


Abstract
Transcriptional activation or ‘rewiring’ of silent genes is an important, yet poorly understood, phenomenon in prokaryotic genomes. Anecdotal evidence coming from experimental evolution studies in bacterial systems has shown the promptness of adaptation upon appropriate selective pressure. In many cases, a partial or complete promoter is mobilized to silent genes from elsewhere in the genome. We term hereafter such recruited regulatory sequences as Putative Mobile Promoters (PMPs) and we hypothesize they have a large impact on rapid adaptation of novel or cryptic functions. Querying all publicly available prokaryotic genomes (1362) uncovered >4000 families of highly conserved PMPs (50 to 100 long with ≥80% nt identity) in 1043 genomes from 424 different genera. The genomes with the largest number of PMP families are Anabaena variabilis (28 families), Geobacter uraniireducens (27 families) and Cyanothece PCC7424 (25 families). Family size varied from 2 to 93 homologous promoters (in Desulfurivibrio alkaliphilus). Some PMPs are present in particular species, but some are conserved across distant genera. The identified PMPs represent a conservative dataset of very recent or conserved events of mobilization of non-coding DNA and thus they constitute evidence of an extensive reservoir of recyclable regulatory sequences for rapid transcriptional rewiring.
INTRODUCTION
Transcriptional rewiring is a term used for defining the modification of transcriptional circuits over evolutionary time, due to changes in transcription factors (TFs) and/or cis-regulatory elements. This concept has been widely used in studies of eukaryotic transcription circuits (1), but much less in prokaryotic systems, mainly because the extent of the phenomenon in bacteria is presently unknown (2,3).
However, transcriptional rewiring may actually play an important role in prokaryotic genome evolution given the large turnover of gene functions. Indeed the prevalence of gene acquisition through horizontal gene transfer (HGT) (4–6) and gene loss from deletion events (7,8) generates highly dynamic genomes that differ even between closely related species or strains. As an example of such a large turnover of genes, it has been estimated that 61 genomes of Escherichia coli strains share only ∼20% of gene functions (9).
Transcriptional rewiring can result in activation of silent genes, such as HGT-derived genes without a compatible promoter (10), or in modification of the expression of already present genes. Such activation requires as a first step the evolution of a functional promoter, i.e. −10, −35 boxes and TF-binding sites that can be recognized by the cell’s transcriptional machinery (11). In principle, a promoter could evolve by two different mechanisms. It can evolve de novo by the creation of cis-regulatory elements through point mutations and indels (12). Alternatively, it can evolve in a single ‘quantum leap’ through the recruitment or mobilization of already existing promoters from elsewhere in the genome (13).
Experimental evolution studies in Pseudomonas putida (14), Lactoccocus lactis (15,16) and E. coli (17–19) have found promoter recruitment to be the main mechanism driving transcriptional activation or rewiring of silent genes, through mobilization of partial or complete promoters by transposable elements (20).
Furthermore, recent advances in understanding the function of DNA repeats in intergenic regions have shown that they can have important regulatory roles in transcription or translation (21); and given their ability to propagate, DNA repeats can also be involved in transcriptional rewiring. Miniature inverted terminal repeat elements (MITEs) are non-autonomous mobile elements, that is, they only transpose if a suitable transposase is provided in trans by an autonomous IS element. Examples of MITEs that can influence transcription are the Neisseria CREE element (22,23) and the Yersinia ERICS (24), both of which carry partial promoters at their termini.
Based on these observations, it seems that intragenomic promoter propagation could represent a major force driving transcriptional activation or rewiring in prokaryotes. In the present study, the extent of promoter propagation in archaea and bacteria was assessed by in silico analysis of all publicly available genomes. Evidence for promoter propagation events was found in more than 4000 families of conserved homologous sequences upstream of non-homologous coding sequences (CDSs). These ‘Putative Mobile Promoters’ (PMPs) present examples of reported insertion sequences (IS) and riboswitches, but notably also a large fraction of novel families of dynamic elements with potential influence on transcription. We hypothesize that PMPs may represent a vast recyclable reservoir of regulatory potential for rapid transcriptional recruitment or rearrangement.
