


Abstract
ATP-dependent nucleic acid helicases and translocases play essential roles in many aspects of DNA and RNA biology. In order to ensure that these proteins act only in specific contexts, their activity is often regulated by intramolecular contacts and interaction with partner proteins. We have studied the bacterial Mfd protein, which is an ATP-dependent DNA translocase that relocates or displaces transcription ECs in a variety of cellular contexts. When bound to RNAP, Mfd exhibits robust ATPase and DNA translocase activities, but when released from its substrate these activities are repressed by autoinhibitory interdomain contacts. In this work, we have identified an interface within the Mfd protein that is important for regulating the activity of the protein, and whose disruption permits Mfd to act indiscriminately at transcription complexes that lack the usual determinants of Mfd specificity. Our results indicate that regulation of Mfd occurs through multiple nodes, and that activation of Mfd may be a multi-stage process.
INTRODUCTION
Proteins that move on nucleic acids participate in a broad range of cellular pathways including DNA repair, replication, recombination, transcription and chromatin remodelling. Their activity is often regulated by interactions with accessory proteins, or interdomain contacts within the proteins, which ensure that nucleic acid translocation occurs only in the correct macromolecular context. The control of DNA motor activity by autoinhibition has been observed in a number of DNA translocases, such as the bacterial proteins Rep, UvrB and Mfd (1–4). In the case of the Mfd protein the N- and C-terminal domains of the protein form an inbibitory clamp around the domains that couple ATP hydrolysis to movement of DNA (2,3,5). The autoinhibitory effect of this clamp is relieved when the Mfd protein binds to RNA polymerase (RNAP), but the mechanism by which this occurs is unclear.
Mfd is the prokaryotic transcription-repair coupling factor (TRCF), and is a member of helicase superfamily 2 (6). It plays a central role in transcription-coupled DNA repair, during which it binds to RNAP that has been stalled by a DNA lesion in the template strand, and translocates the DNA immediately upstream of the stalled transcription complex (7,8). Mfd displaces RNAP from the damaged DNA by pushing the polymerase forward, allowing the lesion to be accessed by repair proteins (8,9). It also recruits the repair protein UvrA to the site of the DNA damage and so accelerates the repair process (5,7).
Mfd is a 130 kDa monomer made up of eight domains (10) (Figure 1A). Domain 4 is the RNAP interaction domain (RID), which makes a specific interaction with the β1 domain of RNAP (11,12). Domains 5 and 6 contain the 7 motifs characteristic of a superfamily 2 helicase and constitute the ATP-dependent DNA translocase activity of the protein. In isolated Mfd, these central domains, which are essential for the RNAP-displacement activity of the protein, are surrounded by a ‘clamp’ made up of the N-terminal and C-terminal regions of the protein (3). The N-terminal part of the clamp is composed of domains 1a, 2 and 1b, and is termed the UvrB homology module (BHM) since it is structurally homologous to part of UvrB (13). The other part of the clamp is domain 7 at the C-terminus of the protein. Interactions between conserved surfaces on domain 2 and 7 hold the clamp closed, and several lines of evidence suggest that this interaction must be broken in order for the activities of Mfd to be upregulated during transcription-coupled repair.
Figure 1.

The Mfd protein. (A) Crystal structure of E. coli Mfd (PDB ID 2EYQ) (10). Residues changed to alanine in the derepressed mutants Mfd D7AAA (E1045, D1048, R1049) and Mfd D2AAA (R165, R181, F185) together with other key residues discussed in the text are shown in space-filling representation. (B) Alternative view of part of the structure, showing the juxtaposition of the RID, relay helix and hook helices.
In the absence of accessory proteins Mfd is in an autoinhibited form and exhibits very low ATPase and DNA translocase activity (2). The surface of D2 that interacts with D7 in the crystal structure of Escherichia coli apo-Mfd partially overlaps with the surface that interacts with UvrA, suggesting that Mfd must undergo major conformational changes in order to bind to UvrA during transcription-coupled repair (5,10,14). Autoinhibition of the ATP-dependent DNA translocase activity is relieved when Mfd binds to stalled transcription elongation complexes (ECs) (2,14). Substitutions that disrupt the interface between D2 and D7 relieve the autoinhibition of the protein in the absence of RNAP (5) (Figure 1), and these derepressed derivatives of Mfd are more susceptible to proteolytic cleavage than WT Mfd, suggesting that the activation of Mfd involves substantial conformational changes within the protein (15). Autoinhibition can also be overcome by deletion of either D7, or the BHM (2,3,5). SAXS analysis of Mfd in the presence of different nucleotides indicates that conformational changes occur during nucleotide binding and hydrolysis, and that these involve movement of D7 away from the remainder of the protein (14). On the basis of these results it has been proposed that binding to RNAP switches Mfd from a ‘closed’, autoinhibited form to a more open and flexible active conformation, stimulating the DNA translocation activity and allowing recruitment of the DNA repair enzymes involved in transcription-coupled repair (TCR). However, the extent of domain movement that occurs, or is required, at each stage of the TCR process is currently unclear: a recent study showed that the ATPase activity of an Mfd derivative in which D2 was cross-linked to D7 was still stimulated somewhat by binding to RNAP, and the cross-linked protein retained the ability to displace stalled transcription complexes from DNA (14).
