


Abstract
Antigenic variation in African trypanosomes involves monoallelic expression and reversible silencing of variant surface glycoprotein (VSG) genes found adjacent to telomeres in polycistronic expression sites (ESs). We assessed the impact on ES silencing of five candidate essential chromatin-associated factors that emerged from a genome-wide RNA interference viability screen. Using this approach, we demonstrate roles in VSG ES silencing for two histone chaperones. Defects in S-phase progression in cells depleted for histone H3, or either chaperone, highlight in particular the link between chromatin assembly and DNA replication control. S-phase checkpoint arrest was incomplete, however, allowing G2/M-specific VSG ES derepression following knockdown of histone H3. In striking contrast, knockdown of anti-silencing factor 1A (ASF1A) allowed for derepression at all cell cycle stages, whereas knockdown of chromatin assembly factor 1b (CAF-1b) revealed derepression predominantly in S-phase and G2/M. Our results support a central role for chromatin in maintaining VSG ES silencing. ASF1A and CAF-1b appear to play constitutive and DNA replication-dependent roles, respectively, in the recycling and assembly of chromatin. Defects in these functions typically lead to arrest in S-phase but defective cells can also progress through the cell cycle leading to nucleosome depletion and derepression of telomeric VSG ESs.
INTRODUCTION
The African trypanosome, Trypanosoma brucei, is a divergent protozoan pathogen of major medical and economic importance. These parasites are transmitted by tsetse flies and circulate in the mammalian host bloodstream. In order to establish a persistent infection in an immunocompetent host, T. brucei undergoes phenotypic and clonal variation of the abundant Variant Surface Glycoprotein (VSG) (1). This involves monoallelic VSG expression and reversible silencing of other VSGs. These genes are found adjacent to telomeres in polycistronic expression sites (ESs) (2) that are, unusually, under the control of promoters that recruit RNA polymerase I (RNAP-1). Beyond the VSGs found in the ESs, T. brucei also possess a subtelomeric and silent archive of up to 2000 VSGs and VSG pseudogenes (3).
The epigenetic mechanisms underlying VSG gene silencing and allelic exclusion are not fully understood. RNAP-I is thought to facilitate the high rate of transcription at the active ES, which is found in an extranucleolar region enriched for RNAP-I known as the ES body (ESB) (4). All VSG ES promoters appear to initiate RNAP-I-mediated transcription, but processivity is poor at silent ESs (5), resulting in a vast differential between VSG transcription at silent and active ESs. Recently, the single active VSG ES was shown to be specifically depleted of nucleosomes (6,7) and, in the past 5 years, several chromatin-associated factors required to maintain effective silencing at other ESs have been identified: the chromatin remodeller, ISWI (8); the histone (H3K76) methyltransferase, DOT1B (disruption of telomeric silencing) (9); the telomere-binding protein, RAP1 (repressor activator protein) (10); a histone deacetylase, DAC3 (11); the chromatin remodelling chaperone, FACT (facilitates chromatin transcription) (12); a nucleoplasmin-like protein, NLP (13); and the lamin-like NUP-1 (nuclear periphery protein-1) (14). All, but the methyltransferase are essential for growth, and their depletion typically reveals pronounced derepression close to the ES promoter, linking chromatin structure to VSG ES silencing. An impact on the VSG itself has been detected following ISWI, DOT1B, RAP1 or NUP-1 depletion, but in every case, derepression is far from complete.
Taken together, the findings detailed above show that chromatin-associated factors play an important role in defining active and silent ESs. However, incomplete derepression suggests incomplete disruption of repressive chromatin when these factors are depleted. Using data derived from a genome-scale RNA interference viability screen (15), we selected five putative essential genes with the potential to play a role in chromatin-based gene silencing. These genes were characterized and assessed in a VSG ES silencing assay, revealing both histone chaperones, CAF-1b and ASF1A, as cell cycle progression regulators and VSG ES regulators. Knockdown of these chaperones or a core histone (H3) resulted in different cell cycle stage-regulated patterns of derepression at a silent VSG ES. Our findings provide insights into the functions of these conserved chromatin regulators and strongly support a role for chromatin in maintaining silent VSG ESs. In particular, the findings also illuminate cell cycle checkpoints that limit progression to the derepressed state.
MATERIALS AND METHODS
Strains
MiTat 1.2, clone 221a (16) bloodstream-form T. brucei, 2T1 cells and 2T1 cells modified with a tetracycline-regulated green fluorescent protein:neomycin phospotransferase (GFP:NPT) cassette immediately downstream of the VSG221 ES promoter (11), were maintained, transfected and differentiated to the insect stage as previously described (17). The modified ES was maintained in the fully active state in the presence of 1 μg/ml tetracycline (‘active ES’) or rendered inactive following the removal of tetracycline (ES2GN); this latter cell line was used in all subsequent experiments. Transformants were selected with blasticidin (10 μg/ml) or hygromycin (2.5 μg/ml), as appropriate. For growth assays, cultures were seeded at ∼105/ml, diluted back and counted daily using a haemocytometer.
Plasmid construction
The pRPaiSL and pNAT constructs (18) were modified to target CAF-1b, ASF1A, C/EBP-NOC1, BDF5, TDP-1 and histone H3 for stem–loop RNAi and to engineer 12 cMYC epitope tags at the C-terminus of a native allele for the first five genes. For RNAi, ORFs were subjected to the RNAit primer prediction algorithm (19), designed to minimize potential off-target effects. In the case of histone H3, the entire open reading frame (ORF) served as the RNAi target. This sequence (see Tb927.1.2430 for example) is not expected to target the histone H3 variant sequence (Tb927.10.15350); <60% identity, longest perfect match = 8 nt, no 25 nt stretch with less than six mismatches. All primer sequences are available upon request. Constructs for RNAi and/or cMYC-tagging were transferred to ES2GN cells using a nucleofector apparatus (Lonza) in conjunction with cytomix or T-cell nucleofection solutions.
Protein analysis
Immuno-blotting was carried out following SDS–PAGE of whole-cell lysates. Protein separation and electroblotting were carried out according to standard protocols and an enhanced chemiluminescent kit (GE Healthcare) was used according to the manufacturer’s instructions. Immunofluorescence was carried out as previously described (20). cMYC fusions were detected using monoclonal mouse anti-cMYC (9E10, Source BioScience; 1:5000 WB, 1:400 IF), GFP-NPT was detected with polyclonal rabbit anti-NPT (Europa; 1:2000 WB).