MATERIALS AND METHODS
Identification of intra-genomic promoter propagation
To identify PMPs in a bacterial genome we looked for conserved homologous sequences upstream of non-homologous CDSs (Figure 1). The promoter of each CDS was assumed to be contained in the first 150 to 100 nt upstream of the translation start site (TLS) of predicted transcriptional units. This assumption builds on the finding that bacterial promoters are relatively compact with 100-nt regions generally containing the regulatory signals needed for initiating transcription (2). Furthermore, those regulatory signals are usually located immediately upstream of CDSs. For example, the majority of transcriptional start sites in E. coli K12 are located between 20 and 40 nt from the TLS, and most of the TF-binding sites are located 50 nt upstream of the transcriptional start site (25). Therefore it can be reasonably assumed that the method deals with sequences probably involved in transcriptional regulation.
Figure 1.

Identification of PMPs. Dashed boxes represent 100 nt defined promoters (green arrows), downstream CDSs (dark blue and pink arrows) and upstream CDSs (orange and blue arrows) used for BLAST alignments. Those regions were taken ±50 nt of the TLS of the downstream CDSs. Two promoters are considered mobile if they align over >50 nt with at least 80% identity (green shadow), while their upstream CDSs (A and C) and downstream CDSs (B and D) do not align.
We took 100-nt fragments from all promoters and CDSs found in a genome starting at 50 nt upstream or downstream, respectively, of the TLS as depicted (Figure 1). The sequences were extracted with an in-house developed Perl script using the annotation (.ptt) and the FASTA files (.fna) of 1362 complete prokaryote genomes (archaea- and eubacteria; 971 species; 503 genera; see Supplementary Table S1 for complete list) reported at the NCBI website (May 2011). The collected sequences from different chromosomes and/or plasmids of the same genome were stored in one file and formatted as a BLAST database. The BLAST (26) alignments were performed within each genome using an E-value cutoff of 0.0001 and the filter for low complexity regions off. A hit between promoters was considered relevant if the alignment was at least 50 nt long with 80% identity (i.e. at least 40 out of 50 nt were identical) while all hits between coding regions were considered indicative of homology. All filtered pair-wise hits were clustered with the NetClust (score cutoff of zero) program (27) to obtain the unfiltered families (we call pre-clusters) of homologous sequences per genome. A pre-cluster was discarded if (i) it contained both promoters and CDSs, since these sequences could represent misannotated TLSs, or (ii) the whole gene was duplicated (promoter region and CDS), since we are interested only in promoter mobilization. Pre-clusters passing the filters became families of PMPs. In each family, the promoter showing homology to the most members was selected as representative. If the representative was homologous to all members in its family, then it was said to be a central node and it indicated the presence of a highly conserved core in the family.
Identification of inter-genomic promoter propagation
CD-HIT-EST (28) was used to cluster all representatives at 80% identity over 50 nt (program parameters: -c 0.8 –G 0 –aL 50). The clustering removed redundancy in the dataset and identified PMPs in different strains of the same species, different species of the same genus or bacteria from different genera.
Control dataset: randomized sequences
To estimate the number of duplicated promoters that one could expect to find by chance, we generated a mock dataset with shuffled sequences having the same promoter and CDS nucleotide compositions for each genome. Sequences were re-shuffled 10–20 times with an in-house developed Perl script and then run through the pipeline.
Functional analyses of the propagated promoters
Quantitative analyses were carried out to investigate the incidence, conservation and possible function of mobile promoters. The non-redundant dataset was used to query RFAM (29), IS Finder (30) and published MITEs datasets (21,22) to assess how many of the identified promoters are actually known RNA regulatory elements, IS or non-autonomous mobile elements. The cmsearch program of the INFERNAL suite (31) was used to search against the 1973 RFAM calibrated models (14 June 2011 release) with the trusted cutoff (–tc). The IS Finder web server was used to search for reported IS elements with an E-value cutoff of 0.0001 and with filter for low complexity regions off. To find the more distant members of each PMP family and thus gain insight into the propagation dynamics of PMPs, we extended the families with all BLAST hits having an e-value < 0.0001 that did not pass the alignment length and identity filters.
Finally a comparison of PMPs present in E. coli strains was performed to check for inter-strain variability.
Pipeline
A pipeline script was programmed in Perl to automate every step of the analysis, except for the use of IS Finder. The pipeline runs in a Linux environment and it requires the data and supporting programs to be installed locally. Please contact the authors for the suite of scripts and instructions.
