


Abstract
Peptidyl-tRNA hydrolase (Pth) cleaves the ester bond between the peptide and the tRNA of peptidyl-tRNA molecules, which are produced by aborted translation, to recycle tRNA for further rounds of protein synthesis. Pth is ubiquitous in nature, and its enzymatic activity is essential for bacterial viability. We have determined the crystal structure of Escherichia coli Pth in complex with the tRNA CCA-acceptor-TΨC domain, the enzyme-binding region of the tRNA moiety of the substrate, at 2.4 Å resolution. In combination with site-directed mutagenesis studies, the structure identified the amino acid residues involved in tRNA recognition. The structure also revealed that Pth interacts with the tRNA moiety through the backbone phosphates and riboses, and no base-specific interactions were observed, except for the interaction with the highly conserved base G53. This feature enables Pth to accept the diverse sequences of the elongator-tRNAs as substrate components. Furthermore, we propose an authentic Pth:peptidyl-tRNA complex model and a detailed mechanism for the hydrolysis reaction, based on the present crystal structure and the previous studies’ results.
INTRODUCTION
During the course of protein synthesis, peptidyl-tRNA is prematurely produced from the ribosome as a result of aborted translation (1). For example, amino acid starvation, tRNA starvation and translation of a truncated mRNA will cause ribosome stalling and the release of peptidyl-tRNAs (2–4). Ribosomal recycling factor, elongation factor G and initiation factor 3, as well as other factors, contribute to this release (5–8). The accumulation of the peptidyl-tRNAs thus released is toxic for the cell, because they can either impair the initiation of translation or slow protein synthesis, due to the limitation of the corresponding tRNAs (9–11). However, this situation is prevented by the activity of peptidyl-tRNA hydrolase (Pth) [for review, see (12)]. Pth releases tRNA from peptidyl-tRNA, by cleaving the ester bond between the C-terminal end of the peptide and the 2′- or 3′-hydroxyl of the ribose at the 3′-end of the tRNA, thereby freeing the tRNA for reuse in protein synthesis (13,14). The activity of Pth is essential for the viability of bacteria (10,15).
The biochemical features of Pth have mainly been studied using the enzyme from Escherichia coli. Pth recognizes both the peptide and tRNA moieties of the substrate (16). Both of the moieties consist of diverse sequences depending on the substrate; nevertheless, Pth can accept various types of peptidyl-elongator tRNAs as substrates (14,17). In addition, Pth accepts peptidyl-tRNAHis, which has the peculiar feature of possessing one extra G nucleotide at position −1, as a substrate (18,19). The mechanism for this broad substrate specificity remains unknown. On the other hand, it is known that N-blocked aminoacyl-tRNAs are the shortest substrates of the enzyme (13,14); however, N-formyl-methionyl-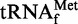 is hardly cleaved by Pth. (14,20). Indeed, this formylated initiator tRNA must be kept intact, to be recruited by initiation factor 2 and to participate in the formation of the ribosomal initiation complex. The resistance of formylated initiator tRNA to Pth is considered to be a consequence of the unpaired 1:72 nucleotides, which are a unique characteristic of the prokaryotic initiators (20). However, the detailed mechanism remains unknown. Moreover, it is not yet clear how the enzyme discriminates peptidyl-tRNAs from aminoacyl-tRNAs as the substrates, which is an essential mechanism for the normal peptide elongation.
is hardly cleaved by Pth. (14,20). Indeed, this formylated initiator tRNA must be kept intact, to be recruited by initiation factor 2 and to participate in the formation of the ribosomal initiation complex. The resistance of formylated initiator tRNA to Pth is considered to be a consequence of the unpaired 1:72 nucleotides, which are a unique characteristic of the prokaryotic initiators (20). However, the detailed mechanism remains unknown. Moreover, it is not yet clear how the enzyme discriminates peptidyl-tRNAs from aminoacyl-tRNAs as the substrates, which is an essential mechanism for the normal peptide elongation.
The Pth enzymes are ubiquitous in nature and can be classified into two types—Pth, sometimes referred to as Pth1 and Pth2. Pth exists in bacteria (21), whereas Pth2 is present in archaea (22,23). There is no sequence or structural similarity between Pth and Pth2. On the other hand, eukaryotes possess multiple types of Pths, including the Pth and Pth2 types and others (12). Considering the rising incidence of bacterial resistance to known antibiotics, Pth is an attractive target and is now in the spotlight, because, as mentioned above, bacteria possess a single type of Pth and its activity is essential for their viability, whereas eukaryotes possess multiple types of Pths (12,24). To develop novel antibiotics, the characterization of the interaction between Pth and its substrate is needed at an atomic level.
The structures of the Pth proteins from several species have been determined, to elucidate the essential translation maintenance and tRNA-recycling functions of this enzyme and to develop novel antibiotics (25–28). However, the molecular mechanism of the reaction remains unclear, because all of these structures have been determined in the substrate-free form. Thus, as the first step, we have determined the crystal structure of E. coli Pth in complex with the CCA-acceptor-TΨC domain of tRNA (Figure 1), the enzyme-binding region of the tRNA moiety (as described later), at 2.4 Å resolution. In addition, we performed site-directed mutagenesis experiments. According to these results, we identified the amino acid residues involved in the tRNA moiety recognition and also revealed the mechanism by which Pth accepts the diverse sequences of the elongator-tRNAs as substrate components. Furthermore, we present an authentic Pth:peptidyl-tRNA complex model and a detailed mechanism for the hydrolysis reaction, which clearly explain the previous studies’ results.
Figure 1.

Full-length tRNA and the CCA-acceptor-TΨC domain. (A) The base sequence of E. coli
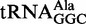 , drawn in the cloverleaf form. The acceptor stem is colored pink, the D loop is lime, the anticodon arm is violet, the variable loop is blue, the TΨC arm is cyan and the CCA terminus is yellow. The CCA-acceptor-TΨC domain is surrounded by a red dashed line. (B) Ribbon diagram of yeast free tRNAPhe (PDB accession code 6TNA) (40), with the same color-coding as in (A). The CCA-acceptor-TΨC domain is surrounded by a red dashed line.
, drawn in the cloverleaf form. The acceptor stem is colored pink, the D loop is lime, the anticodon arm is violet, the variable loop is blue, the TΨC arm is cyan and the CCA terminus is yellow. The CCA-acceptor-TΨC domain is surrounded by a red dashed line. (B) Ribbon diagram of yeast free tRNAPhe (PDB accession code 6TNA) (40), with the same color-coding as in (A). The CCA-acceptor-TΨC domain is surrounded by a red dashed line.
