


Abstract
After the generation of DNA double-strand breaks (DSBs), poly(ADP-ribose) polymerase-1 (PARP-1) is one of the first proteins to be recruited and activated through its binding to the free DNA ends. Upon activation, PARP-1 uses NAD+ to generate large amounts of poly(ADP-ribose) (PAR), which facilitates the recruitment of DNA repair factors. Here, we identify the RNA-binding protein NONO, a partner protein of SFPQ, as a novel PAR-binding protein. The protein motif being primarily responsible for PAR-binding is the RNA recognition motif 1 (RRM1), which is also crucial for RNA-binding, highlighting a competition between RNA and PAR as they share the same binding site. Strikingly, the in vivo recruitment of NONO to DNA damage sites completely depends on PAR, generated by activated PARP-1. Furthermore, we show that upon PAR-dependent recruitment, NONO stimulates nonhomologous end joining (NHEJ) and represses homologous recombination (HR) in vivo. Our results therefore place NONO after PARP activation in the context of DNA DSB repair pathway decision. Understanding the mechanism of action of proteins that act in the same pathway as PARP-1 is crucial to shed more light onto the effect of interference on PAR-mediated pathways with PARP inhibitors, which have already reached phase III clinical trials but are until date poorly understood.
INTRODUCTION
Each day, the cells genome is confronted with up to 50 endogenous DNA double-strand breaks (DSBs). These are extremely hazardous for a cell, as they do not leave an intact complementary strand to serve as a template for repair (1). If left unrepaired, DSBs can have consequences such as cell death or carcinogenesis. Hence, understanding the mechanisms that lead to successful repair of DSBs will further increase the knowledge of cancer progression and treatments. The DNA damage response (DDR) to DSBs is a multilayered process, initiated with sensing and signaling DNA damage, subsequent recruitment of repair proteins and execution of repair (2).
Poly(ADP-ribose) polymerase-1 (PARP-1) is an abundant and ubiquitous nuclear protein that uses NAD+ to synthesize a negatively charged polymer, called poly(ADP-ribose) (PAR), onto a variety of target proteins, such as histones, DSB repair factors and PARP-1 itself. The latter post-translational protein modification has an impact on cellular processes as diverse as transcription (3), cell death (4) and especially DNA repair (5). PARP-1 acts as a strong sensor for DNA damage and rapidly produces PAR at newly generated DNA DSBs, provoking therewith local chromatin relaxation due to its negative charge (3) and facilitating the recruitment of repair factors, such as MRE11 (2,6). The dynamic turnover of PAR within seconds to minutes is executed by poly(ADP-ribose) glycohydrolase (PARG), that possesses endo- and exoglycosidic activities, hence enabling a new round of DNA damage signaling (7).
For subsequent repair, two major DSB repair pathways have evolved, namely nonhomologous end joining (NHEJ) and homologous recombination (HR). Whereas HR is considered as error-free and restricted to the S/G2-phase (8) by its necessity for a homologous template, error-prone NHEJ functions throughout the cell cycle and represents the major pathway for DSB repair in multicellular eukaryotes. Although the NHEJ pathway is highly flexible in terms of substrate ends used for repair, participating repair proteins and possible outcomes, a number of key proteins are indispensable to accomplish classical NHEJ (cNHEJ): Initially, the heterodimeric Ku70/Ku80 complex binds to both ends of the broken DNA molecule (9). Interestingly, Ku has an affinity for PAR (10) and is also a direct target for PARylation (11). The Ku–DNA complex is further bound by the catalytic subunit of DNA–PK (DNA–PKcs) to assemble the end-bridging DNA–PK complex (12). If the two ends are not directly ligatable they have to be processed prior to the final ligation step. A variety of proteins (such as Artemis, PNK, APLF nucleases, TdT, polymerases λ and µ) have been implicated in the end-processing step, emphasizing the mechanistic flexibility of the NHEJ reaction (13–16). The final ligation step is carried out by X4-L4 complex, composed of XRCC4, DNA ligase IV and XLF (17).
Within the last years, growing attention has been drawn to proteins with dual roles in RNA biology and DNA DSB repair. Examples include the Ku protein, which is crucial for the NHEJ pathway but interestingly also for the control of mRNA expression (18,19), the TFHII complex that acts in nucleotide excision repair as well as in transcriptional initiation mediated by RNA polymerase II (20), and recently the RNA-binding protein RBMX and the RNA-splicing factor THRAP3 were implied in the DDR (21–23). About twenty years ago the group of Harris Busch purified and characterized a heterodimer consisting of a 52 and a 100 kDa subunit, most certainly corresponding to what is nowadays known as the 54 kDa nuclear RNA-binding protein (p54nrb/NONO) and the polypyrimidine tract-binding protein-associated splicing factor (PSF/SFPQ). NONO and SFPQ show 71% sequence identity and, together with paraspeckle component 1 (PSPC1), belong to a subfamily of RNA recognition motif (RRM) proteins defined by tandem RRM motifs, flanked by an additional region of sequence similarity predicted to promote formation of heteromeric complexes between each of the proteins (24). NONO and SFPQ have been implicated in nuclear retention of A- to I-edited RNA as paraspeckle components (25), pre-mRNA 3′-end formation (26), cAMP cycling (27) and transcriptional activation (28–30). Interestingly, apart from their functions in RNA biogenesis, NONO and SFPQ were reported to interact with DNA in vitro, which lead to an investigation of their function in the context of DNA repair. Both proteins are transiently recruited with the same kinetics to DNA damage induced by a laser track in human cells (31). Interestingly, a protein complex containing NONO and SFPQ stimulates NHEJ about 10-fold in vitro (32). Furthermore, it has been demonstrated that the attenuation of NONO protein expression, independent of its partner protein SFPQ, delays the resolution of γ-H2AX foci after ionizing irradiation and leads to an accumulation of chromosomal aberrations (33). However, the exact mechanism by which NONO is recruited to DNA damage sites and regulates DSB repair is unclear. Interestingly, a bioinformatics screen from our group for proteins that potentially bind PAR, which is generated within seconds at a new DSB, identified NONO/SFPQ among a variety of NHEJ factors (10,34), leading to the hypothesis that PARP and its associated polymer regulates NONO. In this manuscript, we dissect the role of NONO in DSB repair in the context of PARP activation. We suggest here that NONO is directly implicated in NHEJ, and that its recruitment to DNA damage sites is strictly dependent on activated PARP-1. These results highlight the emerging concept of RNA-binding proteins in DSB repair.
MATERIALS AND METHODS
Cell lines, cell culture, and DNA constructs
HeLa cells and mouse embryonic fibroblasts (MEFs) proficient for PARP-1 and PARP-2 [wild type (WT)], or deficient for either PARP-1 (PARP-1−/−) or PARP-2 (PARP-2−/−) were cultured in DMEM, while MCF-7 cells were cultured in MEM-alpha (air/CO2, 19:1, 37°C). Both media were supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
The NHEJ reporter construct ‘sGEJ’ was kindly provided by Dr. Ralph Scully (35) and stably integrated into the genomic DNA of MCF-7 cells by using G418 disulfate salt (400 µg/ml; Sigma) as a selection marker. The HR reporter construct ‘DR-GFP’ [kindly provided by Dr. Maria Jasin; (36)] was integrated into the genomic DNA of MCF-7 cells by hygromycin selection (400 µg/ml; Invitrogen).
The GFP-NONO construct is a generous gift from Dr. James Patton (Vanderbilt University, Nashville, TN). NONO was cloned for protein purification from the pEGFP vector into a pET-16 b (Novagen) vector using the primers shown in Supplementary Table S1.
Site-directed mutagenesis on the His-NONO and GFP-NONO constructs was carried out with the QuikChange™ Site-Directed Mutagenesis Kit (Stratagene) using the oligos shown in Supplementary Table S1.
Antibodies and siRNAs
For Western blotting analysis and chromatin-immunoprecipitation (ChIP) experiments, polyclonal antibodies for NONO and SFPQ were obtained from Bethyl laboratories. The monoclonal antibody against GAPDH (6C5) was obtained from Fitzgerald Industries. Polyclonal antibodies for RAD51 and PSPC1 were purchased from Santa Cruz. PARP-1 (C2–10) monoclonal antibody was produced in house as described (37).