RESULTS
Identification of intra-genomic promoter propagation
PMPs were identified as highly similar stretches of non-coding DNA located in promoter regions of non-homologous genes in a species (Figure 1). All promoter sequences with minimal length of 150 nt upstream of the start codon (1 142 064) were mined from 1362 prokaryotic genomes and formed 11 821 pre-clusters. Over 60% (7366) of them also shared homology in their corresponding downstream CDSs, and thus cannot be considered as only promoter duplications. This strong reduction to 4455 families indicates that most of the highly conserved duplicated promoters in these bacterial genomes are in fact part of complete gene duplications. We also filtered out cases of homology in the neighbouring upstream CDS and were left with a final dataset of 4071 families (13 111 sequences; see Supplementary Data for FASTA sequences of identified PMPs). Among the discarded data we found several cases (47% = 180/381 pre-clusters) in which the conserved promoters were actually long terminal inverted repeats from transposases present in multiple copies in the genome (e.g. Supplementary Figure S1).
Analysis of the family of 10 members in Treponema brennaborense DSM-12168 (Figure 2 and Supplementary Table S2) showed that the promoters are highly similar to each other (average identity of 95%) over a large stretch (average length of 84 nt). Upon closer inspection it was found that sequence conservation starts around position −5 upstream of the TLS and extends up to position −120 with less conserved sequences up to −170 nt.
Figure 2.

PMP regulating 10 non-homologous CDSs. (A) Alignment of the multiple copies of a PMP in T. brennaborense. A highly conserved core can be observed in the region −120 to −5 upstream of the TLS of the downstream CDSs. Color blocks represent conserved residues. The location of the TLS is indicated. (B) Alignment of the CDSs downstream of the PMP. No sequence conservation is observed. (C) Location of the PMPs (black arrows) along the 3-Mb circular chromosome (Mb are marked as 1, 2 and 3 in the figure). The orientation of each gene is depicted according to the genome annotation.
Identification of inter-genomic promoter propagation
Redundancy in the dataset caused by over-representation of certain bacterial clades in the genomes database (e.g. E. coli) was not purged from the beginning because it was of interest to identify recent promoter propagation events across strains of the same species. To estimate the level of redundancy in the results and to pinpoint cases of PMPs across different species or genera, all identified duplicated promoters were clustered together (see Supplementary Table S3 and Supplementary Data for FASTA sequences of non-redundant inter-genomic PMPs). From the 4074 families in the final dataset, 3216 non-redundant families were formed of which 87% (2791/3216) were formed by single representatives. The rest consisted of homologous promoters between different strains of the same species (168 families), different species of the same genus (146 families) or different genera (75 families). The latter are of particular interest since they could represent cases of HGT-derived promoters present in distant species. For example a PMP was found upstream of eight different CDSs in Herpetosiphon aurantiacus ATCC 23779, Carboxydothermus hydrogenoformans Z 2901, Deinococcus maricopensis DSM 21 211 and Thermotoga lettingae TMO (Figure 3 and Supplementary Table S4). There were also 36 families formed by representatives from the same genome that were not grouped together in the pipeline (due to identity or length filters) but are indeed homologous.
Figure 3.

Alignment of a PMP present in four distant species. (A) Alignment of the promoter region of eight CDSs in Herpetosiphon aurantiacus ATCC23779, Carboxydothermus hydrogenoformans Z 2901, Thermotoga lettingae TMO and Deinococcus maricopensis DSM2121. Color blocks show nucleotide sequence conservation. A variable region of non-conserved sequence (29 to 93 nt long) was observed in all cases between the TLS and the PMP. (B) Alignment of the coding sequence of the eight downstream CDSs. No sequence conservation is observed.
Control dataset: randomized sequences
Using randomized sequences with the same nucleotide composition (mock data), 1457 pre-clusters were obtained through BLAST and Netclust (149 107 in real data). All of the pre-clusters were formed by a mixture of CDSs and promoters sequences, since both types of sequences are used for the clustering in the pipeline. Therefore no pre-cluster made it through to the final families dataset (11 821 did in the real data). Looking at the genomes that were particularly enriched with random pre-clusters, we found four genomes with >20 (Supplementary Table S5). All such genomes have a skewed base composition (<30 or >70% GC), which could explain the high number of random sequence conservation. This is supported by a plot of % GC versus number of families (Supplementary Figure S2). In the real dataset, none of these genomes had a particularly high count of families (all ≤10 families) and the number of families was not correlated to the GC content of the DNA molecule (Supplementary Figure S2). The top five genomes with the largest number of families in the dataset (all ≥21 families) had zero or one family in the mock data.