Two non-exclusive mechanisms have been suggested to explain the autoinhibition of Mfd by interdomain contacts (2,3,10). Firstly, in the autoinhibited form of the protein the N-terminal domains (domains 1–3) form extensive interactions with the translocase domains (primarily domain 6). These interactions, stabilized by the interaction between domains 2 and 7, are proposed to constrain the movements of the translocase domains that are required to couple ATP hydrolysis to DNA translocation. In the simplest interpretation of this model the role of D7 is simply to act as a tether that holds the N-terminal domains in place. An alternative possibility is that, in addition to tethering the N-terminal domains, movement of D7 itself affects the action of the translocase domains by transmitting conformational changes through a network of alpha helices that connect the RID, the translocase domains and D7. Support for a potentially multi-stage mechanism of autoregulation comes from the observation that some mutants in the D2:D7 interface elevate ATPase activity but not DNA translocation activity, indicating that the two activities can be uncoupled (5).
The translocase domains of Mfd, and the regions that immediately flank them, are homologous to those found in RecG, an ATP driven DNA translocase that is involved in the bypass of collapsed replication forks (10,16,17). In both Mfd and RecG the translocase domains are preceded by a long helix (termed the relay helix in Mfd), which in Mfd links the RID to domain 5. The C-terminal translocase domain of both proteins contains a conserved motif termed the TRG (translocation in RecG) motif which is located downstream of the helicase domains and is essential for the DNA translocation activities of both proteins. The TRG motif consists of a helical hairpin, followed in Mfd by a pair of ‘hook’ helices that wrap around the relay helix before linking to D7 (Figure 1B). The relay, TRG and hook helices form a network of connections between the RID, the translocase domains and D7 that makes it likely that movement of any one of these domains will be transmitted to the others. To investigate the role that this network of helices plays in the regulation of Mfd we have examined the effect of disrupting the conserved interaction between the hook helices and the relay helix of Mfd. We show that disrupting this interaction results in a highly active Mfd derivative that is able to displace not only stalled ECs, but also non-specific targets such as transcription initiation complexes (ICs). The mutant protein exhibits altered interactions with DNA, and its translocase domains are more susceptible to proteolysis than previously studied activated Mfd derivatives. Our results indicate that the DNA translocation activity of Mfd is regulated in a multipartite fashion.
MATERIALS AND METHODS
Plasmid construction
The Mfd WA550 expression vector pETMfd-T7 WA550 is a derivative of the WT plasmid pETMfd-T7 (5) and was constructed by Quikchange mutagenesis (Stratagene). The Mfd WA550 RA953 expression vector pETMfd-T7 WA550 RA953 is a derivative of pETMfd-T7 WA550 which was also constructed by Quikchange mutagenesis.
Proteins
His-tagged RNAP holoenzyme (WT and βIA117 KA118 EA119) was purified as described (11). WT Mfd and Mfd D7AAA (Mfd EA1045 DA1048 RA1049) were purified as described in (5). Mfd ΔD7, Mfd WA550 and Mfd WA550 RA953 were purified from BL21(DE3) Δmfd cells transformed with pETMfd1-997-T7 (2), pETMfd-T7 WA550 and pETMfd-T7 WA550 RA953, respectively. Mfd ΔD7 and Mfd WA550 were purified as described for Mfd D7AAA (5), except that cells were grown for 2 h at 30°C after addition of IPTG. Cells expressing Mfd WA550 RA953 were grown in LB containing appropriate antibiotics at 37°C to an A600 of 1.2, expression was induced by the addition of 0.5 mM IPTG and the culture was incubated for 2 h at 30°C before harvesting and protein purification as for Mfd WA550.
ATPase assay
ATPase assays were carried out using an ATP–NADH-coupled ATPase assay essentially as described in (2), with the following modifications: 150 µM (moles of base pairs) herring sperm DNA was used in place of λ DNA and the NADH concentration was 224 µM. Reactions were started by the addition of 1 mM ATP and the change in absorbance at 340 nm was monitored on a lambda 35 spectrophotometer (Perkin Elmer).
TFO displacement assays
TFO displacement assays were carried out as described in (2), except that TFO displacement was monitored from supercoiled, rather than linearized, plasmid pSRTB1.
RNAP displacement assays
The RNAP displacement assays were carried out as described for Mfd E1045A D1048A R1049A in (5).