MATERIALS AND METHODS
Sample preparation and crystallization
Sample production, purification and crystallization of E. coli Pth in complex with the CCA-acceptor-TΨC domain of E. coli tRNAAla (G1 to A7 and A49 to A76 of E. coli
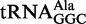 ) were performed as previously described (29). Briefly, E. coli Pth was overexpressed in E. coli strain BL21(DE3) with an N-terminal His-tag. The recombinant protein was purified by a Ni-chelating column and then treated with thrombin to remove the N-terminal His-tag. The protein was further purified using Ni-chelating, benzamidine-conjugated and anion-exchange resins. The CCA-acceptor-TΨC domain of E. coli tRNAAla was synthesized by in vitro transcription using T7 RNA polymerase and purified with an anion-exchange column. For crystallization, E. coli Pth and the CCA-acceptor-TΨC domain were mixed in a molar ratio of 1:2.5 at a final protein concentration of 14 mg/ml, in 10 mM HEPES–KOH (pH 7.6) containing 10 mM MgCl2 and 7 mM β-mercaptoethanol. Crystals were obtained with the sitting-drop vapor-diffusion method using a reservoir solution containing 100 mM sodium acetate buffer (pH 5.2), 20% (w/v) 1,4-butanediol and 30 mM glycyl-glycyl-glycine at 20°C.
) were performed as previously described (29). Briefly, E. coli Pth was overexpressed in E. coli strain BL21(DE3) with an N-terminal His-tag. The recombinant protein was purified by a Ni-chelating column and then treated with thrombin to remove the N-terminal His-tag. The protein was further purified using Ni-chelating, benzamidine-conjugated and anion-exchange resins. The CCA-acceptor-TΨC domain of E. coli tRNAAla was synthesized by in vitro transcription using T7 RNA polymerase and purified with an anion-exchange column. For crystallization, E. coli Pth and the CCA-acceptor-TΨC domain were mixed in a molar ratio of 1:2.5 at a final protein concentration of 14 mg/ml, in 10 mM HEPES–KOH (pH 7.6) containing 10 mM MgCl2 and 7 mM β-mercaptoethanol. Crystals were obtained with the sitting-drop vapor-diffusion method using a reservoir solution containing 100 mM sodium acetate buffer (pH 5.2), 20% (w/v) 1,4-butanediol and 30 mM glycyl-glycyl-glycine at 20°C.
Data collection and structure determination
Prior to data collection, crystals were soaked in reservoir solution containing 10% (v/v) ethylene glycol as a cryoprotectant. Diffraction data were collected at BL41XU of SPring-8 (Harima, Japan) using a Mar 225 CCD detector. The data were processed and scaled using the program XDS (30) and analyzed by the CCP4 program suite (31). Initial phases were obtained by the molecular replacement method with the program MOLREP (32), using the structures of E. coli Pth (PDB accession code 2PTH) as a search model. The electron density of the CCA-acceptor-TΨC domain was then obtained by phase improvement with the program ARP/wARP (33) and could be traced using the program PHENIX (34). The model was subsequently improved by iterative cycles of manual model building with the program COOT (35) and maximum likelihood refinement with the program REFMAC5 (36).
Model analysis
The quality of the model was checked with the program PROCHECK (37). Computer graphic representations were prepared using the program PyMOL (http://www.pymol.org). The surface electrostatic potentials were calculated using the program APBS (38) and visualized with the program PyMOL.
Preparation of N-acetyl-alanyl-tRNAAla
Escherichia coli tRNAAla was synthesized by in vitro transcription as described previously (29). After transcription, the RNA sample was purified by 12% polyacrylamide gel electrophoresis under denaturing conditions with 8 M urea. The tRNAAla was retrieved from gel by soaking crushed gel in 0.5 M ammonium acetate buffer (pH 6.0) containing 1 mM EDTA, 10 mM magnesium acetate and 0.1% SDS, followed by precipitation with 2-propanol. Purified tRNAAla was aminoacylated at 37°C in 22 ml of 50 mM HEPES–KOH (pH 7.6) containing 20 mM KCl, 10 mM MgCl2, 4 mM ATP, 20 mM β-mercaptoethanol, 0.01% BSA, 10 μM tRNAAla, 20 μM [14C]L-alanine (150 Ci/mol) and 150 nM purified E. coli Alanyl-tRNA synthetase (39). After a 1-h incubation, an aminoacylated sample was extracted with phenol and chloroform, followed by precipitation with 2-propanol in the presence of 0.3 M sodium acetate. The tRNA pellet was dissolved in 1.1 ml of 5 mM sodium acetate buffer (pH 5.5) and acetylated at 0°C for 15 min by adding 1.1 ml of dimethylsulfoxide, 0.22 ml of glacial acetic acid and 0.22 ml acetic anhydride, followed by precipitation with 2-propanol. The pellet was dissolved in 0.5 ml of 0.36 M sodium acetate buffer (pH 5.5) containing 10 mM CuSO4 and incubated at 37°C for 30 min. After incubation, to remove copper ions, the sample solution was applied to a PD-10 column (GE Healthcare) equilibrated with 20 mM sodium acetate buffer (pH 5.5) containing 100 mM KCl. The recovered tRNA samples were precipitated with 2-propanol and washed with 80% ethanol. Finally, the pellet was dissolved in 5 mM sodium acetate buffer (pH 5.5) and stored at −80°C.
Peptidyl-tRNA hydrolase assay
Peptidyl-tRNA hydrolase activity was measured at 28°C in 100 μl assays containing 20 mM HEPES–KOH (pH 7.6), 10 mM MgCl2, 3.5 to 25 μM acetyl-[14C]Ala-tRNAAla and 2–6 nM Pth variants. The reaction was quenched by addition of 100 μl of 10% TCA, and 20 μl of 4 mg/ml carrier RNA from yeast. After quenching, the sample was precipitated with 220 μl of 2-propanol in the presence of 0.3 M sodium acetate. 200 μl of the supernatant was added to 1 ml insta-gel plus (PerkinElmer), and the 14C radioactivity was measured by scintillation counting. Km and kcat values were generated by using the program GraphPad Prism version 2.0 (GraphPad Software).
RESULTS AND DISCUSSION
Structure determination
First, we attempted to crystallize E. coli Pth with a full-length tRNA. However, we were unable to obtain such crystals, probably because the flexible structure of tRNA caused problems in the crystallization. Thus, we next tried to crystallize E. coli Pth with the CCA-acceptor-TΨC domain of tRNA (Figure 1), because this region of tRNA is considered to be structurally stable, and the enzyme binds to the tRNA moiety via this region (described in the next section). As a result, we obtained the crystals of Pth in complex with this tRNA construct. The details of the sample preparation and crystallization are described in our previous report (29). The crystals belonged to the hexagonal space group P61, with unit-cell parameters a = b = 55.1, c = 413.1 Å. The structure was solved by molecular replacement at 2.4 Å resolution, with an Rwork of 19.3% and an Rfree of 23.7%. The data collection and structure refinement statistics are provided in Table 1.
Table 1.