Gene silencing was performed using siRNA directed against the following target sequences: 5′-GGAAGCCAGCUGCUCGGAAAGCUCU-3′ against NONO, 5′-GCCAGCAGCAAGAAAGGCAUUUGAA-3′ against SFPQ (Invitrogen). A scrambled siRNA (5′-GACGTCATATACCAAGCTAGTTT-3′) from Dharmacon was used as a negative control. Transfection of 5 nM siRNA per condition was performed for 48 hr using HiPerfect transfection reagent (Qiagen) according to the manufacturer’s protocol. For the siRNA directed against NONO, a second round of transfection (∼36 hr after the first transfection) was performed for another 24 hr.
Colony forming assays
Long-term cell viability of HeLa cells transfected with the indicated siRNAs was assessed by colony forming assays. Briefly, a total of 200 cells per condition were plated into 35-mm dishes. Cells were then exposed to ionizing radiation of 0, 0.5 or 2 Gray using a γ-irradiator (Gammacell-40; MDS Nordion). After 7 to 10 days, colonies were fixed with methanol, stained using a 4 g/L solution of methylene blue in methanol, extensively washed with PBS and counted.
Protein purification
Recombinant wild-type human NONO (NONO-WT) and the RRM1-deletion mutant (NONOΔRRM1) proteins were purified from an Escherichia coli BL-21 strain carrying pET16b-10XHis-NONO or pET16b-10XHis-NONOΔRRM1 expression constructs, grown in 4 L of LB media supplemented with 100 µg/ml ampicillin and 25 µg/ml chloramphenicol. Protein expression was induced for 16 hr at 16°C with 0.1 mM IPTG added to the culture at an OD600 = 0.4. Cells were then harvested by centrifugation and resuspended in 40 ml lysis buffer A (20 mM Tris-HCl pH 8.0, 10% glycerol, 2 mM β-mercapthoethanol, 500 mM NaCl, 5 mM imidazole, 1 mM PMSF, 1 µg/ml leupeptin, 0.019 TIU/ml aprotinin). Samples were lysed with a Dounce homogenizer (10 strokes with the tight pestle), sonicated using a sonicator (Bioruptor; Diagenode) (10 min at the ‘high’ setting, 30 s ON and 30 s OFF) and returned to the Dounce for a second round of lysis. Insoluble material was removed by centrifugation at 40 000 rpm for 1 hr at 4°C and the supernatant subsequently loaded on a 5 ml cobalt-based immobilized metal affinity chromatography resin Talon column (BD Biosciences, Palo Alto, CA). The column was washed and eluted with a linear gradient of imidazole ranging from 5 to 1000 mM prepared in buffer A. Fractions containing His-tagged NONO-WT or NONOΔRRM1 were identified by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), carefully selected, pooled and dialyzed for 1 hr against 20 mM Tris-HCl pH 8.0, 375 mM NaCl, 10% glycerol and 0.05% Tween-20 buffer.
FACS analysis of the cell cycle
Cells were collected by trypsinization, centrifuged and resuspended at 106 cells per 300 µl of PBS and fixed with 700 µl of ice-cold ethanol (100%) while vortexing. Once fixed, cells were washed with PBS and stained with propidium iodide (0.1% sodium citrate, 0.3% Nonidet P40, propidium iodide 50 mg/ml and RNAse A 20 mg/ml). Cell cycle analysis was performed on a Beckman Coulter Epics Elite model ESP by using the Expo2 analysis software.
Pulse-field gel electrophoresis
HeLa cells treated with the indicated siRNA were incubated for 2 hr at 37°C in the presence of 500 ng/ml Neocarzinostatin (NCS). After treatment, cells were released for the indicated time points and trypsinized. One percent agarose plugs containing 5 × 106 cells were prepared with a CHEF-disposable plug mold (Bio-Rad). Cells were lysed by incubation of the gel blocks for 72 hr at 45°C in 1 mg/mL proteinase K, 100 mM ethylenediaminetetraacetic acid (EDTA), 0.2% sodium deoxycholate, 1% N-laurylsarcosyl. Samples were then washed three times for 1 hr each in 20 mM Tris pH 8.0, 50 mM EDTA and embedded into an agarose gel (0.9% agarose in 0.5X filtered TBE). DNA separation was performed at 14°C for 24 hr with a two block pulse linear program (block 1: 0.1 s at 30 s, 5.8 V/cm, 14°C, angle 120°, TBE 0.5X, 12 hr; block 2: 0,1 s at 5 s, 3.6 V/cm, 14°C, angle 110°, TBE 0.5X, 12 hr) in a CHEF-DR III Pulsed Field Electrophoresis System (Bio-Rad). The gel was then dried for 30 min at 55°C and for additional 30 min at room temperature, stained overnight with SYBR green (Molecular Probes) and visualized using a UV lamp. A yeast chromosome PFG marker (NEB 345) served as a ladder for molecular weight.
Nuclear extract preparation
Up to 107 HeLa cells per condition were washed three times with PBS, resuspended and incubated for 15 min on ice in 250 µl hypotonic buffer (10 mM Tris pH 7.4, 10 mM MgCl2, 10 mM KCl and 1 mM DTT). The samples were then passed 5 times through a 1 ml syringe with a 27G needle and centrifuged for 15 min at 3300 × g at 4°C. Pellets were resuspended in 200 µl high salt buffer (hypotonic buffer A with 350 mM NaCl and protease inhibitors) and incubated for 1 hr on ice. After centrifugation for 30 min at 13 000rpm at 4°C, the supernatants were transferred to a clean tube and adjusted to 10% glycerol (v/v) and 10 µM of β-mercapthoethanol.
Cell fractionation and western blot analysis
Cell fractionation was carried out as described in (38) with slight modifications. Briefly, 3 × 106 HeLa cells per condition were collected and resuspended in 200 µL of buffer A (10 mM HEPES pH 8.0, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 1 mM PMSF, 0.1% Triton-X-100, 10 mM NaF, 1 mM Na2VO3, protease inhibitors) and kept for 5 min on ice. The soluble cytoplasmic fraction (S1) was separated from the nuclei (P2) by centrifugation for 4 min at 1300 × g at 4°C. The nuclear fraction P2 was washed twice with 300 µL buffer A then resuspended in 200 µL buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, 1 mM PMSF, 10 mM NaF, 1 mM Na2VO3, protease inhibitors) and kept for 30 min on ice. The insoluble chromatin fraction (P3) was separated from nuclear soluble proteins (S3) by centrifugation for 4 min at 1700 × g at 4 °C. S1 was cleared from insoluble proteins by centrifugation at 14 000rpm for 15 min at 4°C and the supernatant (S2) was kept for analysis. Cell fractions were subsequently analysed by western blotting as described in (39).
ChIP and quantitative polymerase chain reaction
A unique DSB in MCF-7 cells was introduced by electroporating the I-SceI expression vector (pCBASce) into MCF-7 DR-GFP (carrying a chromosomally integrated homology-directed repair site) cells using the Gene Pulser Xcell apparatus (Bio Rad). A total of 2 × 106 cells per electroporation, resuspended in 650 μl PBS, were mixed with 50 μg of circular plasmid and pulsed at 0.25 kV and 1000 μF in 4-mm cuvettes. Cells were then plated onto 10-cm dishes containing fresh medium and kept at 37°C for 12 hr. To crosslink proteins to DNA, cells were treated for 10 min with a 1% formaldehyde solution in PBS. Subsequently, glycine to a final concentration of 0.125 M was added to quench the reaction. Cells were collected in ice cold PBS using a cell scraper, washed twice in cold PBS containing 1 mM PMSF, washed for 10 min in solution I (10 mM HEPES, pH 7.5, 10 mM EDTA, 0.5 mM EGTA, 0.75% Triton X-100) and 10 min in solution II (10 mM HEPES, pH 7.5, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA). Cells were resuspended in lysis buffer (25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate) and kept for 45 min on ice. To shear chromatin to an average size of 0.5 kb, cells were sonicated with a Bioruptor sonicator (Diagenode) for 10 min (high, 30 s ON, 30 s OFF). Samples were then centrifuged at maximum speed in a benchtop centrifuge until clear and the lysate precleared overnight with Sepharose CL-6B beads. Immunoprecipitation was performed for 2 hr in lysis buffer with polyclonal antibodies against NONO. Rabbit anti-human IgG (H+L) antibody (Jackson Immunoresearch Laboratories) was used as a negative control. Protein–antibody complexes were subsequently incubated with protein A/G beads for 1 hr. Complexes were washed twice with RIPA buffer (150 mM NaCl, 50 mM Tris-HCl pH 8.0, 0.1% SDS, 0.5% deoxycholate, 1% NP-40, 1 mM EDTA), once in high salt buffer (50 mM Tris–Cl, pH 8.0, 500 mM NaCl, 0.1% SDS, 0.5% deoxycholate, 1% NP-40, 1 mM EDTA), once in LiCl buffer (50 mM Tris–HCl, pH 8.0, 250 mM LiCl, 1% NP-40, 0.5% deoxycholate, 1 mM EDTA) and twice in TE buffer (10 mM Tris–HCl, pH 8.0, 1 mM EDTA, pH 8.0). Beads were resuspended in TE containing 50 μg/ml RNase A and incubated for 30 min at 37°C. Beads were washed with deionized water and incubated for 15 min in elution buffer (1% SDS, 0.1 M NaHCO3). Crosslinks were reversed by adding 200 mM NaCl followed by an incubation for 6 hr at 65°C. Samples were deproteinized overnight with 300 µg/ml proteinase K and DNA was extracted with phenol–chloroform followed by ethanol precipitation.
