

Abstract
A series of cyclic peptides were designed and prepared to investigate the physicochemical properties that affect oral bioavailabilty of this chemotype in rats. In particular, the ionization state of the peptide was examined by the incorporation of naturally occurring amino acid residues that are charged in differing regions of the gut. In addition, data was generated in a variety of in vitro assays and the usefulness of this data in predicting the subsequent oral bioavailability observed in the rat is discussed.
Introduction
Peptides and peptidomimetics often have molecular weights that exceed 500 Da and hydrogen bonding capabilities that move them outside what has been considered orally bioavailable drug space within the modern pharmaceutical industry. The majority of peptide and protein therapeutics are administered via parenteral routes due to the low oral bioavailability of these compounds. The low oral bioavailability can be attributed to two major problems: poor ability of the peptide/peptidomimetic to cross the gut wall, and short plasma half-life due to proteolytic and P450-mediated oxidative degradation and renal elimination. Thus, designing peptides that cross the gut wall via a passive transcellular mode remains a major challenge in drug discovery.
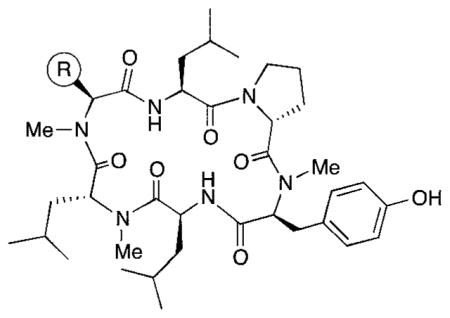
The necessity to solve these challenges is driven by a number of high value molecular targets such as Class B GPCRs and protein–protein interactions that are difficult to modulate using orally bioavailable small molecules that reside within the Rule of Five. Thus the potential of orally bioavailable peptides to engage these targets would provide a competitive advantage to research groups addressing unmet medical needs. There is a consensus that cyclization of peptides improves proteolytic stability and gives the opportunity to modify intramolecular hydrogen bonding networks to deliver gut wall permeable expressions in comparison to their acyclic counterparts. The improved cell permeability of cyclic systems is still an open question with differing conclusions appearing in the literature. We have previously reported1 compound 1 (Table 1) which has a physicochemical profile that lies outside of traditional oral drug space, with a molecular weight of >750 Da and multiple hydrogen bond donors and acceptors which, upon initial inspection, should limit its ability to cross the gut wall. Compound 1 was profiled in a range of in vitro assays and progressed to an in vivo pharmacokinetic (PK) study.ThePK profile of 1 in rats was characterized by low clearance and a moderate ability to partition into tissue, leading to a terminal elimination half-life of 2.8 h. The absolute oral bioavailability (F) of 1 was determined to be ~28%. In this paper we report investigations introducing polar side chains into the cyclic template with the aim of simultaneously understanding the molecular ionization state and physicochemical space that can be tolerated in this chemotype.
Table 1.
In vitro absorption and metabolism data for peptides with natural amino acid side chains
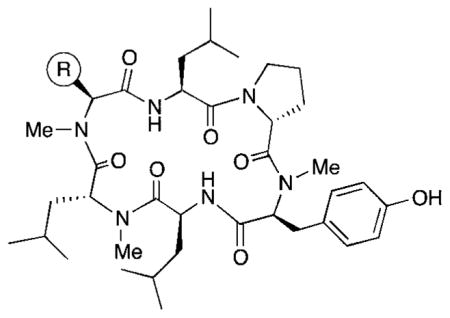
| ||||||
|---|---|---|---|---|---|---|
| Cpd | R | log D (pH 7.4) | RRCK Papp (A to B) × 10−6 cm s−1 | RLM CLint,app,scaled (mL min−1 kg−1) | HLM CLint,app,scaled (mL min−1 kg−1) | Caco-2 Papp (A to B) × 10−6 cm s−1 |
| 1 |
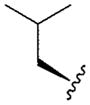
|
4.41 | 4.9 | 36 | 120 | 4.85 |
| 2 |
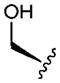
|
2.61 | 1.6 | 96 | 80 | 3.3 |
| 3 |
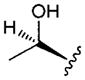
|
3.17 | 4.7 | 44 | 89 | |
| 4 |
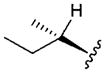
|
4.57 | 3.2 | >300 | ||
| 5 |
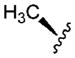
|
3.64 | 5.5 | 190 | ||
| 6 |
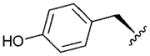
|
3.82 | 1.3 | 29 | ||
| 7 |
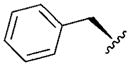
|
4.46 | 5.8 | 170 | ||
| 8 |
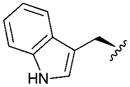
|
4.07 | 0.2 | 56 | ||
| 9 |
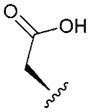
|
0.477 | 0.4 | <9 | <8 | 0.36 |
| 10 |
|
0.678 | 0.3 | <9 | <8 | 0.09 |
Through introduction of basic centers in the side chain, we aimed to modulate a range of parameters such as the ability of the peptide to distribute into tissues and so increase the volume of distribution to greater than the plasma compartment and body water (approx 0.04 and 0.6 L kg−1 respectively). The introduction of acidic groups may also attenuate P450-mediated oxidative clearance and mitigate binding of the molecules to plasma proteins such as albumin. The introduction of either acidic or basic side chains will potentially improve the bio-pharmaceutical properties, in particular aqueous solubility, to simplify formulations and increase chemical diversity for screening in biochemical and biophysical assays.