Cell cycle analysis
Cell cycle analysis was carried out using DNA staining and microscopy or flow cytometry. For microscopy, cells were fixed and settled on slides as for immunofluorescence, mounted in Vectashield (vector Laboratories) containing the DNA counterstain 4,6-diamidino-2-phenylindole (DAPI), and scored for number of nuclei and kinetoplasts. Bloodstream-form cells were exposed to hydroxyurea (10 μg/ml) at a density of 1 × 106/ml and grown for 4–7 h prior to analysis. For flow cytometry, cells were washed in PBS, fixed in 70% methanol 30% PBS overnight at 4°C and stained with propidium iodide (10 μg/ml in PBS containing 10 μg/ml RNAse A) for 45 min at 37°C. Analysis was carried out using a FACScalibur (Becton Dickinson) and Flowjo 7 analysis software; best fit cell cycle models were defined using each uninduced cell line and applied to the matching induced populations.
Chromatin analysis
Micrococcal nuclease treatment was carried out as previously described (6). Briefly, 1 × 108 digitonin-permeabilized bloodstream-form cells were treated with seven gel units of micrococcal nuclease (New England Biolabs) for 10–60 min at 37°C. DNA was isolated using a Qiagen Mini Elute PCR kit and fractionated on a 2% agarose gel. Nuclease-resistant DNA was visualized by staining with ethidium bromide.
RESULTS
Identification of candidate VSG ES regulators
Six of the seven factors known to have an impact on VSG ES silencing are essential for viability in bloodstream-form T. brucei; only DOT1B is dispensable, but VSGs remain repressed and antigenic variation continues to operate in dot1b null cells (9). All of these genes, as well as a second FACT subunit (POB3), reported the expected fitness phenotype in a recent genome-wide RNA interference (RNAi) target sequencing (RIT-seq) analysis (15) (Figure 1). We sought to identify new candidate chromatin regulators that may play a role in VSG ES silencing. To focus on essential functions, we browsed annotations for 1203 genes (16% of a non-redundant gene set) that displayed a highly significant loss-of-fitness (z-score > 5) in the BFT1 RIT-seq screen, involving knockdown in bloodstream-form cells for 3 days (15). We identified a high-mobility group protein, TDP-1 (21), two histone chaperones, CAF-1b and ASF1A (22) and two conserved hypothetical genes encoding a double acetyl–lysine binding bromodomain protein, BDF5, and a protein with homology to both a mammalian CAATT-box enhancer-binding protein and a nucleolar factor, C/EBP-NOC1 (Figure 1). Analysis of CAF-1b from T. brucei, Trypanosoma cruzi and Leishmania major revealed that the trypanosomatid proteins contain the characteristic N- and C-terminal WD-40 domains (data not shown).
Figure 1.

Candidate VSG ES regulators. (A) The plot shows the BFT1 outputs from a genome-scale RNAi screen for viability; RNAi induced in bloodstream-form cells for 3 days. Previously identified VSG ES regulators, new candidate regulators and core histones are highlighted. A z-score >3.3 indicates statistical significance. (B) Heat maps showing previously identified and new candidate VSG ES regulators. BFT1, as above; BFT2, RNAi induced in bloodstream-form cells for 6 days; PF, RNAi induced in procyclic cells; DIF, RNAi induced throughout differentiation from bloodstream to procyclic form. Red indicates a loss-of-fitness. Data derived from previous study (15).
Candidate VSG expression site regulators are nuclear and essential for bloodstream-form growth
For each of the five selected genes, we established strains containing an epitope-tagged allele. Immunofluorescence analysis revealed nuclear localization for CAF-1b, TDP-1 and BDF5 (Figure 2). The nucleolar localization of C/EBP-NOC1 indicated that this is most likely orthologous to the nucleolar factor, NOC1. We were unable to localize ASF1A12MYC.
Figure 2.
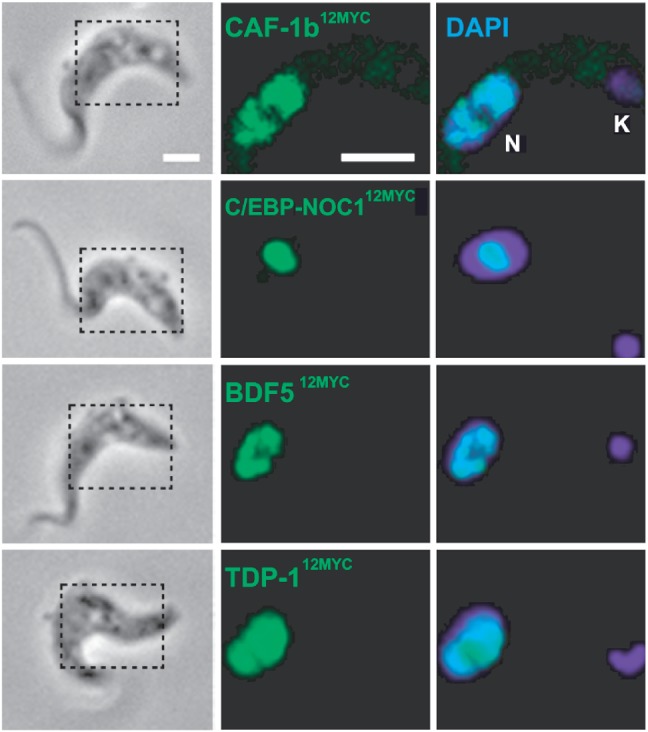
Candidate VSG ES regulators localize to the nucleus. Immunofluorescence detection of epitope-tagged proteins; the regions outlined in the phase panels indicate the regions shown in the (immuno)fluorescence panels. The nucleus (N) and kinetoplast (K, mitochondrial genome) were stained with the DNA intercalating dye, DAPI. Scale bar, 5 µm.