DNA binding activity
DNA binding was analysed by EMSA using either a 570 bp PCR fragment from pAR1707 (5), or a 50 bp oligonucleotide duplex formed by annealing the oligonucleotide 5'-ctcatacgacgctgtcgatccagtcactgtcatgcgctatccgatcctag-3' with its complement. Both the PCR fragment and the annealed oligonucleotide duplex were end labelled using T4 polynucleotide kinase and [γ-32P]ATP. 0.4 nM pAR1707 fragment or 0.5 nM annealed oligonucleotide duplex were incubated with the indicated concentrations of Mfd and its derivatives in 10 µl reactions containing repair buffer (40 mM Hepes pH 8.0, 100 mM KCl, 8 mM MgCl2, 4% v/v glycerol, 5 mM DTT, 100 µg/ml BSA). 2 mM ATP or ATPγS was added where indicated. Reactions were incubated for 30 min at 37°C and analysed by electrophoresis at 4°C through 5% acrylamide/1× TAE/8 mM magnesium acetate gels.
Trypsin proteolysis
WT Mfd and derivatives were digested with trypsin (Sigma). 2 µM protein was digested in a 10 µl reaction with trypsin added at the amounts indicated. The trypsin dilutions were made in 1 mM HCl and the reactions were carried out in repair buffer minus BSA. Reactions were incubated for 20 min at room temperature and stopped by the addition of 10 µl SDS loading buffer (100 mM Tris.HCl pH 6.8, 4% w/v SDS, 20% v/v glycerol, 200 mM DTT, 0.2% w/v bromophenol blue). Samples were heated at 95°C for 5 min and analysed by electrophoresis on a 10% SDS-PAGE gel. Gels were stained with Biosafe Coomassie (Bio-Rad). In order to determine the major site of proteolysis equivalent reactions were run on a 10% SDS-PAGE gel and transferred onto PVDF membrane for 2.5 h at 230 mA using a mini trans-blot module (Bio-Rad). The major proteolysis products were then identified by Edman N-terminal sequencing (Dr Will Mawby, Proteomics facility, University of Bristol).
Multi-angle light scattering
The multi-angle light scattering (MALS) setup consisted of a cross-linked agarose-based size exclusion column (Superdex 200 10/300, GE Healthcare, UK) connected to an HPLC (Agilent Technologies, UK) and two MALS detectors in series: a light scattering (LS) diode array (Dawn Heleos II, Wyatt, USA) and a differential refractive index detector (Optilab rEX, Wyatt, USA). The column was equilibrated overnight in repair buffer (excluding BSA) at 1 ml/min to achieve stable base lines as well as low background scatter (≤0.1 mV). The experiments were performed in repair buffer (excluding BSA) at 1 ml/min. The respective molar mass was calculated from resulting LS and dRI data using the software ASTRA (Wyatt, USA).
RESULTS
Disruption of the hook-relay helix interface in Mfd stimulates its ATPase and DNA translocase activity
When the structure of apo-Mfd was solved, it was noted that several highly conserved hydrophobic residues interact where the hook helices wrap around the relay helix (10). It was suggested that these interactions may play a role in controlling the conformation of the relay helix. One pair of conserved interacting residues is W550 in the relay helix and I970 in the hook (Figure 1). To determine whether the hook-relay interface plays a role in the autoregulation of Mfd activity we disrupted the interaction between W550 and I970 by constructing an Mfd mutant containing an alanine substitution at W550.
The ATPase activity of purified Mfd WA550 was compared with those of the wild-type (WT) protein and the previously characterized derepressed Mfd mutants Mfd ΔD7 (in which D7 is deleted (2)) and Mfd D7AAA (Mfd EA1045 DA1048 RA1049, in which the D7:BHM interface is disrupted (5)). Rates of ATP hydrolysis were determined in the presence and absence of saturating amounts of double stranded DNA in the presence of non-saturating amounts of ATP (Figure 2A). In the absence of DNA, the ATPase activity of Mfd WA550 was ∼10-fold higher than that of the WT protein, and was comparable to the activities of Mfd ΔD7 and Mfd D7AAA. The presence of dsDNA increased the ATPase activity of Mfd WA550 by ∼4-fold, in contrast to the 2-fold stimulation of Mfd ΔD7 and Mfd D7AAA. Mfd WA550 RA953 (in which both the hook:relay helix interface and the TRG motif are disrupted) had the same DNA-independent ATPase activity as Mfd WA550 (Figure 2A), but was stimulated just 1.4-fold by dsDNA, indicating that the integrity of the TRG motif is important for stimulation of Mfd WA550 by dsDNA.
Figure 2.

Mfd WA550 exhibits elevated ATPase and DNA translocase activities. (A) Rates of ATP hydrolysis by Mfd derivatives. ATP hydrolysis rates were measured at 37°C using an ATP/NADH-coupled assay. Reactions were carried out in repair buffer and contained 50 nM protein and 1 mM ATP. 150 µM herring sperm DNA was added where indicated. Data are the average of three independent experiments and are shown with standard deviation. (B) Analysis of DNA translocation by Mfd derivatives. TFO displacement was analysed on 5 nM supercoiled pSRTB1 containing a triplex, which was incubated with 250 nM Mfd or its derivatives. Reactions were initiated with 2 mM ATP or ATPγS and samples were removed at time intervals, quenched and analysed by gel electrophoresis. The data shown are the percentage of TFO displaced normalized to the amount of free TFO at t = 0. The data are the average of three independent experiments shown with standard deviation. The TFO displacement data for WT Mfd and Mfd D7AAA have been published previously (5) and are shown for comparison.