Data collection and refinement statistics
| Data collection | |
|---|---|
| Space group | P61 |
| Unit-cell parameters (Å) | a = b = 55.1, c = 413.1 |
| No. of the Pth:CCA-acceptor–TΨC domain complex/asymmetric unit | 2 |
| VM (Å3/Da) | 2.8 |
| Solvent content (%) | 60.8 |
| Resolution range (Å) | 50.0–2.40 (2.54–2.40)a |
| No. of measured reflections | 296148 (33377) |
| No. of unique reflections | 27129 (4136) |
| Completeness (%) | 98.1 (93.7) |
| Redundancy | 10.9 (8.1) |
| Average I/σ(I) | 24.9 (8.2) |
| Rmergeb | 0.056 (0.187) |
| Refinement | |
| R-factor (%) | 19.3 |
| Rfreec (%) | 23.7 |
| No. of atoms/asymmetric unit | |
| Protein | 2960 |
| RNA | 1532 |
| Water | 139 |
| r.m.s.d. from ideal values | |
| Bond length (Å) | 0.009 |
| Bond angle (°) | 1.366 |
| Ramachandran plot statistics | |
| Most favored regions (%) | 93.8 |
| Allowed regions (%) | 6.2 |
| Generously allowed regions (%) | 0.0 |
| Disallowed regions (%) | 0.0 |
aValues in parentheses are for the highest resolution shell.
bRmerge = ∑hkl∑i |Ii(hkl) − <I(hkl)>|/∑hkl∑i Ii(hkl), where Ii(hkl) is the i-th intensity measurement of reflection hkl, including symmetry-related reflections and <I(hkl)> is its average.
cRfree was calculated by using 5% of randomly selected reflections that were excluded from the refinement.
Overall structure
The crystal contains two complexes of the Pth:CCA-acceptor-TΨC domain in the asymmetric unit, and the two complexes are quite similar. The r.m.s.d.’s over equivalent Cα atoms and backbone phosphorus atoms of the two complexes are 0.08 and 0.09 Å, respectively. Thus, one complex structure of the Pth:CCA-acceptor-TΨC domain in the asymmetric unit is shown in Figure 2. The overall structure of Pth consists of a single α/β globular domain. Seven β-strands form a twisted mixed central β-sheet, surrounded by a total of six helices. The structure of Pth in the complex is similar to that of the substrate-free enzyme (25), except for some loop regions (r.m.s.d. of 1.32 Å over 193 equivalent Cα atoms) (Supplementary Figure S1). On the other hand, the superposition of the CCA-acceptor-TΨC domain and free yeast tRNAPhe (40) showed that, although these structures are essentially similar, there are structural differences at the flexible 3′-CCA terminus and at the acceptor stem, (r.m.s.d. of 3.21 Å over 35 equivalent backbone phosphorus atoms) (Supplementary Figure S1). The structural difference within the acceptor stem part probably arose from the deletion of the D arm, anticodon arm and variable loop. However, a previous report demonstrated that the catalytic efficiency of Pth for the N-acetyl-histidyl-CCA-acceptor-TΨC domain of tRNAHis is comparable to that for the N-acetyl-histidyl-full-length tRNAHis (41), indicating that the deletion does not affect the interaction with Pth.
Figure 2.

Overall structure of Pth in complex with the CCA-acceptor-TΨC domain of tRNA. (A) Two views of the overall structure of the Pth:CCA-acceptor-TΨC domain complex, represented by a ribbon diagram. As in Figure 1, the acceptor stem is colored pink, the TΨC arm is cyan and the CCA terminus is yellow. Pth is colored green. To clarify the configuration for Pth:tRNA binding, the left panel is shown with the same viewing angle of the CCA-acceptor-TΨC domain as that of the full-length tRNA in Figure 1B. The right panel shows the view rotated by ∼90° along the vertical axis from the left panel. (B) Two views of the overall structure of the Pth:CCA-acceptor-TΨC domain complex, showing the surface of Pth colored according to its calculated electrostatic potential (blue, positively charged range +4 kT/e; red, negatively charged −4 kT/e). The viewpoints are the same as those in (A).
Pth interacts with the tRNA acceptor-TΨC domain on the TΨC stem side via two sites (Figure 2). At the first site, the positively charged patch around the active site cavity of Pth interacts with the backbone of the acceptor stem in an electrostatic manner (referred to as the ‘acceptor site’). At the second site, the C-terminal α-helix of Pth interacts with the minor groove of the TΨC stem in a shape-complementary manner (referred to as the ‘TΨC site’). In addition to these two interaction sites, the loop–helix α4 segment over the active site cavity of Pth is proximal to the 3′-CCA terminus, although a direct interaction was not observed (referred to as the ‘CCA site’). Peptidylation or N-acetyl-aminoacylation is probably a prerequisite for the interaction. The details of these three sites are described in a following section. The present structure also indicates that the D arm, anticodon arm and variable loop cannot exist in locations where Pth can interact with them. Therefore, although the CCA-acceptor-TΨC domain used here is a partial construct of tRNA, it is sufficient for the description of all of the aspects of the interaction between Pth and the tRNA moiety of the substrate.
We compared the structure of the Pth:CCA-acceptor-TΨC domain complex with those of the other protein:tRNA complexes determined so far, in which the proteins recognize the acceptor and TΨC stem regions on the TΨC stem side. The structure comparison revealed that, whereas Pth recognizes the acceptor stem and the TΨC stem of the substrate by its single domain, other proteins, such as EF-Tu (42), CCA-adding enzyme (43,44), GatB (45,46) and RNase Z (47), recognize them by multi-domain or multi-subunit systems. These facts indicate that Pth has a unique tRNA recognition feature. For more details, see the Supplementary Results and Discussion and Supplementary Figure S2.
Interaction at the acceptor site
At the acceptor site, the positively charged patch of Pth, which is composed of the highly conserved basic amino acid residues Lys103, Lys105 and Arg133 (Supplementary Figure S3), interacts with the tRNA acceptor stem in an electrostatic manner (Figure 3A and 3D). Specifically, the Nζ atoms of Lys103 and Lys105 form hydrogen bonds with the phosphate groups of G4 and G2, respectively. The Nη1 atom of Arg133 also forms a hydrogen bond with the phosphate group of G3. The Nη2 atom of Arg133 makes a bifurcated hydrogen bond with the O2′ and O3′ atoms of the G2 ribose. The substitution of these basic amino acid residues with neutral ones reduced the substrate-binding efficiency (19,48–50). In addition to these interactions, the main chain amide nitrogen of Gly109 forms hydrogen bonds with the phosphate group of G2 and the O3′ atom of the G1 ribose. The Oδ2 atom of Asp96 also forms a hydrogen bond with the O2′ atom of the G2 ribose. The replacement of Asp96 with alanine, however, has little effect on the catalytic efficiency (41). This replacement may be compensated by Arg133.
Figure 3.

Interaction between Pth and the tRNA moiety of the substrate. (A–C) Close-up view of the acceptor site (A), the TΨC site (B) and the CCA site (C). Pth is represented by a ribbon model, while the tRNA moiety is represented by a phosphate trace model, with the same color-coding as in Figure 2A. The amino acid residues and nucleotides involved in the interaction are depicted by stick models. The catalytic center His20 is also shown by a stick model. Interactions are indicated with dashed lines. Possible hydrogen bonds from Lys142 and Lys182 are indicated with solid lines. (D) Schematic diagram of the interaction between Pth and the tRNA moiety of the substrate. Interactions are indicated by dashed lines. Possible hydrogen bonds from Lys142 and Lys182 are indicated with solid lines. The color-coding of the bases is the same as in (A–C): the bases from the acceptor stem are colored pink, the TΨC arm is cyan and the CCA terminus is yellow. Riboses and phosphate groups are colored light blue and yellow, respectively.