All five genes were then analysed by RNAi. We assembled multiple independent strains with inducible stem–loop RNAi constructs against each gene, and monitored growth following RNAi induction in bloodstream-form cells and in the same strains that had been differentiated to the procyclic or insect-stage. In bloodstream-form cells, we saw a severe growth defect for all five genes (Figure 3A), as predicted by RIT-seq analysis (Figure 1). Three of the genes (CAF-1b, ASF1A and BDF5) were predicted by RIT-seq to be associated with a growth defect following knockdown in insect-stage cells, although only the ASF1A defect was significant, whereas NOC1 and TDP1 knockdowns were predicted to have little effect on growth. All, but NOC1 displayed the expected growth phenotype (Figure 3B). The stage-specific growth defect seen in the TDP-1 knockdown strain suggests that this high-mobility group-related protein plays a particularly important role in bloodstream-stage nuclei.
Figure 3.

Candidate VSG ES regulators are essential for bloodstream stage cell survival. Growth curves for (A) bloodstream and (B) insect stage cells following induction of RNAi (+Tet). In each case, data were derived from four and two independent strains, respectively. Error bars represent 1 SD (standard deviation). RNAi was induced in the presence of 1 μg/ml tetracycline.
CAF-1b and ASF1A maintain VSG expression site silencing
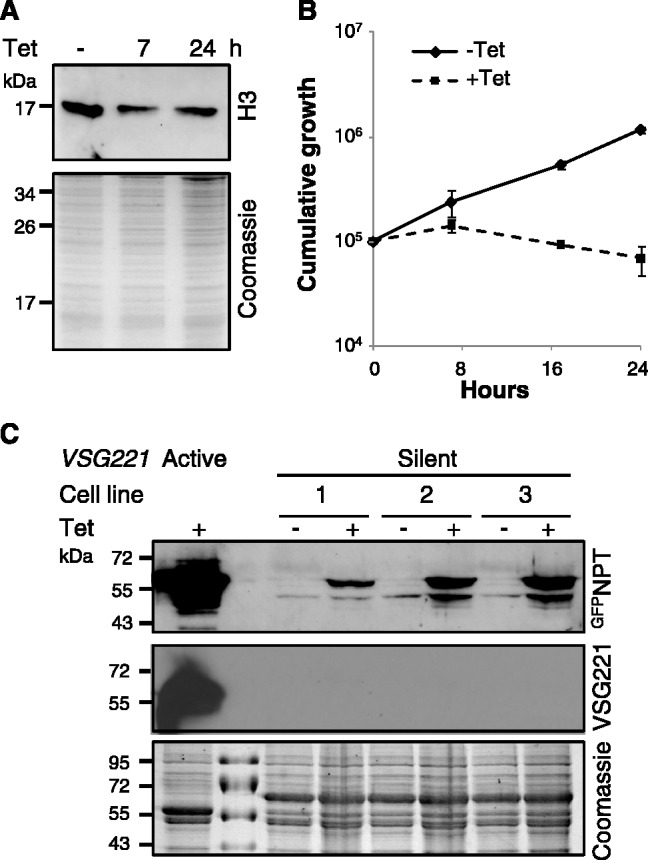
Our next step was to assemble RNAi strains with an epitope-tagged allele and a GFP:NPT-reporter cassette immediately downstream of a repressed VSG ES promoter (Figure 4A); this was achieved as described previously (11,23). Western blot analysis was used to assess knockdown of each tagged candidate regulator and to monitor the VSG ES reporter in parallel. To minimize the possibility of observing ‘secondary’ defects brought on by the toxic effects of the knockdown, we restricted the analysis to the first 24 h following RNAi induction. RNAi strains show little evidence of reduced cell number relative to the control at this point, whereas major growth defects are seen 48 h after induction of RNAi (Figure 3A); hence, we expected to focus on ‘early’ defects linked to the action of each protein at 24 h. Monitoring of the tagged proteins revealed efficient knockdown in each case after 24 h (Figure 4B). The VSG ES reporter revealed pronounced derepression following knockdown of either histone chaperone (CAF-1b or ASF1A), but little or no detectable derepression in the case of any of the other knockdowns (Figure 4B); the derepression phenotype was confirmed in two independent strains for each chaperone. Although the VSG ES promoter-proximal reporter was derepressed, we saw no evidence for derepression of the promoter-distal VSG221 gene (Figure 4B).
Figure 4.

Histone chaperones maintain VSG ES silencing. (A) Schematic map indicating the location of the GFP:NPT reporter downstream of the silent VSG221 ES promoter; these cells express VSGX from a different ES. (B) CAF-1b or ASF1A knockdown coincides with derepression of a silent VSG ES in bloodstream-form cells; data from two independent strains shown for CAF-1b and ASF1A. Coomassie (coo)-stained gels serve as loading controls. RNAi was induced in the presence of 1 μg/ml tetracycline for 24 h.
Histones maintain VSG expression site silencing
The active VSG ES is known to be depleted of nucleosomes (6,7), but it has not been shown that histones or nucleosomes are required for VSG ES repression. Histone chaperones may maintain VSG ES silencing by recycling histones and by loading new histones at silent VSG ESs following DNA replication. They may also recycle histones following low-level RNAP-I transcription adjacent to VSG ES promoters (5). To directly assess the consequences of a deficiency in new histones, we assembled histone H3 knockdown strains with a VSG ES reporter. We then demonstrated histone H3 protein knockdown in these strains (Figure 5A); depletion is likely restricted by the rapid cell-cycle arrest and the persistence of chromatin-associated histones in these cells. As expected, we observed a rapid and severe growth defect associated with histone H3 knockdown (Figure 5B). In this case, to focus on early phenotypes, we analysed cells up to 7 h after induction of RNAi. Three independent clones, in which histone H3 was depleted, all displayed derepression of the VSG ES promoter-proximal reporter but, again, we saw no derepression of the downstream VSG221 gene (Figure 5C). These results indicate for the first time that the histones themselves are required to maintain strong VSG ES silencing.
Figure 5.
Histone H3 depletion leads to VSG ES derepression. (A) Western blot showing depletion of histone H3. (B) Histone H3 RNAi knockdown leads to a severe growth defect and (C) derepression of a previously silent VSG221 ES promoter in bloodstream-form cells after 7-h induction, as determined by western blotting. Coomassie-stained gels serve as loading controls. Data derived from three independent strains. Error bars represent 1 SD. RNAi was induced in the presence of 1 μg/ml tetracycline.