To determine whether Mfd WA550 exhibited derepressed DNA translocase activity in addition to derepressed ATPase activity we analysed triplex dissociation on a supercoiled plasmid DNA template (Figure 2B). Mfd WA550 exhibited RNAP-independent motor activity, and displaced the triplex forming oligonucleotide (TFO) more rapidly than Mfd D7AAA did. To confirm that TFO displacement by Mfd WA550 reflected DNA translocation driven by ATP hydrolysis the assays were repeated using Mfd WA550 RA953, which has a disrupted TRG motif, and also using ATPγS in place of ATP. In both cases the TFO displacement was reduced to the background levels observed in the absence of protein.
Taken together, the ATPase and DNA translocation activity data show that disrupting the hook:relay helix interface of Mfd results in derepression of Mfd motor activity, and that this interface constitutes an additional node through which the autoinhibition of the protein is controlled.
Mfd WA550 is able to displace transcription complexes that are resistant to WT Mfd
The ability of Mfd WA550 to displace stalled transcription ECs was examined in vitro (Figure 3). Transcription complexes initiating from the T7A1 promoter on a radiolabelled DNA fragment were stalled at +20 by the RNA chain terminator 3'dUTP, and the effect of Mfd on the complexes formed was analysed by electrophoretic mobility shift assay (EMSA). Mfd WA550 displaced the stalled transcription EC much more rapidly than WT Mfd did, and this activity required an intact TRG motif, as Mfd WA550 RA953 was unable to displace stalled ECs.
Figure 3.

Mfd WA550 displaces stalled transcription ECs and transcription ICs in vitro. ICs were formed in repair buffer by the incubation of RNAP holoenzyme with a radiolabelled DNA fragment (DNA). ECs stalled at +21 (ECs) were formed by the addition of ApU, ATP, CTP, GTP and 3’dUTP. 250 nM WT Mfd or its derivatives and 2 mM dATP were added and samples loaded on an EMSA gel at the time intervals indicated. (A) Graphical representation of time-course reactions monitoring displacement of stalled ECs by Mfd and its derivatives. The ECs were formed with WT RNAP or RNAP βIA117KA118EA119. Data points are expressed as a percentage of the amount of EC at t = 0 and are an average of three independent experiments, with standard deviation. (B–F) Representative images of EMSA gels showing the effect of WT and mutant Mfd proteins on transcription complexes. Asterisk indicates bands of higher electrophoretic mobility which are thought to be due to the presence of two RNAP molecules on the DNA (one of which is bound to the promoter).
As reported previously, WT Mfd does not displace transcription ICs from DNA (Figure 3B). Surprisingly, Mfd WA550 rapidly displaced the majority of transcription ICs (Figure 3C). The transcription ICs (heparin stable complexes that are observed in the absence of nucleotides) migrated as multiple bands, which presumably represent different conformations of RNAP-promoter complexes. The ability of Mfd WA550 to displace transcription ICs is not a general property of derepressed Mfd derivatives, as neither Mfd ΔD7 nor Mfd D7AAA possess this ability (data not shown).
To further understand the enhanced ability of Mfd WA550 to displace RNAP from DNA we examined its effect on transcription complexes formed with RNAP βIA117 KA118 EA119, which is a derivative of RNAP that is defective in its interaction with the RID of Mfd (11). Stalled transcription ECs formed with RNAP βIA117 KA118 EA119 are not displaced by WT Mfd (Figure 3F and (11)) or Mfd ΔD7 ((2)). However, Mfd WA550 displaced 80% of stalled RNAP βIA117 KA118 EA119 complexes in the first 30 s after its addition (Figure 3A and E), and also displaced the transcription ICs formed by this mutant RNAP.
These results indicate that the RID-RNAP interaction is not required for RNAP displacement by Mfd WA550, and that, in contrast to previously characterized Mfd derivatives, Mfd WA550 might act as a non-specific ‘molecular bulldozer’, displacing proteins from DNA without the need for a specific protein–protein contact.
Disrupting the hook-relay helix interface in Mfd alters the nature of its interaction with DNA
WT Mfd binds stably to DNA fragments in the presence of the poorly hydrolysable ATP analogue ATPγS but not in the presence of ATP or in the absence of nucleotide (18,19) (Figure 4). We used EMSAs to examine the binding of Mfd WA550 to a 570 bp DNA fragment (Figure 4A) and a 50 bp oligonucleotide duplex (Figure 4B). In both cases DNA binding by Mfd WA550 showed the same nucleotide-dependence as the WT protein. However, the complexes formed by Mfd WA550 differed markedly from those formed by WT Mfd.