An objective of this work is to build an in vitro/in vivo correlation of ADME data in order to optimize the predictivity of our in vitro assays for peptides and peptidomimetics. It has previously been shown1,6,7 that computational modeling can accurately predict the relative in vitro permeability of cyclic peptides when accounting for stereochemistry, intramolecular hydrogen-bonding, and N-methylation. An additional objective of this work is to further optimize the effectiveness of computational models to account for the impact of side chain polarity on permeability.
Design and synthesis
Previously,1 we reported that the N-methylated cyclic hexapeptide 1 had an oral bioavailability of 28% in the rat, but had high in vitro clearance in human liver microsomes (HLM) (CLint = 120 mL min−1 kg−1). Examination of the biotransformation pathways of 1 in HLM revealed the tyrosine ring and the leucine side chains as sites for cytochrome P450 mediated oxidations. A similar metabolic profile was discerned in rat liver microsomes (RLM); however, the extent of metabolism was significantly lower than in human liver microsomes (HLM). Several serine variations were also reported, including compound 2.
The design of compound 1 was inspired by a class of lipophilic cyclic peptide natural products that, despite their non-“drug-like” appearance, can show drug-like PK properties, including oral bioavailability. These natural products are characterized by backbone N-methylation, extensive intramolecular hydrogen bonding, and a predominance of aliphatic amino acids. In a previous study we evaluated the effect of stereochemistry and the pattern of N-methylation on backbone geometry, and the resulting impact on membrane permeability.
In order to understand more fully the impact of individual amino acids substitutions on permeability and clearance properties for this cyclic hexapeptide template, we replaced R with eight additional standard amino acids side chains (compounds 3–10), spanning a range of ionization states and log D values. These compounds were profiled in vitro to assess their lipophilicity, permeability, and clearance properties. Lipophilicity was measured using shake-flask log D, permeability was measured using RRCK (Ralph Russ canine kidney cell line, derived from a Madin–Darby canine kidney cell line selected for low-efflux properties2), and clearance was measured using HLM.3 Results for the previously reported compounds 1 and 2, plus the eight new compounds, are shown in Table 1. In order to expand our knowledge of the ADME property of this chemistry space further, we synthesized six non-natural N-methylated amino acids, incorporating them into the cyclic hexapeptide to produce compounds 11–16 (Table 2).
Table 2.