Immunoprecipitated DNA was quantified by quantitative polymerase chain reaction (q-PCR) using the Light Cycler Fast Start DNA Master SYBR Green I (Roche Applied Sciences), which is composed of Fast Start Taq DNA polymerase and SYBR Green Dye. Oligonucleotides [Supplementary Table S1; (40)] flanking the break site were designed and optimized for linearity range and efficiency using a light cycler (Roche). Immunoprecipitated DNA samples were amplified in triplicate and values calculated as fold-enrichment compared with the IgG ChIP control and versus GAPDH as a control locus.
PAR-binding assay
PAR-binding properties of purified proteins were analysed as described in (34). Briefly, 500 ng of the indicated protein were either spotted onto a 0.2 -µm pore size nitrocellulose membrane using a slot blot manifold (Bio Rad) or transferred onto a nitrocellulose membrane following separation on an 8% SDS-PAGE. For both conditions, the membranes were washed three times in TBS-T (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.05% Tween) and incubated for 1 hr at room temperature in TBS-T to allow proper refolding of the protein. Subsequently, the membrane was incubated with 250 nM [32P]-PAR [synthesized as described in (41)] in TBS-T with or without 100-fold of unlabeled competitor RNA (yeast RNA mix, Ambion). The membrane was then washed extensively in TBS-T, air-dried and subjected to autoradiography.
Surface plasmon resonance spectroscopy
Interaction of 10X-His-tagged NONO with PAR was investigated using surface plasmon resonance (SPR) spectroscopy. The binding experiments were carried out on a ProteOn XPR36 (Bio-Rad) biosensor at 25°C using the HTE sensor chip (Bio-Rad). The flow cells of the sensor chip were loaded with a nickel solution to saturate the Tris–NTA surface with Ni2+-ions. Purified His-tagged wild-type NONO diluted in 10 mM MOPS [pH 8.0] was injected in one of six channels of the chip at a flow rate of 30 µl/min, until approximately a 5000 resonance unit (RU) level was reached. After a wash with running buffer (PBS [pH 7.4] with 0.005% (v/v) Tween-20), PAR binding to the immobilized substrates was monitored by injecting a range of concentrations of PAR (500, 250 and 125 nM) along with a blank at a flow rate of 50 µl/min. When the injection of PAR was completed, running buffer was allowed to flow over the immobilized substrates for PAR to dissociate with an association and dissociation phase of 300 and 600 s, respectively. Following dissociation of PAR, the chip surface was regenerated with an injection of 1 M NaCl at a flow rate of 100 µl/ml followed by 100 mM HCl and 300 mM EDTA at a flow rate of 30 µl/min. Interspot channel reference was used for non-specific binding corrections and the blank channel used with each analyte injection served as a double reference to correct for possible baseline drift. Data were analysed using ProteOn Manager Software version 3.1. The Langmuir 1:1 binding model was used to determine the KD values.
Live-cell microscopy and laser micro-irradiation
Recruitment experiments were carried out as described in (6). Briefly, cells were grown on glass-bottom dishes (MatTek Corp.) and transfected using Effectene reagent (Invitrogen) with the indicated constructs. Twelve hours post-transfection with GFP-NONO, GFP-NONOΔRRM1 and mCherry-PARG, cells were placed in fresh medium, treated with 10 µM ABT-888 (Enzo Life Sciences; 5 mM stock solution prepared in H2O) for 2 hr and sensitized with 1 μg/ml Hoechst 33 342 for 30 min prior to irradiation and live cell analysis of recruitment to DNA damage sites. A 37°C preheated stage with 5% CO2 perfusion was used for the time-lapse on a Zeiss LSM-510 META NLO laser-scanning confocal microscope. Localized DNA damage was generated along a defined region across the nucleus of a single living cell by using a bi-photonic excitation of the Hoechst 33 342 dye, generated with a near-infrared 750-nm titanium:sapphire laser line (Chameleon Ultra, Coherent Inc.). The laser output was set to 3%, and we used 10 iterations to generate localized DSB clearly traceable with a 40× objective. Protein accumulation within the laser path was compared with an undamaged region within the same microirradiated cell. We generally selected cells with low expression levels and normalized the fluorescence intensity in the microirradiated area to the initial fluorescence in the whole nucleus to compensate for photobleaching during acquisition. The average accumulation ± S.E. of fluorescently tagged proteins from at least 10 cells from three independent experiments was plotted.
Immunofluorescence
Laser-irradiated HeLa cells from earlier process were analysed by immunofluorescence (IF) for protein-co-localization with PAR as recently published by our group (42). Briefly, cells were washed three times with ice-cold PBS, fixed for 15 min at room temperature in 4% formaldehyde diluted in PBS, washed five times with PBS prior to permeabilization with 0.5% Triton X-100 in PBS for 5 min. After three washes with PBS, cells were incubated with the first antibody diluted in PBS containing 2% FBS for 90 min at room temperature. Following one wash with 0.1% Triton-X in PBS and four washes with PBS, cells were incubated with a secondary antibody diluted in PBS containing 2% FBS for 45 min. Subsequently, cells were washed once with 0.1% Triton X-100 in PBS, four times with PBS and then mounted in Fluoromount-G mounting media (Southern Biotech, Birmingham, AL). Images were acquired using a Leica 6000 microscope. Volocity software v5.5 (Perkin-Elmer Improvision) was used for image acquisition.
NHEJ/HR in vivo reporter assays
To analyse I-SceI induced GFP+-expression in NHEJ or HR reporter MCF7 cells, cell lines were plated onto cover-slips, treated with the indicated siRNAs for 36 hr and subsequently infected with an adenovirus coding for I-SceI. Cells were fixed 24 hr post-infection with 4% paraformaldehyde for 30 min. To enhance the GFP signal-to-noise ratio and therewith enhance the difference in signal intensity between GFP+ and GFP− cells, immunofluorescence was conducted as follows. Cells were permeabilized for 5 min with 0.5% Triton-X/PBS, washed twice with 0.1% Triton-X/PBS and incubated with 1% goat serum/PBS for 1 hr to block unspecific antibody binding. Cells were incubated for 1 hr with a polyclonal GFP antibody (Abcam ab290). The percentage of GFP+ cells per condition was calculated by counting the GFP+ cells over the total number of cells (2500 cells were counted based on DAPI nuclear staining). The percentage was expressed as fold-change normalized to the control siRNA condition.
RESULTS
NONO knockdown leads to a decrease in survival of IR-treated cells and deficient NHEJ repair
It has been previously shown that miRNA-mediated knockdown of NONO in HTC 116 cells left cell survival unaffected but sensitized the latter cells to ionizing irradiation (33). Here, we verified the necessity of NONO for cell proliferation by measuring the impact of attenuated NONO on the long-term survival of HeLa cells with and without ionizing irradiation. We used siRNA-mediated knockdown to attenuate the NONO protein expression level in HeLa cells. Immunoblotting confirmed that the expression level of NONO was reduced by more than 90%, whereas the attenuation of NONO did not affect the expression level of its partner protein SFPQ and vice versa (Figure 1A). A knockdown of NONO had no effect on long-term survival (Figure 1B). However, attenuated NONO sensitizes HeLa cells to ionizing irradiation at low (0.5 Gray) and intermediate doses (2.0 Gray), strongly suggesting a defect in DNA DSB repair (Figure 1C).
Figure 1.