Quantitative analysis of the PMPs distribution
A total of 4074 families were mined from 1043 prokaryotic genomes representing 424 genera. The genera with most families were the ones with more sequenced representatives, e.g. Clostridium (149 families; 31 genomes), Escherichia (141 families; 31 genomes), Streptococcus (125 families; 52 genomes) and Bacillus (104 families; 37 genomes). Normalizing the number of families by the number of genomes in the database showed that four cyanobacterial genera and one bacteroidetes had the highest enrichment of families relative to the number of available genomes. The species with the largest number of PMP families are Anabaena variabilis ATCC29413 (28 families), Geobacter uraniireducens Rf4 (27 families), Cyanothece PCC7424 (25 families), Trichodesmium erythraeum IMS101 (22 families) and Psychromonas ingrahamii 37 (21 families). All five species are free-living organisms and have large circular chromosomes (Supplementary Table S5) suggesting that genome size could be correlated with the number of duplicated promoters, a similar correlation has been reported for gene paralogs (32) and regulatory potential (33). However no correlation was observed when plotting the size of the DNA molecule (chromosome or plasmid) versus the number of families, nor the number of mock pre-clusters (Supplementary Figure S3). The same result was observed when plotting the total genome size versus the number of families (data not shown).
About 80% of the analyzed genomes contain less than six families of duplicated promoters (78% = 812/1043 genomes; Figure 4A) and the majority have only one family. This overall low count of propagated promoters suggests that either mobilized promoters diverge very fast and the present methodology is too conservative to find more cases, or that promoter mobilization independent of CDS duplication is a rare event.
Figure 4.

Quantification of PMP propagation in prokaryotic genomes. (A) Number of families per genome (total = 4074 families in 1043 genomes). (B) Number of promoters per family (total = 13 111 promoters in 4074 families); please note the logarithmic scale on the y-axis.
Small family sizes were obtained with the majority having only two members (68% = 2771/4074 families; Figure 4B). These pairs were on average highly conserved (mean identity of 92%, Figure 5A) and the majority were of the minimal allowed alignment length (50 nt, Figure 5B). Interestingly, the most frequent case was that of identical promoters, which again implies the pipeline is finding predominantly very recent or conserved duplications. The largest family (93 promoters) was found in the anaerobic sulphur-reducer Desulfurivibrio alkaliphilus AHT2.
Figure 5.

Two-members families features (2771/4074 families). (A) Identity distribution (mean = 92%). (B) Alignment length distribution (mean = 66 nt).
Search of riboswitches, IS elements and MITES
Riboswitches and IS are known elements with possible regulatory functions. In order to examine the fraction of PMPs that are in fact such reported elements; we queried representative sequences from the non-redundant dataset (3216 sequences) against the RFAM and IS Finder databases.
Searching the RFAM database resulted in 125 hits (∼4% =125/3216 representatives) with 33 RNA models of RFAM (out of 1973 present in the database). The most frequent hit was with tRNAs (42/125 hits), which are known integration sites for genomic islands (34).
The method effectively purged IS elements from the dataset by restricting sequence conservation only in the promoter regions and not in their neighbouring CDSs. However IS elements can leave behind direct repeats when they excise and insert in another location. Searching against the IS Finder web server to find traces of similarity to IS elements, 210 hits (∼7% = 210/3,216) with 177 different IS were retrieved. Methylobacterium extorquens AM1 had most hits with the database (5/14 families).
Two PMPs had hits both with RNA-regulatory elements and IS elements. One is a pair present in Stenotrophomonas maltophilia that presented similarity to the mraW RNA motif, associated with peptidoglycan synthesis (RFAM, http://rfam.sanger.ac.uk) and to the ISStma8 transposase (IS110 family). The other doublet is present in Glaciecola agarilytica and was similar to the antisense RNA-OUT that regulates transposition and the ISPat1 (IS4 family). Thus, the resemblance to IS elements could provide the RNA elements with mobility. This is interesting since the mechanism by which riboswitch families expand or shrink is presently unknown. However it can be anticipated that the dynamics of mobile elements (e.g. IS, transposases, etc.) can result in different frequencies of the RNA elements, e.g. Streptomyces coelicolor’s genome has nine copies of the adenosyl–cobalamin riboswitch (Ado–CBL) while Streptomyces avermitilis’ has four. The fact that the dataset had a low count of reported riboswitches and IS elements (together ∼11% of families) indicates that our methodology finds mainly new mobile regulatory elements.
To investigate the occurrence of MITEs in the dataset, all representatives were searched against a database of 5′-UTR CREE elements (22). None was found in the dataset. Manual checking confirmed that such repeats were excluded early in the pipeline because they are present both in promoters and CDSs regions.