In vitro absorption and metabolism data for peptides with non-natural N-methyl amino acids
| ||||
|---|---|---|---|---|
| No. | R | log D (pH 7.4) | RRCK Papp (A to B) × 10−6 cm s−1 | HLM CLint,app,scaled (mL min−1 kg−1) |
| 11 |
|
0.723 | 0.2 | 9 |
| 12 |
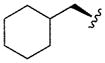
|
5.09 | 0.2 | |
| 13 |
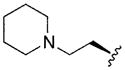
|
2.20 | 0.7 | <8 |
| 14 |
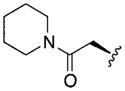
|
3.77 | 1.4 | >300 |
| 15 |
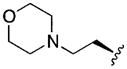
|
2.79 | 1.2 | 16 |
| 16 |
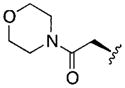
|
2.78 | 0.6 | 57 |
Synthesis of the cyclic peptide derivatives 1–10 and 12 proceeded by incorporating the appropriately protected commercially available amino acids using previously reported procedures.1 Briefly, the peptides were anchored to the solid phase via an ether linkage to 2-chlorotrityl polystyrene resin through the Tyr side chain. The peptides were elongated using standard solid phase peptide synthesis (SPPS) methods, and, after completion of the linear sequence, the C-terminal allyl ester was removed along with the N-terminal Fmoc group in a single step (1.0 eq. Pd(Ph3P)4 in THF with 10% piperidine). Following cyclization (HATU and DIPEA in DMF), the peptides were N-methylated selectively using the conformation-directed N-methylation1 chemistry previously reported for 1, with excess LiOtBu in THF followed by excess CH3I in DMSO. With few exceptions (see below), N-methylation produced a single tri-methyl product, whose regiochemistry was confirmed for selected derivatives by 2D NMR1. For the incorporation of basic side chains (e.g., in 13 and 15), initial attempts to use the corresponding amino acids in the synthesis of the macrocycle failed because the basic residue resulted in complete loss of selectivity in the N-methylation step. Therefore, for compounds 13 and 15, the cyclic peptides were synthesized initially with an Asp(OMe) at the “R” position. After on-resin N-methylation, the ester was converted to the corresponding amine on the solid phase using the sequence shown in Scheme 1. For compounds 14 and 16, Asp(OtBu) was used at the R position and the appropriate amine was coupled using standard amide coupling conditions onto the free carboxylic acid after resin cleavage and deprotection.
Scheme 1.

On-resin conversion of Asp(OMe) to the tertiary amine in compounds 13 and 15.
Physical properties and in vitro ADME
In vitro properties across this series varied significantly in accordance with their log D values. Incorporation of aromatic or polar aliphatic side chains in this position (compounds 4–8) did not significantly lower log D (their log D’s ranged from 3.64 to 4.57) and HLM values remained high, with CLint,app,scaled values of 29.4 mL min−1 kg−1 and higher. Incorporation of the ionizable aspartic acid and lysine side chains (compounds 9 and 10) dramatically lowered log D and clearance, but at the cost of reduced permeability. The aliphatic alcohol threonine side chain (3) afforded reduced log D (3.17) while maintaining moderate permeability (RRCK Papp = 4.69 × 10−6 cm s−1−1). In spite of its lowered log D, the in vitro HLM value for Compound 3 was high (CLint,app,scaled = 88.9 mL min−1 kg−1), a value similar to that seen for the other aliphatic alcohol, Ser derivative 2.
Of special interest among the non-natural N-methylated amino acids (Table 2) were the four heterocyclic side chains of 13–16, designed to have more favorable log D’s. Of these, compounds 13 and 15 contain basic nitrogens and had log D values below three and low HLM clearance. Their RRCK values remained low but were improved over the previously tested peptides with ionizable groups (10–11). The morpholine-containing compound 15 was of particular interest because of its balance of properties and will be the subject of future studies.
Across the set of sixteen cyclic peptides, there was a good correlation between log D and RRCK permeability (Fig. 1) and between log D and HLM clearance (Fig. 2). A log D near three appears especially favorable, as 3 (log D = 3.17) had moderately good permeability (RRCK Papp = 4.69 × 10 −6 cm s−1) while 15 (log D = 2.7) had moderately good clearance (HLM CLint,app,scaled = 15.8 mL min−1 kg−1). We will continue to further profile N-methylated cyclic peptides in this promising chemistry space.
Fig. 1.

log D vs. RRCK plot. The R2 is shown excluding and including the outliers, compounds 8 and 12, which had poor recoveries.
Fig. 2.

log D vs. log HLM plot. The R2 correlation excludes censored measurements (<8 mL min−1 kg−1 and >300 mL min−1 kg−1).
In vivo pharmacokinetics
Table 3 depicts the pharmacokinetic parameters of select N-methylated cyclic hexapeptides in male Wistar-Han rats. Previously, we reported on the in vivo pharmacokinetics of 1, which are characterized by a low plasma clearance (CLp) of 4.5 mL min−1 kg−1 and a moderate steady state distribution volume (Vdss) of 1.1 L kg−1, resulting in a terminal T1/2 of 2.8 h (Table 3).1 Bioavailability (F) upon oral (p.o.) administration to rats was ~28%, which is similar to that reported for the cyclic peptide cyclosporine.1 The fraction of the dose absorbed (Fa) following p.o. administration of 1 was estimated to be ~30% using the equation Fa = F/(Fg × Fh), where Fg and Fh represent the fraction of the oral dose that escapes gut wall and hepatic metabolism, respectively.4
Table 3.