Only histone deficient cells that proceed to G2/M derepress VSG expression sites
We wanted to explore the role of the histone chaperones during the cell cycle, and also whether VSG ES derepression occurred at different points through the cell cycle. We began by using microscopy and flow cytometry to explore cell cycle defects associated with histone H3 knockdown. Nuclear and mitochondrial (kinetoplast) DNA, stained with DAPI, provide excellent cytological markers that define the position in the cell cycle (24). During normal growth, ∼80% of cells display a single kinetoplast and a single nucleus (1K1N), corresponding to G1/S phase. Two kinetoplasts and a single nucleus (2K1N) correspond to nuclear G2/M, and the presence of two kinetoplasts and two nuclei (2K2N) indicates completion of mitosis. Since kinetoplast and nuclear cycles may become uncoupled in perturbed cells, we also assessed the progress of nuclear DNA replication using flow cytometry. Flow cytometry was additionally used to monitor VSG ES reporter fluorescence levels during cell cycle progression.
Cells depleted for histone H3, and stained with DAPI, displayed a severe cell cycle defect, characterized by a failure to progress to mitosis and indicated by an accumulation of 2K1N cells after 7-h induction (Figure 6A). Flow cytometry revealed that this was due to a failure to complete DNA replication, leading to an accumulation of cells in S-phase (Figure 6B). This DNA-replication defect is evident within 4 h of induction of histone H3 RNAi, when the proportion of S-phase cells is already increased by almost 2-fold. However, no effect on VSG ES reporter levels was seen until 7 h post-induction, when a population of ∼5% GFP-positive cells emerged, exhibiting a mean 11-fold increase in GFP fluorescence. This GFP reporter expression is still substantially less than when the VSG ES is fully active; cells with an active VSG ES exhibit a mean 141-fold increase in GFP fluorescence relative to cells with an inactive VSG ES (Figure 6C). Notably, the GFP derepression we observed, coincided with an increase in the G2/M population, indicating that a proportion of the defective cells completed DNA replication. Indeed, it was these cells that specifically exhibited VSG ES derepression; 80% of the GFP-positive population had completed DNA replication (Figure 6D).
Figure 6.
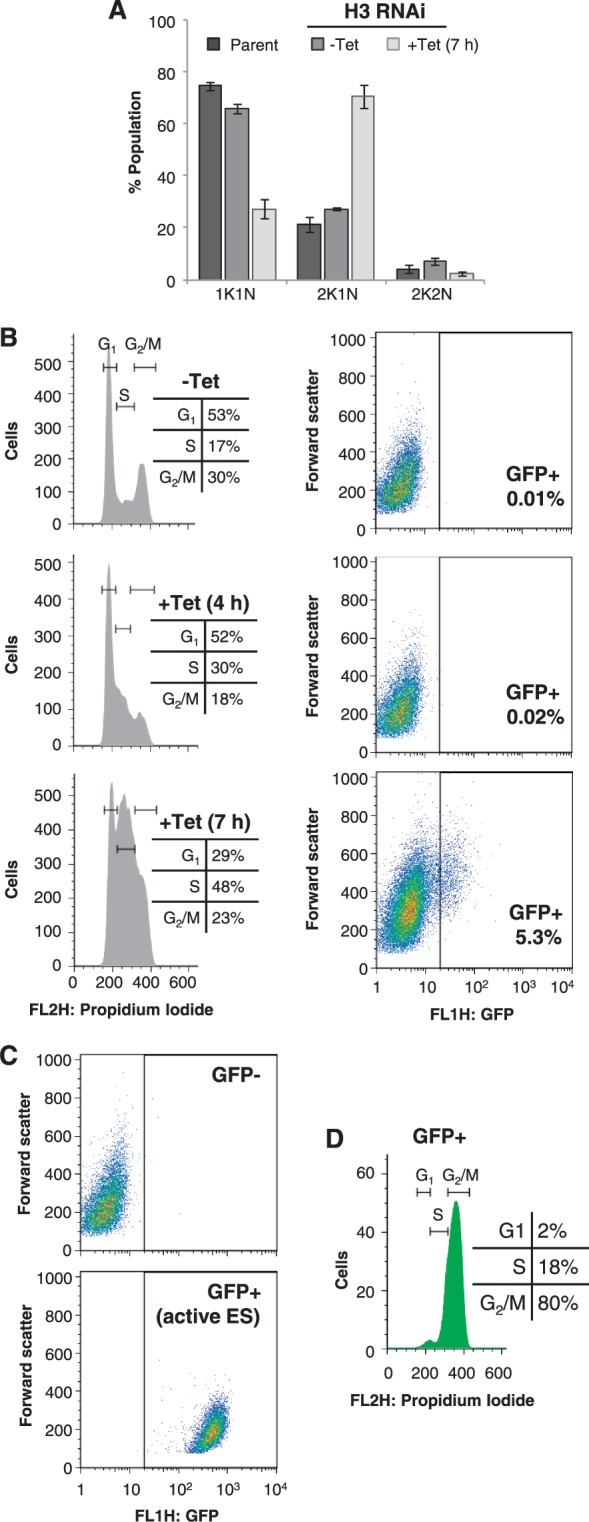
Histone H3 depletion triggers an S-phase defect and G2/M-specific VSG ES derepression. (A) Analysis of DAPI-stained cells after 7-h induction of histone H3 RNAi. K, kinetoplast; N, nucleus. Data derived from three independent strains. Error bars represent 1 SD. (B) Flow cytometry analysis showing cell cycle population distributions (left hand panels) and GFP fluorescence levels (right hand panels) after 4 and 7 h of induction. (C) Flow cytometry analysis showing GFP fluorescence from wild-type cells (upper panel) and cells with the GFP:NPT cassette at the active VSG221 ES promoter (lower panel). (D) Flow cytometry analysis of the GFP positive population from B; histone H3 RNAi for 7 h. Horizontal lines indicate the cell cycle stages, and inset table details the distribution of cells in each stage. RNAi was induced in the presence of 1 μg/ml tetracycline.