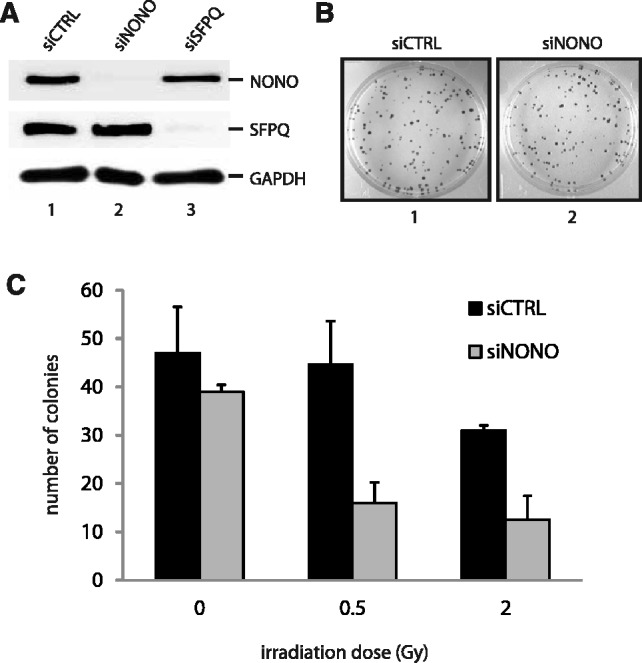
NONO increases cell survival after ionizing irradiation. (A) Western blot analysis demonstrating the efficiency of siRNAs directed against NONO (lane 2) or SFPQ (lane 3) in HeLa cells. (B) The clonogenic survival of HeLa cells treated with a scrambled control siRNA (image 1) and a siRNA directed against NONO (image 2) was analysed using a colony forming assay. (C) Quantitation of cell survival. HeLa cell colonies were counted 10 days after γ-irradiation with 0, 0.5 and 2.0 Gy.
These results suggest that NONO is crucial for survival after ionizing radiation. We therefore analysed the ability of NONO attenuated cells to repair DSBs. Hence, we optimized an assay to assess the sensitivity of these cells to the radiomimetic antibiotic NCS as a means to measure DSB repair kinetics in HeLa cells. NCS consists of an enediyne chromophore, which is tightly bound to a 113 amino acid single chain protein, the active compound responsible for tandem DNA cleavage and highly potent in the induction of DNA single and especially DSBs (43,44). Pulse-field gel electrophoresis (PFGE) was accomplished with HeLa cells 48 hr following transfection with scramble or NONO siRNA and treated for 2 hr with 500 ng/ml NCS to introduce DSBs. Cells were then released for 60 or 120 min and DSB repair kinetics indirectly surveyed by analysing the accumulation of DSBs. We observed that NONO protein knockdown by siRNA impairs the recovery from DNA damage as persistent accumulation of DNA DSBs following a 2-hr NCS treatment is detected by PFGE (Figure 2A). The slower recovery kinetics observed in the context of NONO depletion provides strong indication for the involvement of NONO in DSB repair. However, this observation could also be explained by an effect on cell cycle checkpoints that occurred in NONO knockdown cells. To rule out the possibility that NONO plays in indirect role in repair by affecting cell cycle progression, we analysed the cell cycle phase distribution of siCTRL and siNONO HeLa cells (Supplementary Figure S1). Neither the knockdown of NONO, nor SFPQ, nor the combined knockdown of both affects cell cycle progression. Similarly, cell cycle phase distribution of MCF-7 cells was unaffected by the knockdown of NONO (data not shown).
Figure 2.

Attenuation of NONO decelerates NHEJ. (A) HeLa cells knocked-down with a scrambled siRNA (lanes 3 and 4) or a siRNA directed against NONO (lanes 5 and 6) were treated for 2 hr with NCS (500 ng/ml) and allowed to recover for either 60 or 120 min. Cells were then collected, embedded and lysed in agarose blocks and used for pulse-field gel electrophoresis. (B) A linearized, 32P-end-labeled pBluescript was incubated for the indicated times with a nuclear extract derived from HeLa cells treated with a scrambled siRNA (lanes 2–9) or an siRNA directed against NONO (lanes 10–17). (C) Quantitation of the end joining events using a phosphorimager: The percent end joining represents the total signal intensity per lane normalized to 100% from which is substracted the % intensity of the remaining template (n = 4).
The observed radiosensitivity and accumulation of DSBs in NONO attenuated cells could be a consequence of diminished NHEJ repair activity. Therefore, we set up a cell-free NHEJ assay that measures the ligation of a 32P-labeled linearized plasmid, after incubation with nuclear extracts derived from siRNA control HeLa cells or knocked down for NONO. The knockdown of NONO in HeLa cells delays NHEJ kinetics in vitro, as the end joining reaction with the nuclear extract in which NONO had been knocked-down results in overall less end joining products compared with the control (Figure 2B). In concordance with this observation, less substrate plasmid had been used for the end joining reaction in the absence of NONO. Quantitation of the end joining products at 2 hr revealed a 5-fold decrease in end joining products in the NONO knockdown assay, compared with the assay with control cells (Figure 2C).
NONO is strongly associated with the chromatin and localizes near a unique DSB in vivo
The results mentioned earlier confirmed a function for NONO in DNA DSB repair, and suggested that NONO might play a direct role in DNA repair rather than having an indirect effect through RNA biogenesis. One prediction of such a direct role would be to observe physical association of NONO with DNA damage sites. Following this idea, we used ChIP combined with q-PCR using oligonucleotides flanking a unique I-SceI restriction site in MCF-7 cells to monitor the distribution of NONO relative to a DSB.
To ensure that the RNA-binding protein NONO is localizing to DNA/chromatin in vivo (a prerequisite for ChIP), we fractionated unfixed MCF-7 cells and analysed the chromatin enriched, nuclear soluble and cytoplasmic fractions by western blotting with the indicated antibodies (Figure 3A). Surprisingly, we found that NONO, as its partner proteins SFPQ and PSPC1, is strongly associated with the chromatin and nearly absent in the nuclear soluble and cytoplasmic fractions. PARP-1, RAD51 and GAPDH served as hallmark protein-controls for the nuclear soluble and cytoplasmic fractions, respectively. The results indicate that NONO is associated with the chromatin, even in the absence of exogenous DNA damage and independently of the PARP-1 activation state.
Figure 3.

NONO is a chromatin-associated protein and localizes to a unique DSB in vivo. (A) Unfixed HeLa cells were treated for 1 hr with 10 µM ABT-888, washed with PBS and fractionated into chromatin-enriched, nuclear soluble and cytoplasmic fractions. Fractions were used for an analysis by western blotting. (B) Chromatin immunoprecipitation of NONO from the fixed chromatin of MCF-7 cells, which priorly had been transfected with an I-SceI coding plasmid to generate a unique DSB. An IgG antibody served as a control for the ChIP-experiment. (C) Distribution of primer pairs relative to the DSB created by I-SceI. These primers were used in q-PCR analysis of ChIP shown in (D). Primers for GAPDH served as a control for the PCR. (D) Quantification of NONO relative to the DSB by PCR (n = 3).
Using MCF-7 cells carrying a single I-SceI restriction site, we then combined ChIP with q-PCR to determine the position of NONO relative to a DSB in vivo. We conducted the ChIP experiment 12 hr after transfection with an I-SceI encoding vector, allowing sufficient time for I-SceI expression and generation of the unique DSB. We successfully pulled-down endogenous NONO fixed to the chromatin, as shown in Figure 3B. After purification of the chromatin that has been pulled-down with NONO, we used three sets of primers located at increasing distances from the DSB to evaluate the distribution of NONO (Figure 3C). We were able to detect NONO as close as 464–520 bp from the DSB with a ∼1.5-fold enrichment compared with the IgG control and after normalization with GAPDH (Figure 3D). This localization resembles that of the NHEJ factor and RNA-processing protein Ku80, as we previously reported (39).
NONO is a new PAR-binding protein that binds PAR through its RRM1 motif
The synthesis of PAR that results from the activation of DNA-dependent PARPs is one of the earliest step of DNA damage recognition and signaling in mammalian cells. PARP-1 has notably been shown to localize to DNA damage sites within milliseconds following laser-induced micro irradiation of sub-nuclear regions (6,45).