Figure 4.

Effects of disruption of the hook:relay helix interface on DNA binding in vitro. 0.5 nM radiolabelled DNA was incubated in repair buffer with the indicated concentrations of Mfd or its derivatives for 30 min at 37°C. 2 mM ATP or ATPγS was added where shown. The complexes were analysed by running on a 5% acrylamide TAE/MgAc gel. (A) Image of a typical EMSA gel showing binding of Mfd or Mfd WA550 to a 570 bp PCR-amplified DNA fragment. (B) Images of typical EMSA gels showing binding of WT and mutant Mfd proteins to a 50 bp annealed oligonucleotide duplex. Asterisk indicates complexes of higher electrophoretic mobility observed with Mfd WA550.
On the 570 bp DNA fragment Mfd WA550 formed complexes that ran as a smear on the gel, in contrast to WT Mfd, which formed discrete bands whose mobility was only slightly changed from that of the free DNA. The smeared appearance of the Mfd WA550-DNA complexes suggests that the protein initially formed a complex (or complexes) of low electrophoretic mobility, which subsequently dissociated while the gel was running. We were unable to find electrophoresis conditions that stabilized this complex and allowed it to be resolved as a discrete band(s) (data not shown). The complexes formed by Mfd WA550 on the 50 bp DNA fragment were more discrete than those formed on the longer DNA fragment, and at the higher concentrations of Mfd WA550, protein-DNA complexes of substantially reduced electrophoretic mobility were again observed. The unusual DNA binding properties of Mfd WA550 are dependent on an intact TRG motif, since Mfd WA550 RA953 did not shift the 50 bp oligoduplex in the presence of ATPγS (Figure 4B). This is in contrast to Mfd RA953 which is able to form a stable complex with both a 50 bp oligoduplex (data not shown) and a longer DNA fragment (19). Mfd D7AAA, which has derepressed levels of ATP hydrolysis and DNA translocase activity, bound the 50 bp duplex stably in the presence of ATPγS but not in the presence of ATP or in the absence of nucleotide, and the shifted complexes had similar electrophoretic mobilities to those observed with WT Mfd (Figure 4B).
The formation of low mobility protein-DNA complexes by Mfd WA550 might result from binding of multiple Mfd proteins to the DNA and/or to alterations in the conformation of the DNA caused by Mfd WA550 binding. Our data do not discriminate between these possibilities, but do indicate that the nature of the interaction between Mfd WA550 and DNA differs from that of WT Mfd and Mfd D7AAA. Disruption of the hook-relay interface thus apparently alters the manner in which Mfd engages with DNA.
The C-terminal domains of Mfd are subject to increased proteolysis when the hook-relay helix interactions are disrupted
Derepressed Mfd derivatives that contain disruptions in the interface between D2 and D7, are more susceptible to proteolytic cleavage than WT Mfd (15). The major sites of tryptic cleavage occur in the linker between D3 and the RID and in the flexible hinge between translocase domains 1 and 2 (Figure 5A). The N-terminal domains 1–3 maintain a protease-resistant form in both repressed and derepressed forms of Mfd, supporting the proposal that the N-terminal half of Mfd acts as rigid clamp.
Figure 5.

Effects of disruption of the hook:relay helix interface on the proteolysis of Mfd. (A) Schematic of the domain architecture of Mfd. Asterisks indicate the major sites of trypsin cleavage previously observed in derepressed Mfd mutants in which the D2–D7 interface is disrupted (15). The bold arrow indicates the major site of trypsin cleavage observed in these experiments, with A and B indicating the major proteolysis products that were obtained. (B) Limited trypsin proteolysis of WT Mfd, Mfd D7AAA and Mfd WA550. 2 µM protein was incubated with the indicated amounts of trypsin for 20 min at room temperature and resolved on a 10% SDS-PAGE gel. Gels were stained with Coomassie blue and the images shown are of typical gels. The molecular weights of the molecular weight markers (MW) are indicated in kDa. The position of the full-length protein (FL) and the two major proteolysis products (A and B) are indicated.
In order to investigate the conformational flexibility of Mfd WA550 it was treated with a range of concentrations of trypsin for 20 min at room temperature, and the proteolysis products were analysed by SDS PAGE and Coomassie staining (Figure 5B). WT Mfd and Mfd D7AAA controls were subjected to the same treatment. Consistent with the observations of (15), WT Mfd was much more resistant to proteolysis by trypsin than the two derepressed Mfd derivatives, each of which exhibited different patterns of proteolysis. After proteolysis of Mfd D7AAA two significant bands were observed, designated A and B, which were identified using N-terminal sequencing. Band B was the N-terminal fragment of the protein, and band A was a C-terminal fragment, with cleavage occurring between amino acids 463 and 464 within the linker between D3 and the RID. This corresponds to a major trypsin cleavage site in Mfd derivatives with a disrupted D2–D7 interface reported previously (15). During trypsin digestion of Mfd WA550 only band B was observed as a stable product, with little accumulation of band A. This suggests that in Mfd WA550 the C-terminal fragment containing domains D4–D7 is susceptible to further proteolysis while D1–D3 remains resistant to proteolysis. The increased susceptibility of the translocase domains of Mfd WA550 to proteolysis suggests that that they are in a more open conformation than those of Mfd D7AAA.