Pharmacokinetic parameters in rats for select compounds after a single intravenous (i.v.) or oral (p.o.) dosea
| Compd | Doseb (mg kg−1) | Route | Cmax (ng mL−1) | Tmax (h) | CLp (mL min−1 kg−1) | Vdss (L kg−1) | AUC(0–∞) (ng h mL−1) | T1/2 (h) | F (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.0 | i.v. | N.A. | N.A | 4.53 | 1.13 | 3800 | 2.84 | NA |
| 10 | p.o. | 3800 | 4.0 ± 3.0 | N.A | N.A | 28.1 | |||
| 2 | 1.0 | i.v. | N.A | N.A | 60.4 ± 10.9 | 4.3 ± 1.15 | 282 ± 46.7 | 1.46 ± 0.180 | NA |
| 5.0 | p.o. | 9.86 ± 1.95 | 0.83 ± 1.01 | N.A. | N.A. | 42.3 ± 3.9 | 2.82 ± 1.75 | 2.03 ± 0.76 | |
| 3c | 0.4 | i.v. | N.A | N.A | 63.6 ± 15.0 | 3.8 ± 0.77 | 109 ± 24 | 1.03 ± 0.014 | NA |
| 2.9 | p.o. | 105 ± 81 | 1.17 ± 0.76 | N.A | N.A | 201 ± 66 | 0.92 ± 0.04 | 23.8 ± 7.80 | |
| 9 | 1.48 | i.v. | N.A | N.A | 56.4 ± 6.06 | 0.36 ± 0.11 | 441 ± 45.7 | 0.358 ± 0.109 | NA |
| 10 | p.o. | 17.3 ± 25.2 | 0.78 ± 1.1 | N.A | N.A | 18.7 | 4.73 | 0.469 ± 0.43 | |
| 10 | 1 | i.v. | N.A | N.A | 10.4 ± 1.64 | 0.93 ± 0.07 | 1640 ± 283 | 1.01 ± 0.12 | NA |
| 10 | p.o. | 18.3 ± 13.2 | 1.56 ± 0.86 | N.A | N.A | 18.1 ± 9.85 | 1.56 ± 0.86 | 0.09 ± 0.07 |
Pharmacokinetic studies were conducted in male Wistar-Han rats.
Dosing vehicle used in i.v. and p.o. administration was 1: 1 propylene glycol:20 mM sodium phosphate (pH 7.4). For compound 9, the dosing vehicle was 10% propylene glycol and 90% 20 mM sodium phosphate buffer, pH 7.4. p.o. pharmacokinetic studies were conducted in the fasted state.
In the case of compound 3, the pharmacokinetics of peak 2 are reported. NA = not applicable.
Replacement of the Leu side chain in 1 with a Ser residue yielded 2, which exhibited lower absorptive permeability in the RRCK assay, presumably as a result of its greater polarity. In vivo, the oral F of 2 in the rat (F = 2%) was significantly lower than the oral F of 1. While it is tempting to speculate attenuated permeability as a probable cause for the poor oral F, it is note-worthy that compound 2 was also rapidly cleared (CLp = 60.4 mL min−1 kg−1) in the rat, which is reflected in its higher CLint (relative to 1) in rat liver microsomes (see Table 1). As such, 2 was stable towards oxidative or proteolytic degradation in rat intestinal S-9 fraction and rat brush border membrane vesicles5 suggesting that first pass metabolism in the gut wall was not a limiting factor in the oral absorption of 2 (i.e., Fg was unity). Assuming that 2 is cleared mainly via hepatic metabolism, its CLp of 60.4 mL min−1 kg−1 will translate into a hepatic extraction ratio of ~86% (CLp divided by rat liver blood flow of 70 mL min−1 kg−1), Fh of 14% and Fa of 14.5%.