Functional categories of CDSs downstream of PMPs
To analyze if the PMPs that we find are biased towards certain functional classes of genes, the Cluster of Orthologous Groups (COG) (35) classification from all downstream CDSs was obtained. With respect to the encoded product, most of the genes encode hypothetical proteins (4809/13 111 CDSs) followed by transposases (295/13 111 CDSs) and GCN5-related N-acetyltransferase (57/13 111 CDSs). Only in a minimal fraction of the families (3% = 130/4074 families) all members of the same family belong to the same COG. These could represent genes involved in the same metabolic pathways that would benefit from coregulation.
These data together imply that little information is available for the CDSs found in our study, which is in accordance with our hypothesis that PMPs could be involved in recent events of transcriptional rewiring of species-specific genes rather than housekeeping functions.
Case study: E. coli
Rapid propagation of the PMPs throughout genomes could result in different frequencies of these promoters in closely related strains. An example of intra-species variation was analyzed in E. coli, which is represented in the database by 30 sequenced strains. It was found that even between closely related strains there were substantial differences in the number of families and/or number of promoters in the families (Table 1). Families were found in all 30 reported genomes however the numbers varied from 2 to 8. Differences were found even between isolates of the same strain, for example in E. coli K12 MG1655 (five families) and E. coli K12 DH10B (three families). To validate that the different counts are not an artifact of the set identity and length thresholds, promoter families were made again but taking into account all BLAST hits. Differences in abundance of families and number of promoters were found again thus showing that the PMPs do have different frequencies in closely related strains. For example Table 2 shows the distribution of a PMP across different Enterobacteriales (E. coli, Salmonella enterica, Shigella boydii and Yersinia pestis). The downstream CDSs of the PMP are classified into a large variety of COGs and the degree of sequence conservation is also variable. Diverged copies of the PMP are indicated with gray cells in the table and conserved copies with brown cells. All E. coli CDSs downstream of PMPs were checked to determine the abundance of HGT, by using a dataset of identified HGT events (6). It was found that ∼25% of the CDSs in our dataset present evidence of HGT (Chi square test at P-value = 0.0001), which is about the same as for all E. coli genes (30%). Therefore our dataset of PMPs is involved both in transcriptional activation events for HGT-genes but primarily in transcriptional rewiring of already existing functions. Another interesting observation is that in some cases the number of families and family members did not change or only very little, e.g. E. coli O157 family (Table 2), while in other cases the total number of promoters increased dramatically, e.g. the family in Y. pestis grew from 13 to 100 promoter members (see Supplementary Table S6 for complete list of PMP families, members, riboswitches and IS elements per genome). Such difference in occurrence and conservation could provide information on the mechanism by which the promoters are being mobilized. A promoter with tens or hundreds of copies in a genome could well represent a non-autonomous mobile element that is copied by an active transposase, while a promoter present in two or three copies could be result of random duplication through homologous or non-homologous recombination.
Table 1.
Differences in PMP family number and size in 30 strains of E. coli
| Strain | No. of PMP families | Total number of sequences |
|---|---|---|
| E. coli 536 | 4 | 12 |
| E. coli 55989 | 4 | 12 |
| E. coli APEC O1 | 4 | 8 |
| E. coli ATCC 8739 | 5 | 27 |
| E. coli B REL606 | 6 | 26 |
| E. coli BL21 Gold DE3 pLysS AG | 5 | 21 |
| E. coli BW2952 | 6 | 25 |
| E. coli CFT073 | 3 | 7 |
| E. coli E24377A | 3 | 12 |
| E. coli ED1a | 6 | 15 |
| E. coli HS | 5 | 13 |
| E. coli IAI1 | 3 | 9 |
| E. coli IAI39 | 2 | 7 |
| E. coli K 12 substr DH10B | 3 | 9 |
| E. coli K 12 substr MG1655 | 5 | 24 |
| E. coli K 12 substr W3110 | 5 | 25 |
| E. coli O103 H2 12009 | 6 | 22 |
| E. coli O111 H 11128 | 4 | 18 |
| E. coli O127 H6 E2348 69 | 6 | 13 |
| E. coli O157 H7 EC4115 | 7 | 16 |
| E. coli O157 H7 EDL933 | 6 | 16 |
| E. coli O157 H7 Sakai | 3 | 8 |
| E. coli O157 H7 TW14359 | 7 | 16 |
| E. coli O26 H11 11368 | 5 | 19 |
| E. coli O55 H7 CB9615 | 3 | 8 |
| E. coli S88 | 7 | 15 |
| E. coli SE11 | 8 | 19 |
| E. coli SMS 3 5 | 2 | 4 |
| E. coli UMN026 | 3 | 9 |
| E. coli UTI89 | 3 | 6 |
Table 2.
