is limited so that not all genes can be optimized simultaneously. The three blocks of bars represent three different environmental regimes that differ in the percentage of evolutionary time in which the population was exposed to stress. (b) Measurements of averaged gene tAI values for various sets of genes from the S. cerevisiae genome. The bars show the mean tAI value for house keeping genes (green), ‘good-life’ genes, defined to be the stress-repressed genes (purple), general stress genes (blue) and stress-specific genes, i.e. the union of the gene sets that are specific to each of the stresses (gray).
How well does the simulation predict differences in codon optimization between house-keeping, stress-specific, general-stress and ‘good-life’ genes in the genome? To test the simulation we examined translational efficiency, by the tAI measure in the following S. cerevisiae gene sets, defined based on micro-array expression levels: (i) ‘house-keeping genes’, defined here as genes that maintain constant expression level in all conditions (maximal absolute change in expression of 15%); (ii) ‘good-life’ genes which are repressed under stressful conditions; (iii) general stress genes which are induced in every stress; and (iv) stress-specific genes which are induced in exactly one of the examined stresses (set (iii) and (iv) defined as mentioned above). Figure 5b shows the mean tAI (‘Materials and Methods’ section) values of the genes in each of the four gene sets. The results show a clear rank with significant differences between the gene sets: house-keeping genes show the highest tAI values, followed by the ‘good-life’ genes, followed by the general stress genes, and ending with the stress-specific genes who show the lowest values (all differences are significant P-value < 0.05, t-test). These results are in good agreement with the simulation and they suggest that genes that are rarely used during the life-history would be poorly optimized and display low expression levels.
DISCUSSION
Traditional measures of translational efficiency either consider the codon usage pattern of the coding sequence alone, or additionally weigh the tRNA pool. Yet, even if the tRNA supply is explicitly taken into consideration, translational efficiency should be further evaluated by the actual consumption of tRNAs. Thus, we suggest here that translational efficiency should be thought as a demand versus supply model, in which the supply is given by the tRNAs availability, and the demand is captured by the representation of the ‘consumers’—the codons—in the transcriptome. We show here that the demand varies across conditions, and we anticipate that future investigations will strengthen the notion that the supply too is not constant either in time or between cell types and tissues (10,13).
Our comprehensive model of translational efficiency is based on analysis of the supply-to-demand of the various tRNAs that translate each gene (Figure 6). Both demand and supply may be constant or change across conditions, cell types and developmental stages. Thus, our model of translation efficiency challenges the prevailing simplifying assumption that translation efficiency of a given gene is constant throughout organism life. In turn, our model implies for dynamic range of translation efficiency, suggesting that the interplay between tRNAs availability and codons representation play a role in shaping expression levels of individual genes throughout organism life.
Figure 6.

Putative dynamics of supply and demand for tRNAs and its implication on translational efficiency. The figure illustrates five potential modes of control of changes in the tRNAs supply (blue) and demand (red). In the various regimes the supply and demand might both be constant, or either of them, or both of them might change dynamically. Corresponding to each mode of control is the expected effect on translational efficiency (bottom, green).
Recently, the change in tRNA synthesis was measured across organs and species in several mammals (13) and was shown to be relatively constant within sets of tRNAs that share an anticodon. Such technology may reveal potential changes in the tRNA pool in micro-organisms when they respond to different conditions. A change or lack of a change in the tRNA supply would affect translation provided that the tRNA is in limiting amounts. Utilizing published estimations on the amount of tRNAs molecules and the translated portion of mRNAs in yeast, we calculated (Supplementary Material) a tRNAs-to-codons ratio for codons. Our calculations, although rough, suggest that tRNAs and their respective codons are present in the cell in the same order of magnitudes of copy numbers. This result implies that there is no appreciable surplus of tRNAs which could buffer changes in the demand without a change in the basal tRNA levels.
A main observation of this study is that the stress transcriptome is poorly adapted to the constant tRNA pool. The challenge is thus to explain this somewhat non-intuitive finding. Clearly a fitness advantage could be gained from better adapted stress codon usage. Yet, as we suggested above, a force that counteracts adaptation is genetic drift. Using a computational simulation, we demonstrate that the balance between drift and selection on an evolutionary time scale may explain the low adaptation of stress-specific genes. Real genomes too have been shown to follow such logic, whereby genes that are needed infrequently in the ecology or life-style of a given species remain poorly adapted to its tRNA pool (5,17,44,45). For these genes the eroding effect of drift prohibits optimal adaptation. This scenario is consistent with case no. 2 in Figure 6, which discusses the possibility that the tRNA pool does not change to match, or counteract a change in codon usage of the transcriptome. However, the simulation results of course do not exclude the possibility that the tRNA pool does change in a condition-dependent manner. Indeed our recent observation actually indicate in that direction (9) as they show that during diauxic shift in yeast the tRNA pool does change to a minor extent. Future experimental efforts along these lines would be needed to establish the possibility that the tRNA pool changes dynamically potentially to off-set changes at the demand.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1 and 2.