These results indicate two temporally distinct responses to histone H3 depletion. First, a drop in histone H3 mRNA and protein in S-phase, when it would normally be abundant (25), blocks DNA replication. Second, some cells escape this blockade and, presumably due to nucleosome depletion, derepress the previously silent VSG ES reporter.
To distinguish between derepression as a consequence of perturbed histone deposition rather than S-phase checkpoint activation, we treated cells with hydroxyurea, which is thought to inhibit ribonucleotide reductase, deplete dNTP synthesis and thereby stall DNA replication (26). Hydroxyurea substantially increased the proportion of 2K1N cells after 4 and 7 h, as expected, but failed to induce any detectable derepression of the GFPNPT reporter at the VSG ES (Supplementary Figure S1). Thus, derepression is likely to be a consequence of perturbed histone deposition, rather than of checkpoint activation. We also examined cell cycle progression in cells depleted for the other candidate regulators, BDF5, NOC1 and TDP-1. These knockdowns had little impact on cell cycle distribution as determined by DAPI-staining (Supplementary Figure S2A). In addition, because BDF5 depletion produced a very minor, but detectable, derepression of the GFPNPT reporter at the VSG ES (Figure 4), we also carried out flow cytometry analysis, as described above, for this knockdown (Supplementary Figure S2B and C).
ASF1A deficient cells derepress VSG expression sites throughout the cell cycle
We next carried out a similar analysis to that described above in cells depleted for ASF1A. Analysis of DAPI-stained cells following ASF1A depletion revealed a similar pattern to that seen following histone H3 depletion, namely an increase in 2K1N cells, indicating a failure to progress to mitosis (Figure 7A). Flow cytometry revealed that ASF1A depletion primarily increased the proportion of cells in S-phase (Figure 7B), as previously reported (22). Flow cytometry also confirmed that ASF1A depletion results in VSG ES derepression, and reports a mean 10-fold increase in VSG ES reporter fluorescence in ∼4% of the population. As expected, the ASF1A defect is not associated with a detectable change in steady-state histone H3 levels (Figure 7C). In striking contrast to histone H3 depletion, ASF1A depletion leads to VSG ES reporter derepression at all stages of the cell cycle (Figure 7D), suggesting a replication-independent role for this histone chaperone.
Figure 7.
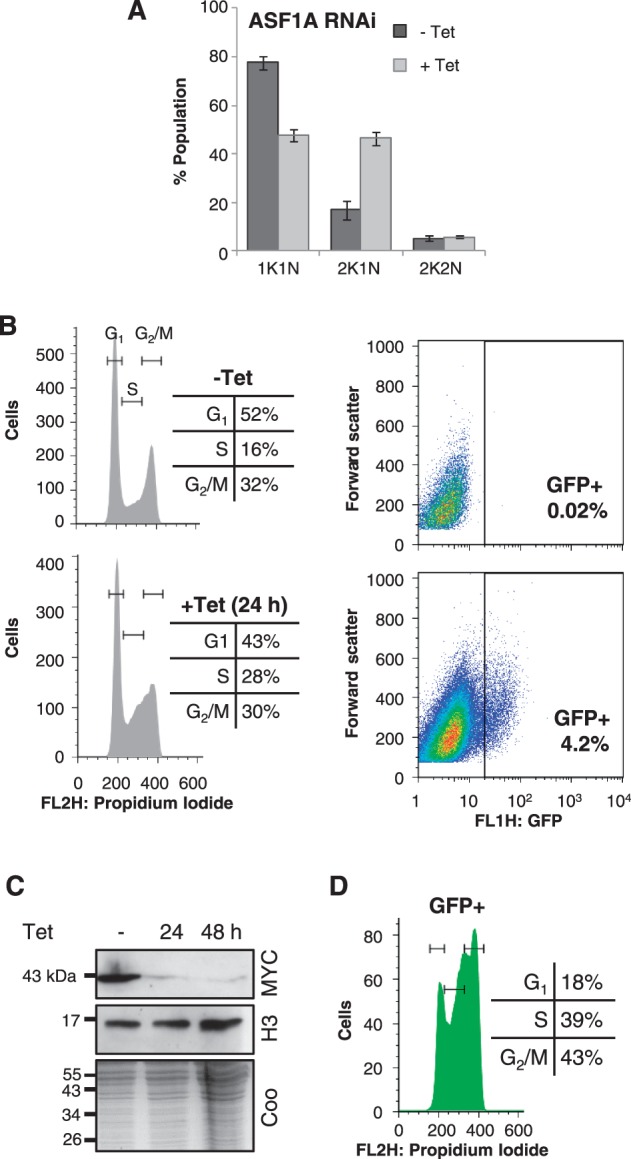
Depletion of ASF1A leads to VSG ES derepression at all cell cycle stages. (A) Analysis of DAPI-stained cells after 24-h induction of ASF1A RNAi. Data were derived from two independent strains. Error bars represent 1 SD. (B) Flow cytometry analysis showing cell cycle population distributions (left hand panels) and GFP fluorescence levels (right hand panels) after 24-h induction. (C) Western blot showing histone H3 levels following ASF1A depletion. The coomassie (coo)-stained gel serves as a loading control. (D) Flow cytometry analysis of the GFP-positive population. Other details as in Figure 6D.
CAF-1b deficient cells derepress VSG expression sites in S-phase and G2/M
Analysis of DAPI-stained cells revealed a similar pattern following RNAi against CAF-1b as seen following histone H3 and ASF1A depletion; an increase in 2K1N cells, indicating a failure to progress to mitosis (Figure 8A). Flow cytometry revealed that CAF-1b depletion increased the proportion of cells in S-phase and G2/M (Figure 8B), again indicating a DNA replication defect. Flow cytometry also confirmed that CAF-1b depletion results in VSG ES derepression, and reports a mean 13-fold increase in VSG ES reporter fluorescence in 29% of the population (Figure 8B). As was the case with ASF1A, CAF-1b depletion and the associated phenotypes were not associated with a detectable change in histone H3 steady state levels (Figure 8C). In the case of CAF-1b-depleted cells, the VSG ES reporter was derepressed in S-phase and G2/M, but displayed little derepression in G1 (Figure 8D), suggesting that CAF-1 is primarily a replication-dependent histone chaperone. Note that only ‘new’ histones are expected to be depleted following RNAi against histone H3, whereas both ‘old’ and ‘new’ histone (re)assembly is expected to be defective in chaperone-depleted cells.