The mutation of Gly100 with aspartic acid is responsible for the Pth (Ts) phenotype, which can grow normally at 30°C, but cannot sustain growth at the non-permissive temperature of 42°C (48). The present structure shows that, although a direct interaction is not observed, Gly100 exists in a loop connected to a β-strand that includes Lys103 and Lys105. Thus, the slight conformational change of the loop, caused by the mutation of Gly100, might perturb the structure around Lys103 and Lys105 at the non-permissive temperature and consequently destabilize the Pth:substrate complex formation, as suggested in a previous report (50).
In the present structure, the phosphate group of G1 is covalently linked to an unexpectedly extended guanosine nucleotide (G-1) (Supplementary Figure S4). This extra G-1 may have been caused by the untemplated incorporation of GMP during in vitro transcription by T7 RNA polymerase, arising from the 5′-terminal consecutive guanosine nucleotides of the RNA sequence (29,51). We identified the transcription products as a mixture of the CCA-acceptor-TΨC domains with and without the extra G-1 and other molecules, by MALDI–TOF MS analyses (Supplementary Figure S5). However, the structure shows that the extra G-1 sustains the flexible 3′-CCA terminus (Supplementary Figure S4), and this situation would result in the preferential crystal formation of the complex including the CCA-acceptor-TΨC domain with the extra G-1. Indeed, we tried to obtain crystals of the complex by using the acceptor-TΨC domain that does not include the extra G-1 by rigorous purification; nevertheless, we could not obtain such a crystal.
Fromant et al. investigated the combined effects of the removal of the 5′-terminal phosphate of the substrate and of various amino acid substitutions in Pth on the kcat/Km values and demonstrated that the removal of the 5′-terminal phosphate exerted no marked combined effect for the K105A and R133A mutants, in contrast to the other mutants (49). In addition, Lys105 and Arg133 are very close together on Pth. Therefore, the 5′-terminal phosphate of the substrate was expected to be clamped by Lys105 and Arg133 (49). On the other hand, in the present crystal structure, as mentioned above, the phosphate group of G1, which corresponds to the intrinsic 5′-terminal phosphate, is covalently linked to the extra G-1 nucleotide, and this extra G-1 is involved in the crystal packing. For this reason, the conformation around the phosphate group of G1 is slightly distorted and lies ∼7 Å away from the surface of Pth. However, the removal of the extra G-1 and a slight conformational change around G1 allowed the original 5′-terminal phosphate to be modeled to bind to the positively charged patch around the active site cavity, which includes Lys105 and Arg133, without disturbing the interaction between Pth and the tRNA moiety, and without disrupting the base pairs (Figure 4A). More specifically, the 5′-terminal phosphate is expected to hydrogen bond with the main chain amide nitrogen of His110. This area is positively charged, and Lys105 and Arg133 would contribute to its net positive charge. Thus, our current model is consistent with the previous mutagenesis study that reported the decreased catalytic efficiency by the alanine substitutions of Lys105 and/or Arg133 (49). In addition, a previous NMR chemical shift perturbation study demonstrated that the spectral properties of the main chain amide nitrogen of His110 are strongly affected by the addition of a duplex RNA that mimics the CCA-acceptor-TΨC stem part of tRNA (41). Further experiments are necessary to determine the precise binding site of the 5′-terminal phosphate group.
Figure 4.

Structure of the Pth:peptidyl-tRNA complex model. (A) Structure around G1, in the case without the extra G-1. (B) Structure around the active site cavity. Left panels in (A) and (B): the amino acid residues involved in the interaction between Pth and the peptidyl-tRNA are depicted by stick models on a transparent ribbon model of Pth. As in Figure 2A, the acceptor stem is colored pink, the CCA terminus is yellow, and Pth is green. In addition, the peptide moiety is represented by an orange stick model. Right panels in (A) and (B): the viewpoint is the same as that of each left panel. Pth is shown in a surface model, colored as in Figure 2B. In (A), to make the structure around G1 more visible, the peptidyl-A76 moiety is omitted. In (B), the ester bond between the peptide and tRNA moiety is indicated by a circle. In all panels, hydrogen bonds are indicated with dashed lines. The removal simulation of the extra G-1 and the peptidyl-A76 docking simulation were performed manually as described in the text, and then the geometry was idealized using the program COOT (35).
Interaction at the TΨC site
At the TΨC site, the C-terminal α-helix of Pth penetrates into and interacts with the minor groove of the TΨC stem of the substrate (Figure 3B and D). Specifically, the Nδ1 atom of the imidazole ring of His188 forms a hydrogen bond with the phosphate group of G63. The involvement of His188 in the interaction was unexpected and thus was not analyzed previously. Therefore, we replaced His188 with alanine and measured the catalytic parameters using N-acetyl-alanyl-tRNAAla as a substrate (Table 2). The results revealed that the H188A mutant resulted in a 5.4-fold decrease in the kcat/Km value, as compared to that of the wild-type enzyme. This reduction was mainly caused by an increase in the Km value. In addition to His188, the crystal structure showed that the Oδ1 and Nδ2 atoms of Asn185 hydrogen bond with the N2 atom of the G53 base and the O2′ atom of the U(T)54 ribose, respectively. These interactions are consistent with a previous NMR chemical shift perturbation study that implied the involvement of Asn185 in the substrate binding (41). As in the case of His188, we substituted Asn185 with alanine and measured the catalytic parameters using N-acetyl-alanyl-tRNAAla as a substrate. The N185A mutant resulted in a 5.7-fold decrease in the kcat/Km value, as compared to that of the wild-type enzyme, whereas the kcat value was essentially unchanged. These results are consistent with those from a previous site-directed mutagenesis study, in which N-acetyl-lysyl-tRNALys was used as a substrate (41). In addition, we measured the catalytic parameters of the double mutant of N185A and H188A. The mutant resulted in a further decrease in the kcat/Km value and a further increase in the Km value, whereas the kcat value was barely influenced. These results indicate that Asn185 and His188 play important roles in facilitating productive substrate binding.
Table 2.
Catalytic parameters of Pth variants
| Enzyme | kcat (/s) | Km (μM) | relative kcat/Kma |
|---|---|---|---|
| WT | 11.7 ± 0.65 | 4.71 ± 0.77 | 100 |
| N185A | 5.80 ± 0.80 | 13.4 ± 2.36 | 17.4 |
| H188A | 7.93 ± 0.53 | 17.1 ± 2.03 | 18.6 |
| N185A-H188A | 8.70 ± 0.83 | 26.9 ± 3.95 | 13 |
aRelative kcat/Km values are shown, given an arbitrary value of 100 with the WT enzyme.
The Nζ atom of Lys182 exists in the proximity of the O3′ atom of U(T)54 and the backbone phosphate group of U(Ψ)55, although a direct interaction is not observed. However, the plasticity of the equivalent segment of Pth from Mycobacterium tuberculosis has been demonstrated by an NMR experiment (27), suggesting that Lys182 interacts with either or both U(T)54 and U(Ψ)55 during the reaction process. Indeed, the Km value of the K182A mutant is >8-fold higher than that of the wild-type enzyme, whereas the kcat value only displayed a minor change (41).