Occurrence of a PMP family in different strains of E. coli, S. enterica, S. boydii and Y. pestis
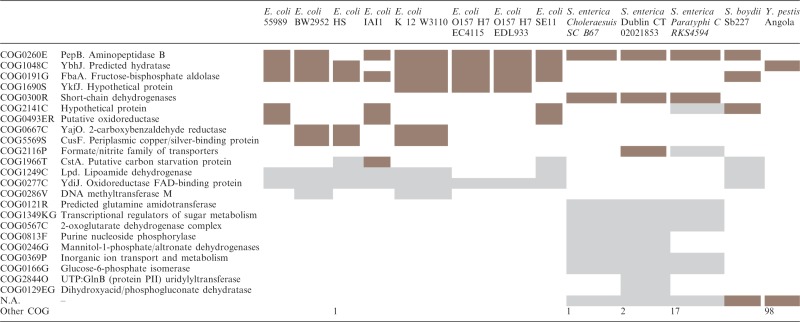 |
The different strains are shown in the columns while rows stand for COG annotations. Brown cells show highly conserved copies of the PMP and gray cells correspond to diverged copies (below length and identity thresholds). Differences in PMP frequencies and conservation can be observed between different genera, species and strains.
DISCUSSION
Treangen et al. (36) provide an operational definition of DNA repeats based on three properties of the copies: (i) the distance between them, (ii) the similarity level and (iii) the length over which they align. Analyses of such properties have produced the guideline that exact repeats >25 nt are statistically significant in most prokaryotic genomes (36). Since the present study required alignments of at least 50 nt with 80% identity, the dataset presented is a conservative investigation of the repeats found in promoter regions throughout the bacterial and archaeal domains. We showed that neither the length nor composition of the DNA molecules is correlated to the presence of PMPs. Our analysis pipeline did not find any family of mobile promoters in a control-randomized dataset (Supplementary Table S5). Therefore we are confident that the data presented in this study indeed represent statistically significant events of promoter propagation.
Bacteria seem to employ various mechanisms to be able to reuse promoter sequences instead of having to evolve them de novo. Based on reported literature and inspection of the dataset, we propose that promoters can be mobilized through four main mechanisms: (i) mobile elements, either as part of the terminal inverted repeats (15), or linked to them (13); (ii) non-autonomous mobile elements, emulating terminal inverted repeats (21); (iii) random duplications mediated by recombination processes; and (iv) HGT, which actually is the result of mobile elements (e.g. conjugative plasmids) or duplications (e.g. minimal mobile elements). Families of promoters that were conserved along with an upstream CDS are probably examples of mobile elements (transposases) that carry promoters (Supplementary Figure S1) in their termini. Families that grew dramatically when all BLAST hits were taken into account probably represent groups of non-autonomous mobile elements or scars from autonomous mobile ones. Pairs found in single species are probably examples of random promoter duplications resulting from homologous or non-homologous recombination. Families present in bacteria from different species or genera could represent HGT-derived promoters (Figure 3 and Supplementary Table S4), potentially capable of being functional in a broad host range. Although there are no reported cases of HGT-derived promoters, it is a plausible scenario since any type of DNA can undergo lateral transfer (37).
This rapid integration of novel gene functions probably is an important factor in the success of HGT and the rapid adaptation to novel niches. It is presently unknown to which extent HGT-derived genes come with a promoter that can be used straightaway. However there are indications that such a promoter-CDS cotransfer is unlikely to occur since expression of the novel gene can be deleterious to fitness or even lethal if the novel CDS product is toxic or poses gene dosage problems (38), plus there is an inherent limitation to HGT regarding the length of simultaneously transferred DNA. Therefore, recycling of appropriate promoters for HGT-derived CDSs seems to be a plausible, economic and biologically significant event in the integration of novel gene functions. This is in agreement with the finding that the evolutionary rate of non-coding upstream sequences is higher for the most recent HGT-derived CDSs in E. coli K12 (12).