In the case of the Thr derivative (3), liquid-chromatography tandem mass spectrometric analysis (monitoring in the multiple reaction monitoring mode using the m/z transitions 743 → 485, 743 → 211, and 743 → 144 which corresponded to the parent molecular weight and diagnostic fragment ions) of rat plasma samples revealed the existence of two isomeric peaks for 3 with distinct chromatographic separation (retention time for peak 1 = 1.28 min, retention time for peak 2 = 1.41 min). Peak 2 was the dominant peak (~4: 1 ratio over peak 1) and was identified as compound 3 based on a retention time match with a neat standard of 3 in acetonitrile. We later determined that conversion of 3 to the unknown isomeric product (corresponding to “peak 1”) was catalysed by the formic acid used in its initial preparative HPLC purification of the crude synthetic material. Purification in the absence of acid allowed us to isolate 3 and unambiguously assign “peak 2” from the in vivo study to the expected compound 3. Consequently, pharmacokinetic parameters for 3 were obtained by specifically monitoring the disappearance of peak 2 in plasma. Compound 3 demonstrated superior oral absorption (oral F = ~23.8%) relative to 2 (oral F = 2%) despite the high CLp, which was similar to that observed with 1. Therefore, it is possible that the increased oral absorption noted with 3 is a reflection of its superior absorptive permeability (and increased lipophilicity). Studies are ongoing to identify the structure of peak 1 that appeared as a minor contaminant in rat plasma samples of 3.
The N-methylated cyclic hexapeptides with pendant ionizable aspartate and lysine side chains also demonstrated interesting pharmacokinetic behavior. The lysine derivative 10 revealed low CLp (10.4 mL min−1 kg−1) and Vdss (0.93 L kg −1) resulting in a T1/2 of ~1.0 h. Despite the low CLp, the corresponding oral F of 10 was low (<1%), suggesting that the poor absorptive permeability of 10 was a rate-limiting factor in its oral absorption. The low CLp of 10 in vivo was consistent with the low CLint,app,scaled (<9.0 mL min−1 kg−1) observed in rat liver microsomal stability studies indicative of a good in vitro/in vivo correlation for clearance (IVIVC). In contrast, despite lack of significant metabolic turnover in rat liver microsomes (CLint,app,scaled < 9.0 mL min kg−1), the aspartate derivative 9 exhibited a relatively high CLp (56.4 mL min−1 kg−1) in vivo. Although urinary excretion of unchanged 9 was insignificant (<1% of the dose), it is possible that alternate elimination mechanisms (e.g., phase II glucuronidation and/or biliary excretion) may play a role in the clearance of 9, which may then explain the weak IVIVC in rat liver microsomal incubations. Coupled with the high in vivo CLp and poor passive permeability, the observed oral F of <1% with 9 was not surprising.
In terms of future direction in the pharmacokinetic optimization of N-methylated hexapeptides, it is interesting to point out that the low hepatic extraction ratio of compound 1 (~0.06) should have led to oral F of >90%, if solubility/permeability (governed by Fa) and gut metabolism (governed by Fg) were not rate-limiting in terms of oral absorption. Thus, it is possible that the oral absorption of 1 (and possibly related N-methylated hexapeptides) could be further improved via the use of solubilizing formulations as has been demonstrated with cyclosporine A.1
Computational modeling
The effects of various substitutions on permeability were prospectively predicted by a physics-based permeability model to provide guidance for experimental efforts and insights into understanding the molecular effects of different modifications. The computational methods have been described in detail previously.1,6–8 Briefly, the approach combines conformational sampling with an estimate of the free energy cost of partitioning into the barrier domain (free energy maximum, thus rate-limiting), which is generally located close to the center of the membrane. The dominant contribution is the free energy cost of desolvating the compounds, a quantity that is correlated to other commonly used measures such as log P or polar surface area. Despite neglecting many aspects of membrane permeation, the minimal free energy of desolvation has been demonstrated to yield predictions in good correlation with experimental measurements and its application aided identification of permeable cyclic peptide scaffolds.1,6,7
Recently we have described improvements to this physics-based approach based on solubility–diffusion theory.8 The theoretical basis and implementation of this model are described in detail elsewhere.8–13 In brief, by treating the membrane as a homogeneous barrier domain that presents a rate-limiting region, the permeability coefficient (Pm) can be estimated from three terms (eqn (1)): the partition coefficient from water to the membrane barrier (Kbarrier), the diffusion coefficient across the membrane barrier (Dbarrier), and the effective length of the membrane barrier (δbarrier).
| (1) |
Building upon prior work and the molecular mechanics-based framework,9–15 the permeability coefficient is computed based on physical principles and does not require prior training on any existing permeability data. The key physical terms are:8
The free energy cost of solvation, where the low dielectric of the membrane is modeled using chloroform as a reference medium.