Interaction at the CCA site
Probably due to either the absence of peptidylation or N-acetyl-aminoacylation, or perhaps the influence of the crystal packing, no direct interaction between Pth and the CCA terminus was observed in the present structure. However, the Nζ atom of Lys142, which resides in the loop–helix α4 segment over the active site cavity, exists in the proximity of the backbone phosphates of C74 and the discriminator A73 (Figure 3C). On the other hand, the plasticity of the loop–helix α4 segment is suggested from the high average temperature factor of this region (Supplementary Figure S6). Moreover, the plasticity of the equivalent segment of Pth from M. tuberculosis has been demonstrated by an NMR experiment (27). These facts suggest that Lys142 interacts with the backbone phosphates of either or both C74 and the discriminator A73 during the reaction process. Indeed, the Km value of the K142A mutant is >5-fold larger than that of the wild-type enzyme, whereas the kcat value was found to be comparable between the two (25). It seems likely that the loop-helix α4 segment captures the flexible CCA terminus of the substrate, by an electrostatic interaction via Lys142, to recruit or stabilize the binding of the peptidyl-adenosine moiety within the active site cavity.
Mechanism of the broad specificity towards tRNA moieties
It has been demonstrated that Pth recognizes both the peptide and tRNA moieties of the substrate (16). Both of these moieties consist of diverse sequences depending on the substrate; nevertheless, Pth can accept many kinds of peptidyl-elongator tRNAs as substrates (14,17). The present structure has revealed the structural basis for the sequence-independent recognition of the tRNA moiety. Specifically, in the acceptor site, the amino acid residues on the positively charged patch of Pth interact with the backbone phosphates and ribose of the acceptor stem, and a base-specific interaction was not observed (Figure 3A and D). Likewise, in the TΨC site, the amino acid residues on the C-terminal α-helix of Pth interact with the backbone phosphate and ribose of the TΨC stem, and a base-specific interaction was also not observed, except for the highly conserved G53 (Figure 3B and D). In the CCA site, as discussed above, Lys142 probably interacts with the discriminator-CCA moiety through the backbone phosphates, but not through the bases (Figure 3C and D). These features allow Pth to accomplish sequence-independent recognition of the tRNA moiety.
Pth accepts peptidyl-tRNAHis, which has the peculiar feature of possessing one extra G nucleotide at position −1 (G-1), as a substrate (18,19). A previous kinetic study demonstrated that the substrate efficiencies of 5′-dephosphorylated and G-1-obliterated N-acetyl-histidyl-tRNAHis molecules were comparable to that of the full-length N-acetyl-histidyl-tRNAHis. However, upon the loss of the 5′-phosphate group of the G-1-obliterated N-acetyl-histidyl-tRNAHis, the kcat/Km value decreased 7-fold (19). These results indicated that the phosphate group of G1 plays a crucial role in the reaction of Pth, in contrast to the dispensability of G-1. On the other hand, in the case of diacetyl-lysyl-tRNALys as a substrate, the removal of the 5′-phosphate group decreased the kcat/Km value by 17-fold (49), as in the case of the removal of the 5′-phosphate group from the G-1-obliterated N-acetyl-histidyl-tRNAHis. Based on these facts, Fromant et al. proposed that the locations of the G1 phosphate group of tRNAHis and the 5′-phosphate group of standard elongator tRNAs on Pth are identical, whereas the G-1 of tRNAHis is in a position that does not hinder the Pth reaction (19). Actually, there is sufficient space on top of G1, on the surface of the enzyme, to accommodate G-1 of tRNAHis (Figure 4A). Therefore, we consider the previous proposition to be structurally reasonable.
Peptidyl-tRNA-binding mode
In the Pth:CCA-acceptor-TΨC domain structure obtained here, the 3′-terminal adenosine A76 did not bind to the active site of Pth. Peptidylation or N-acetyl-aminoacylation may be required to place the 3′-terminal adenosine A76 in the binding site. However, the region on Pth that interacts with A76 can be predicted, using the present structure. In addition, enzymatic analyses and NMR chemical shift perturbation studies have indicated the amino acid residues involved in the substrate binding (19,25,41,49,50,52). Moreover, in the structure of substrate-free Pth from E. coli, three residues (K191–A192–Q193), at the C-terminus of a neighboring enzyme molecule in the crystal, were bound to the active site, and this interaction was assumed to represent the formation of a complex between Pth and the peptide segment of the substrate (25). Taking this information into account, we modeled the peptidyl-A76 moiety in the active site cavity, as discussed below.
There is a pocket suitable for accommodating the adenine ring in the active site cavity (Figure 4B). Due to the conformational change of the CCA terminus backbone on the Pth:CCA-acceptor-TΨC domain structure obtained here, the adenine ring of A76 could be positioned in the pocket, so that the N6 atom of the adenine ring form hydrogen bonds with the main chain carbonyl oxygen of Gly111, and the N3 atom of the adenine ring makes van der Waals contacts with the side chains of Leu95 and Val149 (Figures 4B and 5A). This docking is reasonable, because NMR chemical shift perturbation studies revealed the involvement of Leu95, Gly111 and Val149 in substrate analog binding (41,52). Leu95 has also been demonstrated to be involved in the catalysis by a site-directed mutagenesis study (41). We next placed the K191–A192–Q193 tripeptide of the substrate-free Pth structure into the equivalent position of the Pth:CCA-acceptor-TΨC domain structure. An ester bond between the K191–A192–Q193 tripeptide and the 3′-OH of A76 could be formed, by the slight conformational change of Gln193 (Supplementary Figure S7). Therefore, we rearranged the conformations of the side chains of Asn68 and Asn114 to resemble those in the substrate-free Pth structure, in which the Nδ2 atoms of these amino acid residues form hydrogen bonds with the C-terminal carboxyl groups of the K191–A192–Q193 tripeptide. As a result, in the model, the Nδ2 atoms of Asn68 and Asn114 form hydrogen bonds with the ester carbonyl oxygen of the substrate. In addition, as in the case of the substrate-free Pth, the Nδ2 atom of Asn10 and the main chain amide nitrogen of Asn68 form hydrogen bonds with the main chain carbonyl oxygens of Ala192 and Lys191 of the peptide moiety of the substrate, respectively. Indeed, the involvement of Asn10, Asn68 and Asn114 in substrate binding has been demonstrated by enzymatic analyses and an NMR chemical shift perturbation study (25,49,52).
Figure 5.

Peptidyl-tRNA binding and catalysis. (A) Active site of the Pth:peptidyl-tRNA complex model. Stick models of Pth (green), A76 (yellow) and the peptide moiety (orange) are represented on a transparent ribbon model of Pth. (B) Active site of the acyl-enzyme intermediate of an elastase (PDB accession code 1HAX) (54). Stick models of elastase (green) and the peptide (orange) are represented on a transparent ribbon model of elastase. In (A) and (B), hydrogen bonds are indicated with dashed lines. The water molecules, which are poised to nucleophilically attack the ester carbon, are represented by red spheres.