The oligomeric state of the purified proteins was analysed by MALS analysis (Figure 6), which showed that both WT Mfd and Mfd WA550 were monomeric. This result is consistent with previous studies of E. coli Mfd (7,14), and in contrast to reports of higher order Mfd oligomers in Mycobacterium tuberculosis (20). Mfd WA550 eluted from a size exclusion column slightly earlier than the WT protein, providing further support for the suggestion that Mfd WA550 adopts a more ‘open’ conformation.
Figure 6.
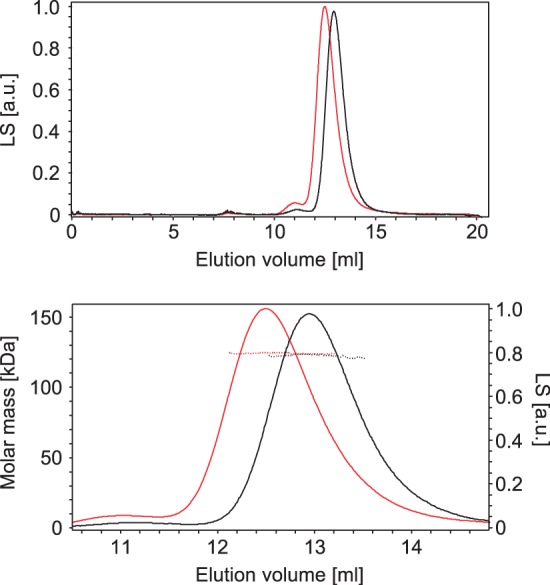
MALS analysis of Mfd oligomeric state. The upper panel shows the elution profile of WT Mfd (black) and Mfd WA550 (red) from a Superdex 200 size exclusion column, monitored by measuring LS (arbitrary units). The lower panel shows a magnified area of this elution profile, displaying the molar masses determined by MALS analysis (dotted lines) across the peaks of eluted protein. Both proteins eluted as a single molecular species, with MALS-determined molar masses of 122 kDa (WT) and 124 kDa (WA550). The theoretical molar mass of monomeric Mfd is 130 kDa.
DISCUSSION
It has previously been suggested that the interdomain contacts between the hook and relay helices of Mfd are ideally positioned to transmit functionally relevant conformational changes within the protein during TCR (10). Comparison of the crystal structure of apo-Mfd with that of RecG (solved in complex with ADP and a three-way DNA junction) reveals interesting differences in the conformation of this region of the two proteins. In the RecG structure the helical hairpin of the TRG motif is in a closed conformation that places positively charged residues at its base in close proximity, and the relay helix adopts a bent conformation (17). In contrast, in the Mfd structure the TRG helical hairpin adopts an open conformation and the relay helix is straight. Deaconescu et al. proposed that the TRG motif forms a ‘spring loaded’ element that opens and closes in response to nucleotide binding and hydrolysis, and that interaction of the hook helices (whose position relies on that of the TRG motif) with the relay helix might control relay helix conformation (10). In this scenario the hook-relay interface might transmit motions of the RID or D7 to the TRG motif and the translocase domains (and vice versa), thus linking DNA translocation activity to conformational changes induced by binding to RNAP. In this work, we have shown that disruption of the hook-relay interface by an alanine substitution of conserved residue W550 does indeed disrupt the regulation of the protein, and results in a derepressed DNA translocase with properties that are distinct from those of Mfd mutants described previously.
The most striking difference between Mfd WA550 and previously characterized mutants is that it is able to displace transcription complexes in the absence of a specific interaction between the RID and the RNAP β subunit. Surprisingly the displacement activity of Mfd WA550 also extends to transcription ICs, which are not a target for WT Mfd or previously characterized derepressed Mfd derivatives (it is thought that interaction of the RNAP σ subunit with DNA prevents Mfd from interacting simultaneously with the RNAP β subunit and the upstream DNA (8)). The WA550 substitution also alters the DNA binding properties of Mfd in a manner that depends on the integrity of the TRG motif, which is essential for DNA translocation activity. Since deregulated Mfd DNA translocase activity alone is not sufficient to allow RNAP displacement in the absence of a RID–RNAP interaction (neither Mfd ΔD7 nor Mfd D7AAA exhibit this property), we suggest that the key to this unusual activity of Mfd WA550 may be increased processivity, due to an altered interaction with DNA. We have previously noted that RNAP may act as a processivity factor for Mfd: acting as an additional tether to the DNA that will allow Mfd to remain engaged even if the translocase domains transiently release the DNA (2). We suggest that disrupting the hook-relay interface allows Mfd to engage with DNA in an altered fashion, which results in increased processivity and enables Mfd WA550 to act as a non-specific ‘bulldozer’ and displace transcription complexes without being physically tethered to them. Our results indicate that untethered DNA translocation by Mfd is capable of displacing stable transcription complexes from DNA, much as the motor activity of the RecBCD helicase/nuclease can displace transcription complexes from its path (21), although we note that this activity of Mfd WA550 must be moderated or tolerated in some way in vivo, as cells expressing the mutant protein exhibit no obvious signs of toxicity.