Our laboratory recently performed a proteome-wide screen for proteins to isolate and identify pADPr-containing multiprotein complexes. Interestingly, the RNA-binding protein NONO was consistently identified together with a variety of DNA DSB repair factors (34,42). A number of DDR factors have been shown to be loaded on DNA damage sites in a PAR-dependent fashion (6,21,46,47). To assess PAR-binding properties of NONO in vitro, His-tagged NONO was expressed in E. coli and purified by affinity purification (Figure 4A). Using a PAR-binding assay developed by our group (10), we determined whether NONO binds PAR. As shown in Figure 4B (lane 1), NONO displays a strong affinity for purified 32P-labeled PAR in vitro. The unlabeled PAR displaced binding of its cognate 32P-labeled polymer (Figure 4B, lanes 2–4). As NONO is a well-established RNA-binding protein and considering that PAR shares some structural features with nucleic acids, we further examined its affinity for PAR in the presence of increasing amounts of unlabeled competitor RNA (Figure 4C, lanes 1–4). Interestingly, binding of 32P-PAR was slightly reduced when cold RNA was added to the binding reactions, suggesting a competition between PAR and RNA. The RNA-binding protein still exhibits PAR-binding in the presence of 100-fold competitor RNA, underpinning its specificity (Figure 4B).
Figure 4.

NONO binds PAR in vitro. (A) SDS-PAGE of 100 ng purified His-NONO protein stained with Coomassie blue (lane 2). (B) In vitro PAR-binding assay. 1 µg of purified His-NONO was loaded on an SDS-PAGE, blotted onto a nitrocellulose membrane and incubated in 250 nM 32P-labeled PAR in TBS-T without (lane 1), with 1-fold (lane 2), 10-fold (lane 3) or 100-fold unlabeled competitor PAR (lane 4). (C) A PAR-binding assay was conducted as in (B) without (lane 1), with 1-fold (lane 2), 10-fold (lane 3) or 100-fold unlabeled competitor RNA (lane 4). (D) Kinetics of PAR binding to purified His-tagged NONO conducted by SPR spectroscopy. To analyse binding kinetics, PAR was injected at three different concentrations (125, 250 and 500 nM). PAR injection was done for 300 s and dissociation data were collected for 600 s. Data were fitted with Langmuir 1:1 interaction plot to calculate rate constants. The sensorgram is representative of three independent experiments.
To further characterize the affinity of NONO for PAR with a label-free approach, we used SPR spectroscopy, such as described in (48). Therefore, purified His-tagged NONO was bound to a HTE sensor chip until a response unit of 5000 RU was reached. Subsequently, purified PAR, produced by PARP-1 in vitro, was injected at three different concentrations (500, 250 and 125 nM) to determine the binding affinity to the immobilized NONO protein. Association and dissociation was allowed to proceed for 300 and 600 seconds, respectively. As shown in Figure 4C, the dissociation rate constant (KD) of NONO was determined at 2.32 × 10−8 M, hence demonstrates a strong affinity for PAR.
As the general model suggests that upon activation by DNA-binding, PARP-1 generates large amounts of long and branched PAR, we tested whether NONO preferentially binds long and complex PAR over shorter PAR molecules. Hence, we fractionated and purified PAR produced in vitro by PARP-1 for our binding analysis. SPR was conducted with two distinct populations of PAR namely complex PAR (60 mer and more average length) and short PAR (less than 30 mers average length). Strikingly, NONO strongly and specifically binds complex PAR, with a KD similar to that observed in Figure 4C but has no affinity for shorter PAR (Supplementary Figure S2A and B).
We next sought to locate the PAR-binding-sites within NONO protein. The NONO Drosophila behavior human splicing (DBHS) protein-core consists of clearly defined structural domains (Figure 5A): two tandem RRM domains and a 100-aa segment of predicted coiled-coil structure, putatively responsible for protein-protein interaction with itself and the other two members of the DBHS family (namely PSPC1 and SFPQ). As it had been shown for other RNA-binding proteins that they bind PAR through their RRM1 (49), we used protein fragments containing either the RRM1, RRM2, both RRMs or none of the RRMs for a PAR-binding assay in vitro (Figure 5B). Interestingly, we observed that NONO binds PAR with its fragment containing the N-terminal RNA-recognition motif 1 (RRM1) (Figure 5C). These results indicate that the RRM1 has a strong affinity for PAR in vitro, and could mediate the interaction between NONO and PAR. We therefore produced and purified a NONO mutant protein, which lacks the RRM1 region (NONOΔRRM1). As the wild-type and mutant NONO proteins were free of contaminants (Figure 5D), we performed an alternative polymer-blot assay without using a detergent-based separation such as SDS-PAGE. By slot-blotting the proteins directly onto a nitrocellulose membrane, we wanted to avoid methods that could disrupt interactions requiring native conformations. Measuring the binding-signal intensity with a phosphor-imager revealed that while the full-length protein shows a strong affinity for PAR (as described earlier), the affinity of the NONOΔRRM1 protein for PAR is reduced by 2.5-fold, indicating that we have successfully deleted a principal PAR-binding-motif (Figure 5E).
Figure 5.

NONO binds PAR through its N-terminal/RNA recognition motif 1 (RRM1). (A) Protein truncations of NONO flanking the protein domains of interest, namely RRM1 and RRM2. (B) SYPRO protein stain of protein fragments loaded on a SDS-PAGE. (C) In vitro PAR-binding assay using 250 nM 32P-labeled PAR in TBS-T. (D) SDS-PAGE of 500 ng His-NONO (lane 2) and His-NONOΔRRM1 (lane 3) each. (E) 1 µg of NONO-WT and NONOΔRRM1 purified proteins were slot blotted onto a nitrocellulose membrane and an in vitro 32P-labeled PAR-binding assay was conducted in TBS-T. Mean values of the radioactivity signal as quantified by a phosphorimager from three independent experiments are presented.
NONO is PAR-dependently recruited to DNA damage sites
An emerging concept in the DDR is that several proteins, such as MRE11 (6), are recruited to DNA damage sites in a PAR-dependent manner to elicit cell cycle arrest or DNA repair. In view of the strong affinity of NONO for PAR in vitro, we analysed whether NONO co-localizes with PARP-1 and PAR at DNA damage sites in cells (Figure 6A). We therefore transfected HeLa cells with GFP-NONO and 24 hr later induced DNA damage in live-cell conditions by laser micro-irradiation. Immediately after irradiation, cells were fixed and subjected to immunofluorescence staining. Evidently, GFP-NONO, PARP-1 and PAR are co-localizing at laser-IR-induced DNA damage sites immediately after introducing DNA lesions.
Figure 6.

NONO is recruited to DNA damage sites in a PAR-dependent manner. (A) Representative images of laser-irradiated HeLa cells expressing GFP-NONO and subjected to IF for detection of PARP-1 and PAR. (B) Representative images of the laser-irradiated cells. HeLa cells were transfected either with the GFP-tagged NONO construct or with a mutant lacking the RRM1. Then cells were either left untreated, treated with 10 µM ABT-888 1 hr before irradiation or cotransfected with mCherry-PARG prior to laser microirradiation. (C) Statistical analysis of the recruitment kinetics. At least 15 cells per condition in three independent experiments were analysed for their fluorescence intensity above the background.
To further analyse whether the recruitment of NONO to DNA damage sites is dependent on PAR, we established the recruitment kinetics of GFP-NONO to DNA damage induced by micro-irradiation in HeLa cells in the presence or absence of the specific PARP inhibitor ABT-888. In these live-cell analysis conditions, NONO is transiently recruited with rapid kinetics to DNA damage sites and reaches a maximum within 120 s following local generation of DNA damage sites (Figure 6B and C). Strikingly, we found that the recruitment of NONO to DNA damage sites completely depends on catalytically active PARP, as in none of the cells the protein is recruited in the presence of the specific PARP inhibitor ABT-888. As another mean to assess the PAR-dependency of recruitment of NONO to DNA damage sites, we co-expressed GFP-NONO with mCherry-PARG, the main PAR-degrading enzyme, to prevent PAR accumulation in laser tracks. We have previously shown that overexpression of PARG prevents PAR accumulation after induction of DNA strand breaks (50). We indeed found that the recruitment of GFP-NONO to laser tracks is completely abolished by PARG overexpression.
This observation is consistent with the finding that PARP inhibition abrogates the recruitment of GFP-NONO and confirms a strict requirement for PAR-binding for its relocation to DNA damage sites. We then sought to define the domain mediating NONO interaction with PAR. Hence, we tested if interaction with PAR occurs through interaction with the RRM1 domain of NONO. As shown in Figure 6A and B, a deletion mutant lacking the RRM1 domain (GFP-NONOΔRRM1) is not recruited to DNA damage sites. This result strongly implicates the RRM1 domain in regulating the interaction with PAR.