Consequently, in the current Pth:peptidyl-tRNA complex model, Pth recognizes only the main chain atoms of the substrate and the side chains of the substrate point toward away from the enzyme (Figures 4B and 5A). Thus, this model explains the sequence independence of the peptide moiety recognition by the enzyme. In addition, this model clearly explains why the enzyme does not digest aminoacyl-tRNAs. Because, Asn10, which is a determinant for discrimination between peptidyl- and aminoacyl-tRNAs (52), forms a hydrogen bond with the first amide oxygen of the peptide moiety, and this interaction cannot be accomplished by aminoacyl-tRNAs.
In the previous docking study, the adenine ring of A76 was expected to stack on Phe66 (41). However, this prediction is not fully consistent with other studies, for the following reasons. First, the degree of the kcat/Km value reduction by the replacement of Phe66 with alanine is very small, as compared to the cases of other putative substrate-binding amino acid residues in the active site cavity (25,41,49,50). Second, the chemical shift variation of Phe66 upon the addition of a substrate analog was not very high, as compared to the cases of other putative substrate-binding amino acid residues in the active site cavity (52). Third, in light of the present complex structure, it seems unlikely that the CCA terminus extends so that the adenine ring of A76 stacks on Phe66, thus positioning the ester bond between the peptide and A76 near His20, in the catalytic center (Figure 4B). If the adenine ring of A76 stacks on Phe66, then this situation would either locate the ester bond farther away from Phe66, or cause steric hindrance between the peptide and tRNA moieties of the substrate. In contrast, in our model, the ester bond exists near His20, as described in the next section. Furthermore, we performed a docking simulation with tripeptidyl-adenosine as a search molecule, using AutoDock Vina (53). The best result fit well to the tripeptidyl-A76 moiety of our model (see ‘Supplementary Materials and Methods’ section and Supplementary Figure S7). No interaction between the adenine ring and Phe66 was observed in all of the candidate docking models suggested by the program.
Hydrolysis reaction of Pth
We found that the configuration around the active site cavity of the Pth:peptidyl-tRNA complex model described above is comparable to that of the acyl-enzyme intermediate of an elastase, in which the C-terminal carboxyl group of a cleaved peptide is linked via an ester bond to the catalytic Ser residue (54). That is, in the active site of Pth (Figure 5A), His20 and Asp93 are present, and a hydrogen bond is formed between the Nδ1 atom of His20 and the Oδ1 atom of Asp93, in a similar manner as the His57 and Asp102 residues of the catalytic triad of the acyl-enzyme intermediate (Figure 5B). Relative to these histidine and aspartic acid residues, the ester groups of the peptidyl-tRNA and the acyl-enzyme intermediate exist in equivalent positions, although these histidine and aspartic acid residues are on opposite sides, relative to their ester bond planes. Hence, we could model a water molecule that can nucleophilically attack the ester carbon of the peptidyl-tRNA, in a similar manner as the acyl-enzyme intermediate. Thereby, this water molecule formed a hydrogen bond with the Nε2 atom of His20 in the Pth:peptidyl-tRNA complex model. In the acyl-enzyme intermediate, the main chain amide nitrogens of the Gly193 and Ser195 residues form hydrogen bonds with the ester carbonyl oxygen in the oxyanion hole. Similarly, in the complex model, the Nδ2 atoms of Asn68 and Asn114 form hydrogen bonds with the ester carbonyl oxygen of the peptidyl-tRNA.
In addition to the structural similarity with the acyl-enzyme intermediate, a previous enzymatic analysis demonstrated that His20 acts as the catalytic base in the hydrolysis reaction of Pth, and Asp93 contributes to the lowering of the pKa value of this His20 (50). Furthermore, a site-directed mutagenesis study revealed that single residue substitutions of Asn68 and Asn114 with alanine caused considerable changes in the kcat values by two orders of magnitude, whereas the Km values were comparable to that of the wild-type enzyme (49). Thus, as also described by Goodall et al. (50), these data suggest that Pth catalyzes peptidyl-tRNA hydrolysis by following a reaction mechanism similar to that of the acyl-enzyme intermediate of an elastase: Asp93 stabilizes the basic form of His20, such that it will accept a proton from a proximal water molecule, which then nucleophilically attacks the ester carbon of the peptidyl-tRNA and generates the tetrahedral oxyanion intermediate. This intermediate is stabilized by the hydrogen bonds from Asn68 and Asn114. The decomposition of the tetrahedral oxyanion intermediate subsequently occurs through general acid catalysis by the Asp93-polarized His20, producing the peptide and the tRNA.
CONCLUSION
In conclusion, we have determined the crystal structure of E. coli Pth in complex with the tRNA CCA-acceptor-TΨC domain, the enzyme-binding region of the tRNA moiety of the substrate. The structure revealed the amino acid residues involved in the tRNA recognition and the mechanism by which Pth accepts the diverse sequences of the elongator-tRNAs as substrate components. Furthermore, we presented the authentic Pth:peptidyl-tRNA complex model and the precise mechanism for the hydrolysis reaction, based on the present crystal structure and the previous studies’ results. This study represents a significant advancement toward the complete understanding of the reaction mechanism of Pth.
ACCESSION NUMBERS
The structure factors and coordinates of the model have been deposited in the Protein Data Bank (PDB accession code: 3VJR).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Materials and Methods, Supplementary Results and Discussion, Supplementary Figures 1–7, and Supplementary References [55–60].
FUNDING
Grant-in-Aid for Young Scientists (Start-up) [20870018 to K.I.] and Grant-in-Aid for Young Scientists (B) [21770108 to K.I.] from the Japan Society for the Promotion of Science (JSPS); the Uchida Energy Science Promotion Foundation grant [22-1-10 to K.I.]; UNION TOOL CO grant (to K.I.); Grant for the Promotion of Niigata University Research Projects [22C017 to K.I.]; Grants-in-Aid for Education and Research at the Institute of Science and Technology from Niigata University (to K.I.). Funding for open access charge: Niigata University.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Shoji Odani and Dr Tomohiro Miyoshi of Niigata University for their helpful advice about this study. The synchrotron-radiation experiments were performed at BL41XU of SPring-8 with the approval of JASRI (Harima, Japan) (Proposal No. 2008B2182). The synchrotron-radiation experiments were also performed at BL17A of PF and at NW12A of PF-AR with the approval of KEK (Tsukuba, Japan) (Proposal No. 2009G178).