FUNDING
Funding for open access charge: European Research Council ‘Starting’ program.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the ‘Ideas’ program of the European Research Council and the Ben May Charitable Trust for grant support.
REFERENCES
- 1.Sharp PM, Li WH. The codon adaptation index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wright F. The ‘effective number of codons' used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 3.Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J. Mol. Biol. 1981;151:389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 4.dos Reis M, Savva R, Wernisch L. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 2004;32:5036–5044. doi: 10.1093/nar/gkh834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Man O, Pilpel Y. Differential translation efficiency of orthologous genes is involved in phenotypic divergence of yeast species. Nat. Genet. 2007;39:415–421. doi: 10.1038/ng1967. [DOI] [PubMed] [Google Scholar]
- 6.Tuller T, Waldman YY, Kupiec M, Ruppin E. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl Acad. Sci. USA. 2010;107:3645–3650. doi: 10.1073/pnas.0909910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gingold H, Pilpel Y. Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 2011;7:481. doi: 10.1038/msb.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cannarozzi G, Schraudolph NN, Faty M, von Rohr P, Friberg MT, Roth AC, Gonnet P, Gonnet G, Barral Y. A role for codon order in translation dynamics. Cell. 2010;141:355–367. doi: 10.1016/j.cell.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 9.Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010;141:344–354. doi: 10.1016/j.cell.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 10.Dittmar KA, Goodenbour JM, Pan T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006;2:e221. doi: 10.1371/journal.pgen.0020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong H, Nilsson L, Kurland CG. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 1996;260:649–663. doi: 10.1006/jmbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- 12.Heyman T, Agoutin B, Fix C, Dirheimer G, Keith G. Yeast serine isoacceptor tRNAs: variations of their content as a function of growth conditions and primary structure of the minor tRNA(Ser)GCU. FEBS Lett. 1994;347:143–146. doi: 10.1016/0014-5793(94)00524-9. [DOI] [PubMed] [Google Scholar]
- 13.Kutter C, Brown GD, Goncalves A, Wilson MD, Watt S, Brazma A, White RJ, Odom DT. Pol III binding in six mammals shows conservation among amino acid isotypes despite divergence among tRNA genes. Nat. Genet. 2011;43:948–955. doi: 10.1038/ng.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sorensen MA. Charging levels of four tRNA species in Escherichia coli Rel(+) and Rel(-) strains during amino acid starvation: a simple model for the effect of ppGpp on translational accuracy. J. Mol. Biol. 2001;307:785–798. doi: 10.1006/jmbi.2001.4525. [DOI] [PubMed] [Google Scholar]
- 15.Elf J, Nilsson D, Tenson T, Ehrenberg M. Selective charging of tRNA isoacceptors explains patterns of codon usage. Science. 2003;300:1718–1722. doi: 10.1126/science.1083811. [DOI] [PubMed] [Google Scholar]
- 16.Begley U, Dyavaiah M, Patil A, Rooney JP, DiRenzo D, Young CM, Conklin DS, Zitomer RS, Begley TJ. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol. Cell. 2007;28:860–870. doi: 10.1016/j.molcel.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaborske JM, Narasimhan J, Jiang L, Wek SA, Dittmar KA, Freimoser F, Pan T, Wek RC. Genome-wide analysis of tRNA charging and activation of the eIF2 kinase Gcn2p. J. Biol. Chem. 2009;284:25254–25267. doi: 10.1074/jbc.M109.000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plotkin JB, Robins H, Levine AJ. Tissue-specific codon usage and the expression of human genes. Proc. Natl Acad. Sci. USA. 2004;101:12588–12591. doi: 10.1073/pnas.0404957101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shalem O, Dahan O, Levo M, Martinez MR, Furman I, Segal E, Pilpel Y. Transient transcriptional responses to stress are generated by opposing effects of mRNA production and degradation. Mol. Syst. Biol. 2008;4:223. doi: 10.1038/msb.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell A, Romano GH, Groisman B, Yona A, Dekel E, Kupiec M, Dahan O, Pilpel Y. Adaptive prediction of environmental changes by microorganisms. Nature. 2009;460:220–224. doi: 10.1038/nature08112. [DOI] [PubMed] [Google Scholar]