Figure 8.

Depletion of CAF-1b leads to VSG ES derepression during S-phase and G2/M. (A) Analysis of DAPI-stained cells after 24-h induction of CAF-1b RNAi. Data were derived from two independent strains. Error bars represent 1 SD. (B) Flow cytometry analysis showing cell cycle population distributions (left hand panels) and GFP fluorescence levels (right hand panels) after 24-h induction. (C) Western blot showing histone H3 levels following CAF-1b depletion. The coomassie (coo)-stained gel serves as a loading control. (D) Flow cytometry analysis of the GFP positive population from B. Other details as in Figure 6D. (E) Nucleosome ladders generated by micrococcal nuclease digestion; equal numbers of cells were analysed in each sample and digestion was carried out between 10 and 60 min.
To directly examine the status of nucleosomes in CAF-1b-depleted cells, we partially digested chromatin from these cells with micrococcal nuclease (MNase). The resulting ladders of digestion products correspond to regularly spaced nucleosomes (Figure 8E). CAF-1b depletion does not appear to have a substantial impact on global nucleosome spacing, but the reduced intensity of the ladder does indicate nucleosome depletion (Figure 8E). These results, confirmed in independent strains (Supplementary Figure S3), are consistent with CAF-1 independent DNA replication through parental nucleosomes (27,28), but reduced nucleosome segregation or assembly on nascent DNA. The results also suggest a direct link between nucleosome assembly and maintenance of VSG ES repression.
DISCUSSION
Evidence has emerged in recent years for chromatin-based control of silent VSG ESs. We sought to identify other genes involved in this process and to further characterize the role of chromatin and histones in maintaining the silent state. Taking outputs from a genetic screen for viability, we selected five essential genes with a putative role in chromatin structure and function. Taken together, our findings reveal surveillance of chromatin integrity in T. brucei and delayed S-phase progression in response to reduced histone or chaperone dosage. We link histone H3 and two new histone chaperones to VSG ES silencing, and reveal derepression in subpopulations of histone-depleted cells that proceed to G2/M phase and in subpopulations of chaperone-depleted cells predominantly in S-phase and G2/M; derepression is notably later in the cell cycle in CAF-1b-depleted cells.
The importance of chromatin structural integrity in the maintenance of the repressed state at VSG ES loci is demonstrated by the consequences of histone H3 knockdown. Cells rapidly accumulate in S-phase within 4 h of RNAi induction, suggesting that S-phase checkpoint surveillance senses reduced histone availability. Importantly though, a silent VSG ES was derepressed only in the small proportion of cells that progressed beyond S-phase, and even in these cells, the silent VSG itself was not derepressed. This indicates that perturbation of chromatin structure that is sufficient to trigger cell cycle arrest and to disrupt monoallelic VSG ES transcription is insufficient to allow transcription to extend though the silent VSG ES.
Histone chaperones disassemble and assemble nucleosomes. The entire genome must be replicated, so this function is equally important at transcribed and non-transcribed loci, and chromatin differences must be reinstated behind the replication fork to maintain cellular phenotypes. These chaperones may also serve replication-independent functions. Recent reviews focusing on histone chaperones detail roles in replication and repair (29), structure–function relationships (30) and crosstalk with histone post-translational modifications (PTMs) and the inheritance of epigenetic states (31). The chaperones we identify are orthologues of ASF1 and CAF-1. The genes encoding these proteins are dispensable in the yeast, Saccharomyces cerevisiae (32), possibly due to redundancy with other related functions, but essential in mammalian or other vertebrate cells. Depletion of either vertebrate ASF1 (33) or CAF-1 (34,35) leads to accumulation in S-phase, and these histone chaperones have also been linked to chromatin-based silencing in yeast (36–39) and mammals (40), similar to the situation we now observe in trypanosomes.
Trypanosoma brucei FACT, a histone H2A-H2B chaperone, was previously linked to VSG ES silencing (12). FACT in other eukaryotes appears to fulfil a primarily replication-independent function by displacing H2A-H2B dimers (41), though there is also evidence supporting a role during replication (42). In contrast, CAF-1 has a primarily replication-associated role, recycling and assembling H3-H4 dimers into chromatin, whereas ASF1 is involved in recycling and assembly of H3-H4 dimers during DNA replication and transcription (31). There are two ASF1-related proteins in T. brucei, both of which play a role in S-phase progression (22 and this study). Trypanosoma brucei CAF-1b also functions in S-phase progression (this study), whereas FACT does not (12), suggesting replication-associated and replication-independent roles, respectively.
Our data now indicate that CAF-1b and ASF1A are important for the maintenance and inheritance of epigenetically determined states at silent telomeric VSG ESs in T. brucei. ASF1 and CAF-1 are highly conserved from yeast to mammals. In these organisms, ASF1 appears to play a role in disassembly ahead of the replication fork and the recycling of old histones and their PTMs, by passing these histones to CAF1, which is thought to participate in chromatin assembly directly behind the passing DNA polymerase (31). ASF1 has a similar function in histone recycling during transcription (31), and has recently been shown to play a role in heterochromatin assembly in Schizosaccharomyces pombe (39). Thus, the inheritance of epigenetic states is thought to depend upon the reassembly of locally displaced nucleosomes (43), thereby re-establishing the prior nucleosomes and their modifications, initially in a hemi-modified form, which could then serve as a signal for the addition of further similar modifications. Histone H3K56 acetylation appears to be central to this process, and for checkpoint signalling (44). Although an equivalent PTM has yet to be identified in trypanosomatids, the cell cycle and loss-of-silencing phenotypes that we identified associated with either ASF1A or CAF-1b knockdown are consistent with these chaperones functioning in conserved transcription/replication and replication-associated pathways, respectively.