REFERENCES
- 1.Menninger JR. Peptidyl transfer RNA dissociates during protein synthesis from ribosomes of Escherichia coli. J. Biol. Chem. 1976;251:3392–3398. [PubMed] [Google Scholar]
- 2.Caplan AB, Menninger JR. Tests of the ribosomal editing hypothesis: amino acid starvation differentially enhances the dissociation of peptidyl-tRNA from the ribosome. J. Mol. Biol. 1979;134:621–637. doi: 10.1016/0022-2836(79)90370-x. [DOI] [PubMed] [Google Scholar]
- 3.Cruz-Vera LR, Magos-Castro MA, Zamora-Romo E, Guarneros G. Ribosome stalling and peptidyl-tRNA drop-off during translational delay at AGA codons. Nucleic Acids Res. 2004;32:4462–4468. doi: 10.1093/nar/gkh784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh NS, Varshney U. A physiological connection between tmRNA and peptidyl-tRNA hydrolase functions in Escherichia coli. Nucleic Acids Res. 2004;32:6028–6037. doi: 10.1093/nar/gkh924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heurgué-Hamard V, Karimi R, Mora L, MacDougall J, Leboeuf C, Grentzmann G, Ehrenberg M, Buckingham RH. Ribosome release factor RF4 and termination factor RF3 are involved in dissociation of peptidyl-tRNA from the ribosome. EMBO J. 1998;17:808–816. doi: 10.1093/emboj/17.3.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karimi R, Pavlov MY, Heurgué-Hamard V, Buckingham RH, Ehrenberg M. Initiation factors IF1 and IF2 synergistically remove peptidyl-tRNAs with short polypeptides from the P-site of translating Escherichia coli ribosomes. J. Mol. Biol. 1998;281:241–252. doi: 10.1006/jmbi.1998.1953. [DOI] [PubMed] [Google Scholar]
- 7.Gong M, Cruz-Vera LR, Yanofsky C. Ribosome recycling factor and release factor 3 action promotes TnaC-peptidyl-tRNA Dropoff and relieves ribosome stalling during tryptophan induction of tna operon expression in Escherichia coli. J. Bacteriol. 2007;189:3147–3155. doi: 10.1128/JB.01868-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh NS, Ahmad R, Sangeetha R, Varshney U. Recycling of ribosomal complexes stalled at the step of elongation in Escherichia coli. J. Mol. Biol. 2008;380:451–464. doi: 10.1016/j.jmb.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 9.Atherly AG. Peptidyl-transfer RNA hydrolase prevents inhibition of protein synthesis initiation. Nature. 1978;275:769. doi: 10.1038/275769a0. [DOI] [PubMed] [Google Scholar]
- 10.Menninger JR. Accumulation of peptidyl tRNA is lethal to Escherichia coli. J. Bacteriol. 1979;137:694–696. doi: 10.1128/jb.137.1.694-696.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menez J, Heurguè-Hamard V, Buckingham RH. Sequestration of specific tRNA species cognate to the last sense codon of an over produced gratuitous protein. Nucleic Acids Res. 2000;28:4725–4732. doi: 10.1093/nar/28.23.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das G, Varshney U. Peptidyl-tRNA hydrolase and its critical role in protein biosynthesis. Microbiology. 2006;152:2191–2195. doi: 10.1099/mic.0.29024-0. [DOI] [PubMed] [Google Scholar]
- 13.Cuzin F, Kretchmer N, Greenberg RE, Hurwitz R, Chapeville F. Enzymatic hydrolysis of N-substituted aminoacyl-tRNA. Proc. Natl. Acad. Sci. USA. 1967;58:2079–2086. doi: 10.1073/pnas.58.5.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kössel H, RajBhandary UL. Studies on polynucleotides. LXXXVI. Enzymic hydrolysis of N-acylaminoacyl-transfer RNA. J. Mol. Biol. 1968;35:539–560. doi: 10.1016/s0022-2836(68)80013-0. [DOI] [PubMed] [Google Scholar]
- 15.Menez J, Buckingham RH, de Zamaroczy M, Campelli CK. Peptidyl-tRNA hydrolase in Bacillus subtilis, encoded by spoVC, is essential to vegetative growth, whereas the homologous enzyme in Saccharomyces cerevisiae is dispensable. Mol. Microbiol. 2002;45:123–129. doi: 10.1046/j.1365-2958.2002.02992.x. [DOI] [PubMed] [Google Scholar]
- 16.Shiloach J, Bauer S, de Groot N, Lapidot Y. The influence of the peptide chain length on the activity of peptidyl-tRNA hydrolase from E. coli. Nucleic Acids Res. 1975;2:1941–1950. doi: 10.1093/nar/2.10.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Groot N, Groner Y, Lapidot Y. Peptidyl-tRNA. VII. Substrate specificity of peptidyl-tRNA hydrolase. Biochim. Biophys. Acta. 1969;186:286–296. doi: 10.1016/0005-2787(69)90006-9. [DOI] [PubMed] [Google Scholar]
- 18.Menninger JR. The accumulation as peptidyl-transfer RNA of isoaccepting transfer RNA families in Escherichia coli with temperature-sensitive peptidyl-transfer RNA hydrolase. J. Biol. Chem. 1978;253:6808–6813. [PubMed] [Google Scholar]
- 19.Fromant M, Plateau P, Blanquet S. Function of the extra 5'-phosphate carried by histidine tRNA. Biochemistry. 2000;39:4062–4067. doi: 10.1021/bi9923297. [DOI] [PubMed] [Google Scholar]
- 20.Dutka S, Meinnel T, Lazennec C, Mechulam Y, Blanquet S. Role of the 1-72 base pair in tRNAs for the activity of Escherichia coli peptidyl-tRNA hydrolase. Nucleic Acids Res. 1993;21:4025–4030. doi: 10.1093/nar/21.17.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brun G, Paulin D, Yot P, Chapeville F. Peptidyl-tRNA hydrolase: demonstration in various organisms. Enzymatic activity in the presence of ribosomes. Biochimie. 1971;53:225–231. doi: 10.1016/s0300-9084(71)80054-8. [DOI] [PubMed] [Google Scholar]
- 22.Rosas-Sandoval G, Ambrogelly A, Rinehart J, Wei D, Cruz-Vera LR, Graham DE, Stetter KO, Guarneros G, Söll D. Orthologs of a novel archaeal and of the bacterial peptidyl-tRNA hydrolase are nonessential in yeast. Proc. Natl Acad. Sci. USA. 2002;99:16707–16712. doi: 10.1073/pnas.222659199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fromant M, Ferri-Fioni ML, Plateau P, Blanquet S. Peptidyl-tRNA hydrolase from Sulfolobus solfataricus. Nucleic Acids Res. 2003;31:3227–3235. doi: 10.1093/nar/gkg428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris SM, McFeeters H, Ogungbe IV, Cruz-Vera LR, Setzer WN, Jackes BR, McFeeters RL. Peptidyl-tRNA hydrolase screening combined with molecular docking reveals the antibiotic potential of Syzygium johnsonii bark extract. Nat. Prod. Commun. 2011;6:1421–1424. [PubMed] [Google Scholar]
- 25.Schmitt E, Mechulam Y, Fromant M, Plateau P, Blanquet S. Crystal structure at 1.2 Å resolution and active site mapping of Escherichia coli peptidyl-tRNA hydrolase. EMBO J. 1997;16:4760–4769. doi: 10.1093/emboj/16.15.4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selvaraj M, Roy S, Singh NS, Sangeetha R, Varshney U, Vijayan M. Structural plasticity and enzyme action: crystal structures of Mycobacterium tuberculosis peptidyl-tRNA hydrolase. J. Mol. Biol. 2007;372:186–193. doi: 10.1016/j.jmb.2007.06.053. [DOI] [PubMed] [Google Scholar]