The results also show how bacteria could recycle genetic material not only at the CDS level for generating paralogs in the process of neofunctionalization but also in non-coding regions to generate (novel) families of regulatory sequences. Since mainly small PMP families are identified, it seems that either they diverge very fast or family expansion is uncommon. Family expansion to include all BLAST hits of the PMP provided examples of both cases. While the doublets (families of two promoters) were highly conserved (∼92% identity, Figure 5), the larger families already presented many variations near the TLS (see Figures 2 and 3 for examples). This could be an illustration of how a generic mobile promoter adapts to produce different transcriptional responses in the downstream CDSs, providing thus flexibility in the type of regulation it provides. This also indicates that most probably the doublets represent the most recent cases of promoter propagation, which is supported by the fact that identical promoters are the most common case (Figure 5). Finally, it can also be argued that the fast divergence of PMPs families also prevents genomic instability by quickly reducing the chance of recombination between identical copies. This could explain why we find highly conserved PMP families at a low frequency in all analyzed genomes (on average three families per genome) with the conservative methodology we followed. It will be interesting to determine which proportion of the PMPs are transcriptional activators, down-regulators or even silencers, and if their function lies at the transcriptional or post-transcriptional level.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–6, Supplementary Figures 1–3 and Supplementary Data.
FUNDING
The Netherlands Organization for Scientific Research (NWO) via a VENI grant (to M.W.J.vP.); the Netherlands Consortium for Systems Biology, which is part of the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research (to H.N.); and the Consejo Nacional de Ciencia y Tecnología (CONACyT) via a graduate scholarship to M.M.G. Funding for open access charge: Science Innovation Grant from the dutch science foundation (NWO) (to M.W.J.vP.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
This work is dedicated to the memory of Professor Jack A.M. Leunissen, one of the first Dutch bioinformaticians.
REFERENCES
- 1.Tuch BB, Li H, Johnson AD. Evolution of eukaryotic transcription circuits. Science. 2008;319:1797–1799. doi: 10.1126/science.1152398. [DOI] [PubMed] [Google Scholar]
- 2.Perez JC, Groisman EA. Transcription factor function and promoter architecture govern the evolution of bacterial regulons. Proc. Natl Acad. Sci. USA. 2009;106: 4319–4324. doi: 10.1073/pnas.0810343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang L, Wang FF, Qian W. Evolutionary rewiring and reprogramming of bacterial transcription regulation. J. Genet. Genomics. 2011;38:279–288. doi: 10.1016/j.jgg.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 4.Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 5.Treangen TJ, Rocha EP. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 2011;7:e1001284. doi: 10.1371/journal.pgen.1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Popa O, Hazkani-Covo E, Landan G, Martin W, Dagan T. Directed networks reveal genomic barriers and DNA repair bypasses to lateral gene transfer among prokaryotes. Genome Res. 2011;21:599–609. doi: 10.1101/gr.115592.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Passel MW, Marri PR, Ochman H. The emergence and fate of horizontally acquired genes in Escherichia coli. PLoS Comput. Biol. 2008;4:e1000059. doi: 10.1371/journal.pcbi.1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Passel MW, Smillie CS, Ochman H. Gene decay in archaea. Archaea. 2007;2:137–143. doi: 10.1155/2007/165723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lukjancenko O, Wassenaar TM, Ussery DW. Comparison of 61 sequenced Escherichia coli genomes. Microb. Ecol. 2010;60:708–720. doi: 10.1007/s00248-010-9717-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pal C, Papp B, Lercher MJ. Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat. Genet. 2005;37:1372–1375. doi: 10.1038/ng1686. [DOI] [PubMed] [Google Scholar]
- 11.Browning DF, Busby SJ. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2004;2:57–65. doi: 10.1038/nrmicro787. [DOI] [PubMed] [Google Scholar]
- 12.Lercher MJ, Pal C. Integration of horizontally transferred genes into regulatory interaction networks takes many million years. Mol. Biol. Evol. 2008;25:559–567. doi: 10.1093/molbev/msm283. [DOI] [PubMed] [Google Scholar]