Three distinct histone chaperones have now been linked to VSG ES silencing in T. brucei. Notably, knockdown of each factor brings about a distinct cell-cycle-regulated pattern of derepression. FACT depletion leads to G2/M specific derepression but no apparent delay in S-phase (12), possibly indicating a post-S-phase role. ASF1A depletion delays S-phase and causes derepression in a subpopulation of cells through the cell cycle, consistent with a constitutive role in histone loading and recycling; the recycling role during G1 is likely to be important to maintain attenuation of transcription at silent VSG ESs (5). CAF-1b depletion delays S-phase and primarily causes derepression during S-phase and G2/M, consistent with a histone loading role during S-phase. Thus, our results indicate conserved histone chaperone functions for both ASF1A and CAF-1b from trypanosomes to humans. We conclude that chaperone-dependent chromatin reassembly following attenuated transcription (ASF1A) and DNA replication (ASF1A and CAF-1b) are essential for the maintenance of VSG ES silencing. In addition, we show that cell cycle checkpoints often limit progression to the derepressed state in chromatin-perturbed cells.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–3.
FUNDING
The Wellcome Trust [089172]. Funding for open access charge: The Wellcome Trust.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Horn D, McCulloch R. Molecular mechanisms underlying the control of antigenic variation in African trypanosomes. Curr. Opin. Microbiol. 2010;13:700–705. doi: 10.1016/j.mib.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hertz-Fowler C, Figueiredo LM, Quail MA, Becker M, Jackson A, Bason N, Brooks K, Churcher C, Fahkro S, Goodhead I, et al. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS One. 2008;3:e3527. doi: 10.1371/journal.pone.0003527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marcello L, Barry JD. Analysis of the VSG gene silent archive in Trypanosoma brucei reveals that mosaic gene expression is prominent in antigenic variation and is favored by archive substructure. Genome Res. 2007;17:1344–1352. doi: 10.1101/gr.6421207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Navarro M, Gull K. A pol I transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature. 2001;414:759–763. doi: 10.1038/414759a. [DOI] [PubMed] [Google Scholar]
- 5.Vanhamme L, Poelvoorde P, Pays A, Tebabi P, Van Xong H, Pays E. Differential RNA elongation controls the variant surface glycoprotein gene expression sites of Trypanosoma brucei. Mol. Microbiol. 2000;36:328–340. doi: 10.1046/j.1365-2958.2000.01844.x. [DOI] [PubMed] [Google Scholar]
- 6.Figueiredo LM, Cross GA. Nucleosomes are depleted at the VSG expression site transcribed by RNA polymerase I in African trypanosomes. Eukaryot. Cell. 2010;9:148–154. doi: 10.1128/EC.00282-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stanne TM, Rudenko G. Active VSG expression sites in Trypanosoma brucei are depleted of nucleosomes. Eukaryot. Cell. 2010;9:136–147. doi: 10.1128/EC.00281-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stanne TM, Kushwaha M, Wand M, Taylor JE, Rudenko G. TbISWI regulates multiple polymerase I (Pol I)-transcribed loci and is present at Pol II transcription boundaries in Trypanosoma brucei. Eukaryot. Cell. 2011;10:964–976. doi: 10.1128/EC.05048-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Figueiredo LM, Janzen CJ, Cross GA. A histone methyltransferase modulates antigenic variation in African trypanosomes. PLoS Biol. 2008;6:e161. doi: 10.1371/journal.pbio.0060161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang X, Figueiredo LM, Espinal A, Okubo E, Li B. RAP1 is essential for silencing telomeric variant surface glycoprotein genes in Trypanosoma brucei. Cell. 2009;137:99–109. doi: 10.1016/j.cell.2009.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang QP, Kawahara T, Horn D. Histone deacetylases play distinct roles in telomeric VSG expression site silencing in African trypanosomes. Mol. Microbiol. 2010;77:1237–1245. doi: 10.1111/j.1365-2958.2010.07284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denninger V, Fullbrook A, Bessat M, Ersfeld K, Rudenko G. The FACT subunit TbSpt16 is involved in cell cycle specific control of VSG expression sites in Trypanosoma brucei. Mol. Microbiol. 2010;78:459–474. doi: 10.1111/j.1365-2958.2010.07350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narayanan MS, Kushwaha M, Ersfeld K, Fullbrook A, Stanne TM, Rudenko G. NLP is a novel transcription regulator involved in VSG expression site control in Trypanosoma brucei. Nucleic Acids Res. 2011;39:2018–2031. doi: 10.1093/nar/gkq950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DuBois KN, Alsford S, Holden JM, Buisson J, Swiderski M, Bart JM, Ratushny AV, Wan Y, Bastin P, Barry JD, et al. NUP-1 is a large coiled-coil nucleoskeletal protein in trypanosomes with lamin-like functions. PLoS Biol. 2012;10:e1001287. doi: 10.1371/journal.pbio.1001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alsford S, Turner DJ, Obado SO, Sanchez-Flores A, Glover L, Berriman M, Hertz-Fowler C, Horn D. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 2011;21:915–924. doi: 10.1101/gr.115089.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alsford S, Kawahara T, Glover L, Horn D. Tagging a T. brucei RRNA locus improves stable transfection efficiency and circumvents inducible expression position effects. Mol. Biochem. Parasitol. 2005;144:142–148. doi: 10.1016/j.molbiopara.2005.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alsford S, Kawahara T, Isamah C, Horn D. A sirtuin in the African trypanosome is involved in both DNA repair and telomeric gene silencing but is not required for antigenic variation. Mol. Microbiol. 2007;63:724–736. doi: 10.1111/j.1365-2958.2006.05553.x. [DOI] [PubMed] [Google Scholar]
- 18.Alsford S, Horn D. Single-locus targeting constructs for reliable regulated RNAi and transgene expression in Trypanosoma brucei. Mol. Biochem. Parasitol. 2008;161:76–79. doi: 10.1016/j.molbiopara.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Redmond S, Vadivelu J, Field MC. RNAit: an automated web-based tool for the selection of RNAi targets in Trypanosoma brucei. Mol. Biochem. Parasitol. 2003;128:115–118. doi: 10.1016/s0166-6851(03)00045-8. [DOI] [PubMed] [Google Scholar]