- 27.Pulavarti SV, Jain A, Pathak PP, Mahmood A, Arora A. Solution structure and dynamics of peptidyl-tRNA hydrolase from Mycobacterium tuberculosis H37Rv. J. Mol. Biol. 2008;378:165–177. doi: 10.1016/j.jmb.2008.02.027. [DOI] [PubMed] [Google Scholar]
- 28.Clarke TE, Romanov V, Lam R, Gothe SA, Peddi SR, Razumova EB, Lipman RS, Branstrom AA, Chirgadze NY. Structure of Francisella tularensis peptidyl-tRNA hydrolase. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 2011;67:446–449. doi: 10.1107/S174430911100515X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ito K, Qi H, Shimizu Y, Murakami R, Miura K, Ueda T, Uchiumi T. Crystallization and preliminary X-ray analysis of Peptidyl-tRNA hydrolase from Escherichia coli in complex with the acceptor-TΨC domain of tRNA. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011;67:1566–1569. doi: 10.1107/S1744309111038383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993;26:795–800. [Google Scholar]
- 31.Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr, D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 33.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 34.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 37.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 38.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimizu Y, Inoue A, Tomari Y, Suzuki T, Yokogawa T, Nishikawa K, Ueda T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001;19:751–755. doi: 10.1038/90802. [DOI] [PubMed] [Google Scholar]
- 40.Sussman JL, Holbrook SR, Warrant RW, Church GM, Kim SH. Crystal structure of yeast phenylalanine transfer RNA. I. Crystallographic refinement. J. Mol. Biol. 1978;123:607–630. doi: 10.1016/0022-2836(78)90209-7. [DOI] [PubMed] [Google Scholar]
- 41.Giorgi L, Bontems F, Fromant M, Aubard C, Blanquet S, Plateau P. The RNA binding site of Escherichia coli peptidyl-tRNA hydrolase. J. Biol. Chem. 2011;286:39585–39594. doi: 10.1074/jbc.M111.281840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nissen P, Kjeldgaard M, Thirup S, Polekhina G, Reshetnikova L, Clark BF, Nyborg J. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. Science. 1995;270:1464–1472. doi: 10.1126/science.270.5241.1464. [DOI] [PubMed] [Google Scholar]
- 43.Xiong Y, Steitz TA. Mechanism of transfer RNA maturation by CCA-adding enzyme without using an oligonucleotide template. Nature. 2004;430:640–645. doi: 10.1038/nature02711. [DOI] [PubMed] [Google Scholar]
- 44.Tomita K, Ishitani R, Fukai S, Nureki O. Complete crystallographic analysis of the dynamics of CCA sequence addition. Nature. 2006;443:956–960. doi: 10.1038/nature05204. [DOI] [PubMed] [Google Scholar]
- 45.Blaise M, Bailly M, Frechin M, Behrens MA, Fischer F, Oliveira CL, Becker HD, Pedersen JS, Thirup S, Kern D. Crystal structure of a transfer-ribonucleoprotein particle that promotes asparagine formation. EMBO J. 2010;29:3118–3129. doi: 10.1038/emboj.2010.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ito T, Yokoyama S. Two enzymes bound to one transfer RNA assume alternative conformations for consecutive reactions. Nature. 2010;467:612–616. doi: 10.1038/nature09411. [DOI] [PubMed] [Google Scholar]
- 47.Li de la Sierra-Gallay I, Mathy N, Pellegrini O, Condon C. Structure of the ubiquitous 3' processing enzyme RNase Z bound to transfer RNA. Nat. Struct. Mol. Biol. 2006;13:376–377. doi: 10.1038/nsmb1066. [DOI] [PubMed] [Google Scholar]
- 48.García-Villegas MR, De La Vega FM, Galindo JM, Segura M, Buckingham RH, Guarneros G. Peptidyl-tRNA hydrolase is involved in lambda inhibition of host protein synthesis. EMBO J. 1991;10:3549–3555. doi: 10.1002/j.1460-2075.1991.tb04919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fromant M, Plateau P, Schmitt E, Mechulam Y, Blanquet S. Receptor site for the 5'-phosphate of elongator tRNAs governs substrate selection by peptidyl-tRNA hydrolase. Biochemistry. 1999;38:4982–4987. doi: 10.1021/bi982657r. [DOI] [PubMed] [Google Scholar]
- 50.Goodall JJ, Chen GJ, Page MG. Essential role of histidine 20 in the catalytic mechanism of Escherichia coli peptidyl-tRNA hydrolase. Biochemistry. 2004;43:4583–4591. doi: 10.1021/bi0302200. [DOI] [PubMed] [Google Scholar]
- 51.Pleiss JA, Derrick ML, Uhlenbeck OC. T7 RNA polymerase produces 5' end heterogeneity during in vitro transcription from certain templates. RNA. 1998;4:1313–1317. doi: 10.1017/s135583829800106x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giorgi L, Plateau P, O'Mahony G, Aubard C, Fromant M, Thureau A, Grøtli M, Blanquet S, Bontems F. NMR-Based substrate analog docking to Escherichia coli peptidyl-tRNA hydrolase. J. Mol. Biol. 2011;412:619–633. doi: 10.1016/j.jmb.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 53.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilmouth RC, Edman K, Neutze R, Wright PA, Clifton IJ, Schneider TR, Schofield CJ, Hajdu J. X-ray snapshots of serine protease catalysis reveal a tetrahedral. Nat. Struct. Biol. 2001;8:689–694. doi: 10.1038/90401. [DOI] [PubMed] [Google Scholar]
- 55.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller DL, Weissbach H. Aminoacyl-tRNA transfer factors. In: Pestka S, Weissbach H, editors. Molecular Mechanisms of Protein Biosynthesis. New York: Academic Press; 1977. pp. 323–373. [Google Scholar]
- 57.Deutscher MP. tRNA nucleotidyltransferase. In: Boyer PD, editor. The Enzymes. 1982. Vol. XV, part B, Academic Press, New York, pp. 183–215. [Google Scholar]
- 58.Raczniak G, Becker HD, Min B, Söll D. A single amidotransferase forms asparaginyl-tRNA and glutaminyl-tRNA in Chlamydia trachomatis. J. Biol. Chem. 2001;276:45862–45867. doi: 10.1074/jbc.M109494200. [DOI] [PubMed] [Google Scholar]
- 59.Oshikane H, Sheppard K, Fukai S, Nakamura Y, Ishitani R, Numata T, Sherrer RL, Feng L, Schmitt E, Panvert M, et al. Structural basis of RNA-dependent recruitment of glutamine to the genetic code. Science. 2006;312:1950–1954. doi: 10.1126/science.1128470. [DOI] [PubMed] [Google Scholar]
- 60.Nashimoto M. Distribution of both lengths and 5' terminal nucleotides of mammalian pre-tRNA 3' trailers reflects properties of 3' processing endoribonuclease. Nucleic Acids Res. 1997;25:1148–1154. doi: 10.1093/nar/25.6.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.