- 21.Marks VD, Ho Sui SJ, Erasmus D, van der Merwe GK, Brumm J, Wasserman WW, Bryan J, van Vuuren HJ. Dynamics of the yeast transcriptome during wine fermentation reveals a novel fermentation stress response. FEMS Yeast Res. 2008;8:35–52. doi: 10.1111/j.1567-1364.2007.00338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kristell C, Orzechowski Westholm J, Olsson I, Ronne H, Komorowski J, Bjerling P. Nitrogen depletion in the fission yeast Schizosaccharomyces pombe causes nucleosome loss in both promoters and coding regions of activated genes. Genome Res. 2010;20:361–371. doi: 10.1101/gr.098558.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park SK, Tedesco PM, Johnson TE. Oxidative stress and longevity in Caenorhabditis elegans as mediated by SKN-1. Aging Cell. 2009;8:258–269. doi: 10.1111/j.1474-9726.2009.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crick FH. Codon–anticodon pairing: the wobble hypothesis. J. Mol. Biol. 1966;19:548–555. doi: 10.1016/s0022-2836(66)80022-0. [DOI] [PubMed] [Google Scholar]
- 26.Watanabe K, Osawa S. tRNA sequences and variation in the genetic code. In: Söll D, RajBhandary U, editors. tRNA: Structure, Biosynthesis and Function. Washington, DC: AMS Press; 1995. pp. 225–250. [Google Scholar]
- 27.Yokoyama S, Nishimura S. Modified nucleosides and codon recognition. In: Söll D, RajBhandary U, editors. tRNA: Structure, Biosynthesis and Function. Washington, DC: AMS Press; 1995. pp. 207–223. [Google Scholar]
- 28.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piganeau G, Eyre-Walker A. Estimating the distribution of fitness effects from DNA sequence data: implications for the molecular clock. Proc. Natl Acad. Sci. USA. 2003;100:10335–10340. doi: 10.1073/pnas.1833064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crow JF, Kimura M. An Introduction to Population Genetics Theory. New York: Harper & Row; 1970. p. 406. [Google Scholar]
- 31.Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA. Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell. 2001;12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D, Brown PO. Precision and functional specificity in mRNA decay. Proc. Natl Acad. Sci. USA. 2002;99:5860–5865. doi: 10.1073/pnas.092538799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernstein JA, Lin PH, Cohen SN, Lin-Chao S. Global analysis of Escherichia coli RNA degradosome function using DNA microarrays. Proc. Natl Acad. Sci. USA. 2004;101:2758–2763. doi: 10.1073/pnas.0308747101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Molin C, Jauhiainen A, Warringer J, Nerman O, Sunnerhagen P. mRNA stability changes precede changes in steady-state mRNA amounts during hyperosmotic stress. RNA. 2009;15:600–614. doi: 10.1261/rna.1403509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amorim MJ, Cotobal C, Duncan C, Mata J. Global coordination of transcriptional control and mRNA decay during cellular differentiation. Mol. Syst. Biol. 2010;6:380. doi: 10.1038/msb.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shalem O, Groisman B, Choder M, Dahan O, Pilpel Y. Transcriptome kinetics is governed by a genome-wide coupling of mRNA production and degradation: a role for RNA Pol II. PLoS Genet. 2011;7:e1002273. doi: 10.1371/journal.pgen.1002273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mazel D, Marliere P. Adaptive eradication of methionine and cysteine from cyanobacterial light-harvesting proteins. Nature. 1989;341:245–248. doi: 10.1038/341245a0. [DOI] [PubMed] [Google Scholar]
- 39.Kudla G, Lipinski L, Caffin F, Helwak A, Zylicz M. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 2006;4:e180. doi: 10.1371/journal.pbio.0040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Percudani R, Pavesi A, Ottonello S. Transfer RNA gene redundancy and translational selection in Saccharomyces cerevisiae. J. Mol. Biol. 1997;268:322–330. doi: 10.1006/jmbi.1997.0942. [DOI] [PubMed] [Google Scholar]
- 41.Kanaya S, Yamada Y, Kudo Y, Ikemura T. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene. 1999;238:143–155. doi: 10.1016/s0378-1119(99)00225-5. [DOI] [PubMed] [Google Scholar]
- 42.Konigsberg W, Godson GN. Evidence for use of rare codons in the dnaG gene and other regulatory genes of Escherichia coli. Proc. Natl Acad. Sci. USA. 1983;80:687–691. doi: 10.1073/pnas.80.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang SP, Zubay G, Goldman E. Low-usage codons in Escherichia coli, yeast, fruit fly and primates. Gene. 1991;105:61–072. doi: 10.1016/0378-1119(91)90514-c. [DOI] [PubMed] [Google Scholar]
- 44.Bahir I, Fromer M, Prat Y, Linial M. Viral adaptation to host: a proteome-based analysis of codon usage and amino acid preferences. Mol. Syst. Biol. 2009;5:311. doi: 10.1038/msb.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Botzman M, Margalit H. Variation in global codon usage bias among prokaryotic organisms is associated with their lifestyles. Genome Biol. 12:R109. doi: 10.1186/gb-2011-12-10-r109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.