How does disruption of the hook:relay interface activate the ATPase and DNA translocase activity of the Mfd protein? Like substitutions in the D2–D7 interface, the WA550 substitution results in an ‘open’ conformation of Mfd, as judged by proteolysis. Although our data do not allow us to determine whether or not the D2–D7 interaction is disrupted in Mfd WA550, the simplest explanation for the increased proteolytic sensitivity of the linker between domains 3 and 4 is that the interaction between the hook and the relay helix is important for maintenance of the inhibitory clamp around the translocase domains in the autoinhibited form of the protein. Weakening of the hook-relay interface likely allows D7 more flexibility of motion and shifts the equilibrium between the closed and open forms of the D2–D7 clamp. However, the WA550 substitution also increases the proteolytic sensitivity of the C-terminal fragment of the protein, indicating that disruption of the hook-relay interface also allows the RID-translocase-D7 fragment to adopt a more open and/or flexible conformation than seen in Mfd derivatives in which the D2–D7 interface is disrupted directly.
Our results suggest that multiple interdomain contacts regulate the ATPase and DNA translocase activities of Mfd by routes that are at least partially separable: mutagenesis studies have identified substitutions in D2 that elevate ATPase but not DNA translocase activitities, substitutions in D7 that elevate both activities, and now substitutions in the hook-relay interface that deregulate ATPase and DNA translocase activities and the specificity of RNAP displacement (5). Presumably, in the context of the WT protein, rearrangements at the various nodes involved in autoinhibition are induced by interaction with its partner proteins during TCR. This may occur either in a concerted fashion, or as part of a multi-stage event in which different activities are regulated temporally. Our results do not enable us to tell whether the hook-relay interface has evolved as a permanently applied ‘brake’ that prevents optimal engagement of the translocase domains with the DNA in order to restrict their modes of action, or whether the conformation induced by the WA550 substitution is an ‘on pathway’ state for WT Mfd during RNAP displacement (i.e. whether interaction of the RID with RNAP alters the hook-relay interface and allows the translocase domains to engage with DNA in a more processive manner).
The possibility that movement of D7 can regulate Mfd activity via the hook/relay/TRG helices as well as by holding or releasing the N-terminal clamp around the translocase domains has implications for the regulation of atypical Mfd proteins in some organisms. The obligate endosymbiotic bacteria Buchnera aphidicola and Wigglesworthia glossinidia have mimimalist genomes and lack the NER proteins UvrA, B and C (22,23). The chloroplasts of Arabidopsis thaliana similarly contain an Mfd homologue but lack UvrA, B and C (24). It is not clear what the function of Mfd is in these organisms since they lack the critical repair components of the TCR pathway, but analysis of their genome sequence reveals that the Mfd gene does not encode the N-terminal BHM that forms a key part of the autoinhibitory clamp in E.coli Mfd. It seems unlikely that Mfd would exist in a permanently activated form in the cell, and as D7 and the hydrophobic hook-relay interface are conserved in these atypical Mfd proteins our results suggest that their activity might be regulated by interactions between the RID, D7 and the hook/relay/TRG helices.
FUNDING
Research grants from the Biotechnology and Biological Sciences Research Council [BB/F007361/1, BB/I003142/1] and the Wellcome Trust [078794]. Funding for open access charge: Wellcome Trust.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We are grateful to Prof. M. Szczelkun for comments on the manuscript and Dr. G.H. Thomas for helpful discussions.