Although our results underscore the importance of PAR for NONO dynamics in the DDR, they leave open the question which PARP family member generates the PAR that mediates the recruitment of NONO to DNA damage sites. It is well accepted, that PARP-1 is responsible for ∼95% of all PARylation events after DNA damage, whereas PARP-2 carries out almost all of the remaining 5%. Therefore, we overexpressed GFP-NONO in wild-type and PARP-1−/− MEFs. Recruitment of GFP-NONO was detected in the PARP-1-proficient MEFs with similar kinetics to those in HeLa cells, whereas GFP-NONO was not recruited to the laser track in PARP-1−/− cells, highlighting the necessity of PARP-1 to generate PAR at the DNA damage sites (Figure 7). The specificity for PARP-1 is further highlighted by the observation that GFP-NONO is recruited with fast and transient kinetics in PARP-2−/− MEFs similar to that in the WT-MEFs and HeLa cells (Figure 7). Hence, PARP-1 is required to recruit NONO to DNA damage sites, whereas PARP-2 is rather dispensable. Collectively, these results show that the recruitment of NONO is PARP-1 and PAR-dependent, and mediated by the RRM1 region of NONO.
Figure 7.
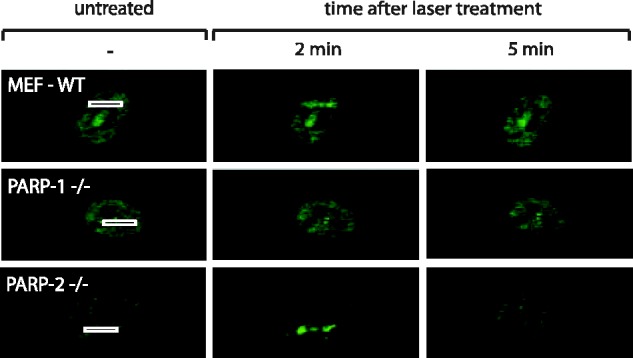
Representative images of laser-irradiated MEFs that were either proficient for PARP-1 and PARP-2 (MEF-WT) or deficient for either PARP-1 (PARP-1−/−) or PARP-2 (PARP-2−/−). Cells had been transfected with a GFP-NONO construct 24 hr before laser micro-irradiation. At least 20 cells per condition were tested in two independent experiments. Recruitment has been observed in none of the PARP-1−/− MEFs.
NONO promotes NHEJ and represses HR in vivo in the same pathway as PARP-1
As a consequence of the results described above, we hypothesize that NONO plays important regulatory role in the DDR by stimulating DSB repair. Indeed, we showed that NONO promotes cell survival and DSB repair through NHEJ, localizes near a unique DSB site and accumulates to sites of DNA damage in a pADPr-dependent fashion. However, a direct implication of NONO in NHEJ has not been shown in vivo and the question as whether NONO also influences the other DSB repair pathway, namely HR, has not been answered yet.
To address these two key questions, we generated two stable reporter cell lines enabling us to monitor both, NHEJ and HR repair (Figure 8A and B). Each of these cells lines has an integrated cassette comprising an I-SceI cleavage site that, upon repair by either NHEJ or HR, restores GFP expression, as previously described (35,36). Cells with normal or knocked down expression of NONO were assessed for each repair mechanism as indicated by the percentage of cells that express GFP. In the NHEJ reporter system assay, we found that the knockdown of NONO decreases NHEJ by more than 50% (Figure 8C). In this same assay, PARP inhibition, with the potent and specific PARP inhibitor ABT-888 also significantly reduced NHEJ repair. Knowing that NONO is PAR-dependently recruited to DNA DSBs, we combined the siRNA directed against NONO with PARP inhibitor to confirm our findings above. As expected, the siRNA-mediated knockdown of NONO combined with the inhibition of PARP does not have an additive effect in inhibiting NHEJ, indicating that PARP and NONO function in the same pathway and hence supporting the idea of PAR-dependent recruitment. Interestingly, an attenuation of NONO does not only decrease NHEJ but also facilitates repair by HR ∼40% (Figure 8D). Again here, when combining siRNA directed against NONO with the PARP-1 inhibitor ABT-888, no additive effect was observed, supporting the same conclusion regarding PAR-dependent recruitment.
Figure 8.

Attenuation of NONO decreases NHEJ and increases HR. (A) Schematic representation of the I-SceI-based NHEJ in vivo reporter system. (B) Schematic representation of the I-SceI-based HR in vivo reporter system. (C) NHEJ repair rates in percent with siCTRL or siNONO and with or without 10 μM of the PARP-inhibitor ABT-888. The siCTRL condition was normalized to 100% (n = 3). (D) Diagram of the HR repair rates after treatment with siCTRL or siNONO and with or without 10 μM ABT-888. The siCTRL condition was normalized to 100% (n = 3).
DISCUSSION
Although the RNA binding properties of NONO related to RNA biogenesis and the architecture of paraspeckles have been subject of an abundant literature, [reviewed in (51)], little is known on the functions of NONO in the context of DNA DSB repair. We have conducted a detailed molecular and cellular analysis of NONO in the context of the DDR and our data establish NONO as a PARP-1-dependent regulator of DSB repair by facilitating NHEJ and promoting cell survival after irradiation.
In the past few years, the list of proteins that possess dual roles in gene regulation and genomic stability through RNA biology and DNA repair, respectively, has largely expanded. Examples include the catalytic subunit of DNA-PK, a core complex of NHEJ that is necessary to arrest RNA-polymerase II transcription after the induction of DSBs (52) and the Ku protein that has dual roles in transcriptional reinitiation and NHEJ (19). In addition, the heterogeneous nuclear ribonucleoprotein (hnRNP) RBMX acts in alternative splicing and accumulates at DNA damage sites in a PARP-dependent manner (21). Also, the heterogeneous nuclear ribonucleoprotein hnRNPU influences end resection (53). Another study highlights the role of the splicing-associated protein THRAP3 in the DNA damage signaling network (22). Even PARP-1 itself functions in promoter/enhancer regulation (54), single-strand break repair and the alternative NHEJ pathway (55,56).
Because of its possible role in RNA biogenesis, it came as a surprise to find that NONO is mostly associated to the chromatin. Moreover, we show here for the first time that NONO is localized with close proximity to a unique DSB in vivo. In an earlier study (39), we have detected the NHEJ-related protein Ku80 within the same distance to the break-site as NONO (∼ 400 bps), suggesting a direct implication for NONO in DNA DSB repair. In line with these findings, the Shiloh group has detected NONO in a protein complex composed of Ku70, Ku80 and Ligase IV (31). Here, we are giving further evidence for a direct implication of NONO in DSB repair by showing that down-regulation of NONO protein expression by siRNA sensitizes HeLa cells to ionizing irradiation and decreases NHEJ in vitro and in vivo. Hence, the data presented complement the recent findings that attenuation of NONO delays the resolution of γ-H2AX foci and results in an increase of chromosomal aberrations following radiation exposure (33). The fact that cells with attenuated NONO are still viable and capable of NHEJ might be explained by a possible backup through its homologous protein partner PSPC1. The expression level of PSPC1 in the presence of NONO in HeLa cells is very low and increases upon siRNA-mediated knock-down of NONO (data not shown).
PARP-1 is an abundant nuclear chromatin-associated protein, well characterized for its high DNA damage sensing ability. Once encountering free DNA ends, PARP-1 is catalytically activated and generates large amounts of PAR serving as a scaffold for the recruitment of a variety of DNA repair proteins. We performed a large scale analysis of proteins bound to PAR following MNNG exposure. NONO was identified in SILAC experiments with an enrichment ratio (control versus DNA damage), which is one of the strongest in the pADPr immunoprecipitates (42). Also after neocarzinostatin treatment, we observed a complex between PAR and NONO as well as PARP-1 and NONO (Supplementary Figure S3). We have previously shown that key DNA repair proteins share a high affinity for PAR, the product of catalytically active PARP-1, with many RNA-binding proteins (41). To date, many proteins have been shown to be recruited in a PAR-dependent manner by cell imaging techniques: MRE11 (6), NBS1 (6), APLF (46), XRCC1 (57), CHD4 (58), NuRD (59) and ALC1 (47). However, in none of these studies PAR-dependent recruitment has been directly shown by deleting the PAR-binding module that is necessary for recruitment. We present here for the first time that the RNA-binding protein NONO has a strong and specific affinity for complex PAR in vitro, interestingly through its RRM1. We provide several lines of evidence that the recruitment of NONO to DNA lesions is strictly dependent on the presence of PAR. Indeed, we show that NONO relocation to DNA damage sites is suppressed by (I) PARP-1 inhibition with ABT-888; (II) PARG overexpression (the antagonist of PARP-1); (III) loss of PARP-1 expression in PARP-1−/− MEFs and (IV) deleting the PAR-binding motif located within the RRM1. Actually, the characterization of the RRM1 domain of NONO as a PAR-interacting module is consistent with previous studies that also established a similar role for the RRM1 domains of the splicing factor ASF/SF2 (49) and hnRNP A1 (41). It has been suggested that bound PAR competes out RNA-binding properties, therewith modulating the proteins splicing activity. The idea of a direct competition between PAR and RNA for the same site within a protein might also be applicable for RNA-binding proteins in the context of DNA repair. As it is of physiological importance for a cell to stop transcriptional activity (60) and initiate repair in response to excessive DNA damage, the PAR, which is largely generated at DNA damage sites, might serve as a molecular switch to direct proteins from RNA biogenesis toward DNA repair.