- 20.Alsford S, Horn D. Elongator protein 3b negatively regulates ribosomal DNA transcription in African trypanosomes. Mol. Cell Biol. 2011;31:1822–1832. doi: 10.1128/MCB.01026-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erondu NE, Donelson JE. Differential expression of two mRNAs from a single gene encoding an HMG1-like DNA binding protein of African trypanosomes. Mol. Biochem. Parasitol. 1992;51:111–118. doi: 10.1016/0166-6851(92)90206-y. [DOI] [PubMed] [Google Scholar]
- 22.Li Z, Gourguechon S, Wang CC. Tousled-like kinase in a microbial eukaryote regulates spindle assembly and S-phase progression by interacting with Aurora kinase and chromatin assembly factors. J. Cell Sci. 2007;120:3883–3894. doi: 10.1242/jcs.007955. [DOI] [PubMed] [Google Scholar]
- 23.Glover L, Alsford S, Beattie C, Horn D. Deletion of a trypanosome telomere leads to loss of silencing and progressive loss of terminal DNA in the absence of cell cycle arrest. Nucleic Acids Res. 2007;35:872–880. doi: 10.1093/nar/gkl1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siegel TN, Hekstra DR, Cross GA. Analysis of the Trypanosoma brucei cell cycle by quantitative DAPI imaging. Mol. Biochem. Parasitol. 2008;160:171–174. doi: 10.1016/j.molbiopara.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ersfeld K, Docherty R, Alsford S, Gull K. A fluorescence in situ hybridisation study of the regulation of histone mRNA levels during the cell cycle of Trypanosoma brucei. Mol. Biochem. Parasitol. 1996;81:201–209. doi: 10.1016/0166-6851(96)02709-0. [DOI] [PubMed] [Google Scholar]
- 26.Forsythe GR, McCulloch R, Hammarton TC. Hydroxyurea-induced synchronisation of bloodstream stage Trypanosoma brucei. Mol. Biochem. Parasitol. 2009;164:131–136. doi: 10.1016/j.molbiopara.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonne-Andrea C, Wong ML, Alberts BM. In vitro replication through nucleosomes without histone displacement. Nature. 1990;343:719–726. doi: 10.1038/343719a0. [DOI] [PubMed] [Google Scholar]
- 28.Kamakaka RT, Bulger M, Kaufman PD, Stillman B, Kadonaga JT. Postreplicative chromatin assembly by Drosophila and human chromatin assembly factor 1. Mol. Cell. Biol. 1996;16:810–817. doi: 10.1128/mcb.16.3.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ransom M, Dennehey BK, Tyler JK. Chaperoning histones during DNA replication and repair. Cell. 2010;140:183–195. doi: 10.1016/j.cell.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das C, Tyler JK, Churchill ME. The histone shuffle: histone chaperones in an energetic dance. Trends Biochem. Sci. 2010;35:476–489. doi: 10.1016/j.tibs.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Avvakumov N, Nourani A, Cote J. Histone chaperones: modulators of chromatin marks. Mol. Cell. 2011;41:502–514. doi: 10.1016/j.molcel.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Ramey CJ, Howar S, Adkins M, Linger J, Spicer J, Tyler JK. Activation of the DNA damage checkpoint in yeast lacking the histone chaperone anti-silencing function 1. Mol. Cell Biol. 2004;24:10313–10327. doi: 10.1128/MCB.24.23.10313-10327.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanematsu F, Takami Y, Barman HK, Fukagawa T, Ono T, Shibahara K, Nakayama T. Asf1 is required for viability and chromatin assembly during DNA replication in vertebrate cells. J. Biol. Chem. 2006;281:13817–13827. doi: 10.1074/jbc.M511590200. [DOI] [PubMed] [Google Scholar]
- 34.Quivy JP, Gerard A, Cook AJ, Roche D, Almouzni G. The HP1-p150/CAF-1 interaction is required for pericentric heterochromatin replication and S-phase progression in mouse cells. Nat. Struct. Mol. Biol. 2008;15:972–979. doi: 10.1038/nsmb.1470. [DOI] [PubMed] [Google Scholar]
- 35.Takami Y, Ono T, Fukagawa T, Shibahara K, Nakayama T. Essential role of chromatin assembly factor-1-mediated rapid nucleosome assembly for DNA replication and cell division in vertebrate cells. Mol. Biol. Cell. 2007;18:129–141. doi: 10.1091/mbc.E06-05-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dohke K, Miyazaki S, Tanaka K, Urano T, Grewal SI, Murakami Y. Fission yeast chromatin assembly factor 1 assists in the replication-coupled maintenance of heterochromatin. Genes Cells. 2008;13:1027–1043. doi: 10.1111/j.1365-2443.2008.01225.x. [DOI] [PubMed] [Google Scholar]
- 37.Le S, Davis C, Konopka JB, Sternglanz R. Two new S-phase-specific genes from Saccharomyces cerevisiae. Yeast. 1997;13:1029–1042. doi: 10.1002/(SICI)1097-0061(19970915)13:11<1029::AID-YEA160>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 38.Sharp JA, Fouts ET, Krawitz DC, Kaufman PD. Yeast histone deposition protein Asf1p requires Hir proteins and PCNA for heterochromatic silencing. Curr. Biol. 2001;11:463–473. doi: 10.1016/s0960-9822(01)00140-3. [DOI] [PubMed] [Google Scholar]
- 39.Yamane K, Mizuguchi T, Cui B, Zofall M, Noma K, Grewal SI. Asf1/HIRA facilitate global histone deacetylation and associate with HP1 to promote nucleosome occupancy at heterochromatic loci. Mol. Cell. 2011;41:56–66. doi: 10.1016/j.molcel.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tchenio T, Casella JF, Heidmann T. A truncated form of the human CAF-1 p150 subunit impairs the maintenance of transcriptional gene silencing in mammalian cells. Mol. Cell. Biol. 2001;21:1953–1961. doi: 10.1128/MCB.21.6.1953-1961.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;301:1090–1093. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- 42.Tan BC, Chien CT, Hirose S, Lee SC. Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J. 2006;25:3975–3985. doi: 10.1038/sj.emboj.7601271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Radman-Livaja M, Verzijlbergen KF, Weiner A, van Welsem T, Friedman N, Rando OJ, van Leeuwen F. Patterns and mechanisms of ancestral histone protein inheritance in budding yeast. PLoS Biol. 2011;9:e1001075. doi: 10.1371/journal.pbio.1001075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fillingham J, Greenblatt JF. A histone code for chromatin assembly. Cell. 2008;134:206–208. doi: 10.1016/j.cell.2008.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.