REFERENCES
- 1.Brendza KM, Cheng W, Fischer CJ, Chesnik MA, Niedziela-Majka A, Lohman TM. Autoinhibition of Escherichia coli Rep monomer helicase activity by its 2B subdomain. Proc. Natl Acad. Sci. USA. 2005;102:10076–10081. doi: 10.1073/pnas.0502886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith AJ, Szczelkun MD, Savery NJ. Controlling the motor activity of a transcription-repair coupling factor: autoinhibition and the role of RNA polymerase. Nucleic Acids Res. 2007;35:1802–1811. doi: 10.1093/nar/gkm019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murphy MN, Gong P, Ralto K, Manelyte L, Savery NJ, Theis K. An N-terminal clamp restrains the motor domains of the bacterial transcription-repair coupling factor Mfd. Nucleic Acids Res. 2009;37:6042–6053. doi: 10.1093/nar/gkp680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H, DellaVecchia MJ, Skorvaga M, Croteau DL, Erie DA, Van Houten B. UvrB domain 4, an autoinhibitory gate for regulation of DNA binding and ATPase activity. J. Biol. Chem. 2006;281:15227–15237. doi: 10.1074/jbc.M601476200. [DOI] [PubMed] [Google Scholar]
- 5.Manelyte L, Kim Y-IT, Smith AJ, Smith RM, Savery NJ. Regulation and rate enhancement during transcription-coupled DNA repair. Mol. Cell. 2010;40:714–724. doi: 10.1016/j.molcel.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Savery N. Prioritising the repair of DNA damage that is encountered by RNA polymerase. Transcription. 2011;2:168–172. doi: 10.4161/trns.2.4.16146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selby CP, Sancar A. Molecular mechanism of transcription-repair coupling. Science. 1993;260:53–58. doi: 10.1126/science.8465200. [DOI] [PubMed] [Google Scholar]
- 8.Park JS, Marr MT, Roberts JW. E. coli transcription repair coupling factor (Mfd Protein) rescues arrested complexes by promoting forward translocation. Cell. 2002;109:757–767. doi: 10.1016/s0092-8674(02)00769-9. [DOI] [PubMed] [Google Scholar]
- 9.Park JS, Roberts JW. Role of DNA bubble rewinding in enzymatic transcription termination. Proc. Natl Acad. Sci. USA. 2006;103:4870–4875. doi: 10.1073/pnas.0600145103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deaconescu AM, Chambers AL, Smith AJ, Nickels BE, Hochschild A, Savery NJ, Darst SA. Structural basis for bacterial transcription-coupled DNA repair. Cell. 2006;124:507–520. doi: 10.1016/j.cell.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 11.Smith AJ, Savery NJ. RNA polymerase mutants defective in the initiation of transcription-coupled DNA repair. Nucleic Acids Res. 2005;33:755–764. doi: 10.1093/nar/gki225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westblade LF, Campbell EA, Pukhrambam C, Padovan JC, Nickels BE, Lamour V, Darst SA. Structural basis for the bacterial transcription-repair coupling factor/RNA polymerase interaction. Nucleic Acids Res. 2010;38:8357–8369. doi: 10.1093/nar/gkq692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Assenmacher N, Wenig K, Lammens A, Hopfner KP. Structural basis for transcription-coupled repair: the N terminus of Mfd resembles UvrB with degenerate ATPase motifs. J. Mol. Biol. 2006;355:675–683. doi: 10.1016/j.jmb.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 14.Deaconescu AM, Sevostyanova A, Artsimovitch I, Grigorieff N. Nucleotide excision repair (NER) machinery recruitment by the transcription-repair coupling factor involves unmasking of a conserved intramolecular interface. Proc. Natl Acad. Sci. USA. 2012;109:3353–3358. doi: 10.1073/pnas.1115105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srivastava DB, Darst SA. Derepression of bacterial transcription-repair coupling factor is associated with a profound conformational change. J. Mol. Biol. 2011;406:275–284. doi: 10.1016/j.jmb.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahdi AA, Briggs GS, Sharples GJ, Wen Q, Lloyd RG. A model for dsDNA translocation revealed by a structural motif common to RecG and Mfd proteins. EMBO J. 2003;22:724–734. doi: 10.1093/emboj/cdg043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singleton MR, Scaife S, Wigley DB. Structural analysis of DNA replication fork reversal by RecG. Cell. 2001;107:79–89. doi: 10.1016/s0092-8674(01)00501-3. [DOI] [PubMed] [Google Scholar]
- 18.Selby CP, Sancar A. Structure and function of transcription-repair coupling factor. I. Structural domains and binding properties. J. Biol. Chem. 1995;270:4882–4889. doi: 10.1074/jbc.270.9.4882. [DOI] [PubMed] [Google Scholar]
- 19.Chambers AL, Smith AJ, Savery NJ. A DNA translocation motif in the bacterial transcription-repair coupling factor, Mfd. Nucleic Acids Res. 2003;31:6409–6418. doi: 10.1093/nar/gkg868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prabha S, Rao DN, Nagaraja V. Distinct properties of hexameric but functionally conserved Mycobacterium tuberculosis transcription-repair coupling factor. PLoS ONE. 2011;6:e19131. doi: 10.1371/journal.pone.0019131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finkelstein IJ, Visnapuu ML, Greene EC. Single-molecule imaging reveals mechanisms of protein disruption by a DNA translocase. Nature. 2010;468:983–987. doi: 10.1038/nature09561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shigenobu S, Watanabe H, Hattori M, Sakaki Y, Ishikawa H. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature. 2000;407:81–86. doi: 10.1038/35024074. [DOI] [PubMed] [Google Scholar]
- 23.Akman L, Yamashita A, Watanabe H, Oshima K, Shiba T, Hattori M, Aksoy S. Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat. Genet. 2002;32:402–407. doi: 10.1038/ng986. [DOI] [PubMed] [Google Scholar]
- 24.Arabidopsis genome initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]