Finally, we show that NONO not only facilitates NHEJ but also represses the other major DSB repair pathway, HR, therewith channeling the DSB repair pathway decision between NHEJ and HR. Interestingly, we find that PARP activity has effects similar to NONO on both pathways and the combination of siRNA-mediated knock-down of NONO with an ABT-888 treatment does not show any additive effect. The latter suggests that NONO and PARP act in the same pathway, pointing towards the model of PAR-dependent recruitment of NONO. PARP-1 itself has also been shown to play a role in the back-up NHEJ pathway, but exclusively in the absence of classical NHEJ factors such as Ku80 (56). The role of PARP-1 in recruiting NONO in our system can be seen independent of its role in the backup-NHEJ pathway as we and others have observed that a knock-down of NONO leaves the expression level of proteins acting in the classical NHEJ pathway (Ku70/Ku80, DNA-PK, Ligase IV) unaffected suggesting that our system monitors exclusively classical NHEJ (33). Conclusively, our results place NONO in the very early steps of the DDR after PARP activation, promoting the error-prone NHEJ pathway over error-free HR.
Underpinning the fact that NONO promotes NHEJ over HR, which is an error-prone repair pathway that facilitates mutagenesis, we found by an Oncomine-based search that NONO is over-expressed in a variety of cancer types, such as colorectal and lung cancer. Within the latter two cancer types, NONO is among the top 1% over-expressed genes and therefore a promising candidate to investigate in the context of carcinogenesis. Moreover it has been published only very recently that NONO is implicated in the development and progression of malignant melanoma (61). Further investigation is needed to clarify NONOs possible role as a factor that promotes carcinogenesis.
Collectively, our study strengthens the suggested role for NONO in NHEJ and adds another layer of complexity by showing PAR-dependent recruitment through its principal RNA-binding motif. We have much to learn on NONO, a factor potentially promoting carcinogenesis in the context of PARP-activation as it sheds more light onto the mechanism of action of PARP inhibitors, which have already reached phase III clinical trials but are still poorly understood.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–3.
FUNDING
Funding for open access charge: Canadian Institutes of Health Research (to J.-Y.M. and G.G.P.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank members of the Masson and Poirier labs for critical reviewing of the manuscript, Mikaël Bédard for providing us with NONO protein fragments and Yan Coulombe for precious help in microscopy-based studies. They also acknowledge Valérie Schreiber (Université de Strasbourg, France) for the kind gift of Parp2−/− cells. Jana Krietsch is a Fonds de Recherche du Québec Nature et technologies scholar and Julien Vignard is a Canadian Institutes of Health Research postdoctoral fellow. Guy G. Poirier holds a Canada Research Chair in Proteomics and Jean-Yves Masson is a Fonds de Recherche en Santé du Québec senior investigator.
REFERENCES
- 1.Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA. 2003;100:12871–12876. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wacker DA, Frizzell KM, Zhang T, Kraus WL. Regulation of chromatin structure and chromatin-dependent transcription by poly(ADP-ribose) polymerase-1: possible targets for drug-based therapies. Subcell Biochem. 2007;41:45–69. doi: 10.1007/1-4020-5466-1_3. [DOI] [PubMed] [Google Scholar]
- 4.Bouchard VJ, Rouleau M, Poirier GG. PARP-1, a determinant of cell survival in response to DNA damage. Exp. Hematol. 2003;31:446–454. doi: 10.1016/s0301-472x(03)00083-3. [DOI] [PubMed] [Google Scholar]
- 5.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol. Cell. 2010;39:8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ, Poirier GG. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008;283:1197–1208. doi: 10.1074/jbc.M706734200. [DOI] [PubMed] [Google Scholar]
- 7.Slade D, Dunstan MS, Barkauskaite E, Weston R, Lafite P, Dixon N, Ahel M, Leys D, Ahel I. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature. 2011;477:616–620. doi: 10.1038/nature10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17:5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev. Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gagne JP, Isabelle M, Lo KS, Bourassa S, Hendzel MJ, Dawson VL, Dawson TM, Poirier GG. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 2008;36:6959–6976. doi: 10.1093/nar/gkn771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li B, Navarro S, Kasahara N, Comai L. Identification and biochemical characterization of a Werner’s syndrome protein complex with Ku70/80 and poly(ADP-ribose) polymerase-1. J. Biol. Chem. 2004;279:13659–13667. doi: 10.1074/jbc.M311606200. [DOI] [PubMed] [Google Scholar]
- 12.Meek K, Dang V, Lees-Miller SP. DNA-PK: the means to justify the ends? Adv. Immunol. 2008;99:33–58. doi: 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Pluth JM, Cooper PK, Cowan MJ, Chen DJ, Yannone SM. Artemis deficiency confers a DNA double-strand break repair defect and Artemis phosphorylation status is altered by DNA damage and cell cycle progression. DNA Repair (Amst) 2005;4:556–570. doi: 10.1016/j.dnarep.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 14.Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J. 2002;21:2827–2832. doi: 10.1093/emboj/21.11.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capp JP, Boudsocq F, Bertrand P, Laroche-Clary A, Pourquier P, Lopez BS, Cazaux C, Hoffmann JS, Canitrot Y. The DNA polymerase lambda is required for the repair of non-compatible DNA double strand breaks by NHEJ in mammalian cells. Nucleic Acids Res. 2006;34:2998–3007. doi: 10.1093/nar/gkl380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Capp JP, Boudsocq F, Besnard AG, Lopez BS, Cazaux C, Hoffmann JS, Canitrot Y. Involvement of DNA polymerase mu in the repair of a specific subset of DNA double-strand breaks in mammalian cells. Nucleic Acids Res. 2007;35:3551–3560. doi: 10.1093/nar/gkm243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124:301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 18.Giffin W, Torrance H, Rodda DJ, Prefontaine GG, Pope L, Hache RJ. Sequence-specific DNA binding by Ku autoantigen and its effects on transcription. Nature. 1996;380:265–268. doi: 10.1038/380265a0. [DOI] [PubMed] [Google Scholar]
- 19.Woodard RL, Lee KJ, Huang J, Dynan WS. Distinct roles for Ku protein in transcriptional reinitiation and DNA repair. J. Biol. Chem. 2001;276:15423–15433. doi: 10.1074/jbc.M010752200. [DOI] [PubMed] [Google Scholar]
- 20.Beck BD, Hah DS, Lee SH. XPB and XPD between transcription and DNA repair. Adv. Exp. Med. Biol. 2008;637:39–46. doi: 10.1007/978-0-387-09599-8_5. [DOI] [PubMed] [Google Scholar]
- 21.Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012;14:318–328. doi: 10.1038/ncb2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beli P, Lukashchuk N, Wagner SA, Weinert BT, Olsen JV, Baskcomb L, Mann M, Jackson SP, Choudhary C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell. 2012;46:212–225. doi: 10.1016/j.molcel.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell. 2009;35:228–239. doi: 10.1016/j.molcel.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng R, Dye BT, Perez I, Barnard DC, Thompson AB, Patton JG. PSF and p54nrb bind a conserved stem in U5 snRNA. RNA. 2002;8:1334–1347. doi: 10.1017/s1355838202022070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z, Carmichael GG. The fate of dsRNA in the nucleus: a p54(nrb)-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell. 2001;106:465–475. doi: 10.1016/s0092-8674(01)00466-4. [DOI] [PubMed] [Google Scholar]