- 13.Stavrinides J, Ma W, Guttman DS. Terminal reassortment drives the quantum evolution of type III effectors in bacterial pathogens. PLoS Pathog. 2006;2:e104. doi: 10.1371/journal.ppat.0020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kasak L, Horak R, Kivisaar M. Promoter-creating mutations in Pseudomonas putida: a model system for the study of mutation in starving bacteria. Proc. Natl Acad. Sci. USA. 1997;94:3134–3139. doi: 10.1073/pnas.94.7.3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bongers RS, Hoefnagel MH, Starrenburg MJ, Siemerink MA, Arends JG, Hugenholtz J, Kleerebezem M. IS981-mediated adaptive evolution recovers lactate production by ldhB transcription activation in a lactate dehydrogenase-deficient strain of Lactococcus lactis. J. Bacteriol. 2003;185:4499–4507. doi: 10.1128/JB.185.15.4499-4507.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Visser JA, Akkermans AD, Hoekstra RF, de Vos WM. Insertion-sequence-mediated mutations isolated during adaptation to growth and starvation in Lactococcus lactis. Genetics. 2004;168:1145–1157. doi: 10.1534/genetics.104.032136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee DH, Palsson BO. Adaptive evolution of Escherichia coli K-12 MG1655 during growth on a nonnative carbon source, L-1,2-propanediol. Appl. Environ. Microbiol. 2010;76:4158–4168. doi: 10.1128/AEM.00373-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Posfai G, Plunkett G, 3rd, Feher T, Frisch D, Keil GM, Umenhoffer K, Kolisnychenko V, Stahl B, Sharma SS, de Arruda M, et al. Emergent properties of reduced-genome Escherichia coli. Science. 2006;312:1044–1046. doi: 10.1126/science.1126439. [DOI] [PubMed] [Google Scholar]
- 19.Stoebel DM, Dorman CJ. The effect of mobile element IS10 on experimental regulatory evolution in Escherichia coli. Mol. Biol. Evol. 2010;27:2105–2112. doi: 10.1093/molbev/msq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, Saier MH., Jr A novel mechanism of transposon-mediated gene activation. PLoS Genet. 2009;5:e1000689. doi: 10.1371/journal.pgen.1000689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delihas N. Impact of small repeat sequences on bacterial genome evolution. Genome Biol. Evol. 2011;3:959–973. doi: 10.1093/gbe/evr077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snyder LA, Cole JA, Pallen MJ. Comparative analysis of two Neisseria gonorrhoeae genome sequences reveals evidence of mobilization of Correia repeat enclosed elements and their role in regulation. BMC Genomics. 2009;10:70. doi: 10.1186/1471-2164-10-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siddique A, Buisine N, Chalmers R. The transposon-like Correia elements encode numerous strong promoters and provide a potential new mechanism for phase variation in the meningococcus. PLoS Genet. 2011;7:e1001277. doi: 10.1371/journal.pgen.1001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Gregorio E, Silvestro G, Petrillo M, Carlomagno MS, Di Nocera PP. Enterobacterial repetitive intergenic consensus sequence repeats in yersiniae: genomic organization and functional properties. J. Bacteriol. 2005;187:7945–7954. doi: 10.1128/JB.187.23.7945-7954.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mendoza-Vargas A, Olvera L, Olvera M, Grande R, Vega-Alvarado L, Taboada B, Jimenez-Jacinto V, Salgado H, Juarez K, Contreras-Moreira B, et al. Genome-wide identification of transcription start sites, promoters and transcription factor binding sites in E. coli. PLoS One. 2009;4:e7526. doi: 10.1371/journal.pone.0007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuzniar A, Dhir S, Nijveen H, Pongor S, Leunissen JA. Multi-netclust: an efficient tool for finding connected clusters in multi-parametric networks. Bioinformatics. 2010;26:2482–2483. doi: 10.1093/bioinformatics/btq435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, et al. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37:D136–D140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gevers D, Vandepoele K, Simillon C, Van de Peer Y. Gene duplication and biased functional retention of paralogs in bacterial genomes. Trends Microbiol. 2004;12:148–154. doi: 10.1016/j.tim.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 33.Konstantinidis KT, Tiedje JM. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl Acad. Sci. USA. 2004;101:3160–3165. doi: 10.1073/pnas.0308653100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ou HY, Chen LL, Lonnen J, Chaudhuri RR, Thani AB, Smith R, Garton NJ, Hinton J, Pallen M, Barer MR, et al. A novel strategy for the identification of genomic islands by comparative analysis of the contents and contexts of tRNA sites in closely related bacteria. Nucleic Acids Res. 2006;34:e3. doi: 10.1093/nar/gnj005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 36.Treangen TJ, Abraham AL, Touchon M, Rocha EP. Genesis, effects and fates of repeats in prokaryotic genomes. FEMS Microbiol. Rev. 2009;33:539–571. doi: 10.1111/j.1574-6976.2009.00169.x. [DOI] [PubMed] [Google Scholar]
- 37.Ragan MA, Beiko RG. Lateral genetic transfer: open issues. Philos. Trans. R Soc. Lond B Biol. Sci. 2009;364:2241–2251. doi: 10.1098/rstb.2009.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sorek R, Zhu Y, Creevey CJ, Francino MP, Bork P, Rubin EM. Genome-wide experimental determination of barriers to horizontal gene transfer. Science. 2007;318: 1449–1452. doi: 10.1126/science.1147112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.