- 26.Kaneko S, Rozenblatt-Rosen O, Meyerson M, Manley JL. The multifunctional protein p54nrb/PSF recruits the exonuclease XRN2 to facilitate pre-mRNA 3′ processing and transcription termination. Genes Dev. 2007;21:1779–1789. doi: 10.1101/gad.1565207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amelio AL, Miraglia LJ, Conkright JJ, Mercer BA, Batalov S, Cavett V, Orth AP, Busby J, Hogenesch JB, Conkright MD. A coactivator trap identifies NONO (p54nrb) as a component of the cAMP-signaling pathway. Proc. Natl. Acad. Sci. USA. 2007;104:20314–20319. doi: 10.1073/pnas.0707999105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathur M, Tucker PW, Samuels HH. PSF is a novel corepressor that mediates its effect through Sin3A and the DNA binding domain of nuclear hormone receptors. Mol. Cell Biol. 2001;21:2298–2311. doi: 10.1128/MCB.21.7.2298-2311.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong X, Sweet J, Challis JR, Brown T, Lye SJ. Transcriptional activity of androgen receptor is modulated by two RNA splicing factors, PSF and p54nrb. Mol. Cell Biol. 2007;27:4863–4875. doi: 10.1128/MCB.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishitani K, Yoshida T, Kitagawa H, Ohta H, Nozawa S, Kato S. p54nrb acts as a transcriptional coactivator for activation function 1 of the human androgen receptor. Biochem. Biophys. Res. Commun. 2003;306:660–665. doi: 10.1016/s0006-291x(03)01021-0. [DOI] [PubMed] [Google Scholar]
- 31.Salton M, Lerenthal Y, Wang SY, Chen DJ, Shiloh Y. Involvement of Matrin 3 and SFPQ/NONO in the DNA damage response. Cell Cycle. 2010;9:1568–1576. doi: 10.4161/cc.9.8.11298. [DOI] [PubMed] [Google Scholar]
- 32.Bladen CL, Udayakumar D, Takeda Y, Dynan WS. Identification of the polypyrimidine tract binding protein-associated splicing factor.p54(nrb) complex as a candidate DNA double-strand break rejoining factor. J. Biol. Chem. 2005;280:5205–5210. doi: 10.1074/jbc.M412758200. [DOI] [PubMed] [Google Scholar]
- 33.Li S, Kuhne WW, Kulharya A, Hudson FZ, Ha K, Cao Z, Dynan WS. Involvement of p54(nrb), a PSF partner protein, in DNA double-strand break repair and radioresistance. Nucleic Acids Res. 2009;37:6746–6753. doi: 10.1093/nar/gkp741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gagne JP, Haince JF, Pic E, Poirier GG. Affinity-based assays for the identification and quantitative evaluation of noncovalent poly(ADP-ribose)-binding proteins. Methods Mol. Biol. 2011;780:93–115. doi: 10.1007/978-1-61779-270-0_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 2009;16:814–818. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duriez PJ, Desnoyers S, Hoflack JC, Shah GM, Morelle B, Bourassa S, Poirier GG, Talbot B. Characterization of anti-peptide antibodies directed towards the automodification domain and apoptotic fragment of poly (ADP-ribose) polymerase. Biochim. Biophys. Acta. 1997;1334:65–72. doi: 10.1016/s0304-4165(96)00077-3. [DOI] [PubMed] [Google Scholar]
- 38.Zou L, Cortez D, Elledge SJ. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002;16:198–208. doi: 10.1101/gad.950302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodrigue A, Lafrance M, Gauthier MC, McDonald D, Hendzel M, West SC, Jasin M, Masson JY. Interplay between human DNA repair proteins at a unique double-strand break in vivo. EMBO J. 2006;25:222–231. doi: 10.1038/sj.emboj.7600914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ismail IH, Gagne JP, Caron MC, McDonald D, Xu Z, Masson JY, Poirier GG, Hendzel MJ. CBX4-mediated SUMO modification regulates BMI1 recruitment at sites of DNA damage. Nucleic Acids Res. 2012;40:5497–5510. doi: 10.1093/nar/gks222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gagne JP, Hunter JM, Labrecque B, Chabot B, Poirier GG. A proteomic approach to the identification of heterogeneous nuclear ribonucleoproteins as a new family of poly(ADP-ribose)-binding proteins. Biochem. J. 2003;371:331–340. doi: 10.1042/BJ20021675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gagne JP, Pic E, Isabelle M, Krietsch J, Ethier C, Paquet E, Kelly I, Boutin M, Moon KM, Foster LJ, et al. Quantitative proteomics profiling of the poly(ADP-ribose)-related response to genotoxic stress. Nucleic Acids Res. 2012;40:7788–7805. doi: 10.1093/nar/gks486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith BL, Bauer GB, Povirk LF. DNA damage induced by bleomycin, neocarzinostatin, and melphalan in a precisely positioned nucleosome. Asymmetry in protection at the periphery of nucleosome-bound DNA. J. Biol. Chem. 1994;269:30587–30594. [PubMed] [Google Scholar]
- 44.Povirk LF. DNA damage and mutagenesis by radiomimetic DNA-cleaving agents: bleomycin, neocarzinostatin and other enediynes. Mutat. Res. 1996;355:71–89. doi: 10.1016/0027-5107(96)00023-1. [DOI] [PubMed] [Google Scholar]
- 45.Tartier L, Spenlehauer C, Newman HC, Folkard M, Prise KM, Michael BD, Menissier-de Murcia J, de Murcia G. Local DNA damage by proton microbeam irradiation induces poly(ADP-ribose) synthesis in mammalian cells. Mutagenesis. 2003;18:411–416. doi: 10.1093/mutage/geg015. [DOI] [PubMed] [Google Scholar]
- 46.Rulten SL, Cortes-Ledesma F, Guo L, Iles NJ, Caldecott KW. APLF (C2orf13) is a novel component of poly(ADP-ribose) signaling in mammalian cells. Mol Cell Biol. 2008 Jul;28:4620–4628. doi: 10.1128/MCB.02243-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahel D, Horejsi Z, Wiechens N, Polo SE, Garcia-Wilson E, Ahel I, Flynn H, Skehel M, West SC, Jackson SP, et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science. 2009;325:1240–1243. doi: 10.1126/science.1177321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fahrer J, Kranaster R, Altmeyer M, Marx A, Burkle A. Quantitative analysis of the binding affinity of poly(ADP-ribose) to specific binding proteins as a function of chain length. Nucleic Acids Res. 2007;35:e143. doi: 10.1093/nar/gkm944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malanga M, Czubaty A, Girstun A, Staron K, Althaus FR. Poly(ADP-ribose) binds to the splicing factor ASF/SF2 and regulates its phosphorylation by DNA topoisomerase I. J. Biol. Chem. 2008;283:19991–19998. doi: 10.1074/jbc.M709495200. [DOI] [PubMed] [Google Scholar]
- 50.Haince JF, Ouellet ME, McDonald D, Hendzel MJ, Poirier GG. Dynamic relocation of poly(ADP-ribose) glycohydrolase isoforms during radiation-induced DNA damage. Biochim. Biophys. Acta. 2006;1763:226–237. doi: 10.1016/j.bbamcr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 51.Shav-Tal Y, Zipori D. PSF and p54(nrb)/NonO—multi-functional nuclear proteins. FEBS Lett. 2002;531:109–114. doi: 10.1016/s0014-5793(02)03447-6. [DOI] [PubMed] [Google Scholar]
- 52.Pankotai T, Bonhomme C, Chen D, Soutoglou E. DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat. Struct. Mol. Biol. 2012;19:276–282. doi: 10.1038/nsmb.2224. [DOI] [PubMed] [Google Scholar]
- 53.Polo SE, Blackford AN, Chapman JR, Baskcomb L, Gravel S, Rusch A, Thomas A, Blundred R, Smith P, Kzhyshkowska J, et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol. Cell. 2012;45:505–516. doi: 10.1016/j.molcel.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kraus WL. Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell. Biol. 2008;20:294–302. doi: 10.1016/j.ceb.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mansour WY, Rhein T, Dahm-Daphi J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res. 2010;38:6065–6077. doi: 10.1093/nar/gkq387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31:5526–5533. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010;29:3130–3139. doi: 10.1038/emboj.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, Gygi SP, Colaiacovo MP, Elledge SJ. A chromatin localization screen reveals poly(ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. USA. 2010;107:18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2010;141:970–981. doi: 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schiffner S, Zimara N, Schmid R, Bosserhoff AK. p54nrb is a new regulator of progression of malignant melanoma. Carcinogenesis. 2011;32:1176–1182. doi: 10.1093/carcin/bgr103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.