The permeant’s ionization state, i.e. the free energy cost of neutralizing the molecule.
A size-selective factor that incorporates anisotropic characteristics of the membrane missing from the isotropic organic reference medium (chloroform), which depends on the size of the permeant and the pressure of the membrane environment, essentially penalizing large permeants.
The diffusion coefficient is computed using the simple size-dependent Stokes–Einstein model with the assumption that diffusion across membrane is similar to that in liquid.
The effective barrier length of the membrane is assumed to be identical for all permeants and is thus defined as a constant. This physical permeability model has been employed to predict permeability for diverse compounds sets that include drug-like small molecules and peptidomimetics, producing good correlation with PAMPA and cell-based permeability assays.8
The computational workflow (Fig. 3) closely follows the recently published protocol with a few adjustments made for modeling cyclic peptides.8 Assuming that the permeability rate of charged species would be negligible, only the neutral form of the permeant was evaluated and a state penalty was imposed on those with ionizable functional groups for deionization. Based on the pKa of the substituent, which was predicted by EPIK (Schrodinger) if not known, i.e. for non-standard amino acids,16 the deionization penalty was calculated using the Henderson–Hasselbach equation at pH 7.4. Conformer generation was carried out using the conformational search protocol previously reported by which the torsional space was sampled in a semi-exhaustive manner.1 Libraries of observed backbone and side chain rotamers were employed to enhance sampling efficiency by focusing on the relevant conformational space. The energy of each conformer was subsequently evaluated in water and chloroform, which were represented by implicit solvent models. Conformations within 5 kcal mol−1 from the lowest-energy conformer in either medium were selected to constitute the low-energy conformational “ensemble” and to undergo permeability calculations. The conformational penalty computed in the prior study for small molecules was not calculated here due to the difficulty of accurately assessing conformational entropy of cyclic systems.8 The permeant’s volume required for the size-selective factor was calculated as overlapped spheres defined by van der Waals’ parameters. Finally, the optimal Pm value was determined from the ensemble and the corresponding conformation was identified as the “membranephilic” conformer that permeates across membrane. Conformational search and all energy calculations were performed using the PLOP program and OPLS force field parameters.17,18
Fig. 3.

Computational workflow.
The permeability predictions are summarized in Table 4 and the linear regression model between calculated permeability coefficients and RRCK permeability data is shown in Fig. 4. Overall, the physics-based predictions are in reasonable agreement with the experimental measurements. Compounds 8 (Trp) and 12 (CyclohexylAla) were not included in the regression analysis due to the low % recovery of their measurements, which could be related to hydrophobicity as indicated by their high log D values. Factors that might lead to a low recovery include low solubility, membrane retention, nonspecific binding to plate material, sequestration within the cells, and pipetting variability. These potential issues help explain the overestimated permeability rates by the in silico model.
Table 4.
Permeability and percentage recovery from RRCK assays, and calculated permeability coefficients
| Cpd. | log Papp(RRCK) | % Recovery | z-Scorea | log Pm(calc) |
|---|---|---|---|---|
| 1 | −5.31 | 53.7 | 0.0 | 6.49 |
| 2 | −5.81 | 84.3 | 1.9 | 4.65 |
| 3 | −5.33 | 64.2 | 0.7 | 4.63 |
| 4 | −5.49 | 39.7 | −0.8 | 6.68 |
| 5 | −5.26 | 47.1 | −0.4 | 4.96 |
| 6 | −5.88 | 72.6 | 1.2 | 5.94 |
| 7 | −5.24 | 59.8 | 0.4 | 6.82 |
| 8 | −6.66 | 37.1 | −1.0 | 5.83 |
| 9 | −6.34 | 74.8 | 1.3 | 2.46 |
| 10 | −6.56 | 56.5 | 0.2 | 2.34 |
| 11 | −6.63 | 59.7 | 0.4 | 3.50 |
| 12 | −6.74 | 12.2 | −2.5 | 7.50 |
| 13 | −6.15 | 47.9 | −0.3 | 4.37 |
| 14 | −5.84 | 45.6 | −0.5 | 5.62 |
| 15 | −5.92 | 48.7 | −0.3 | 6.83 |
| 16 | −6.20 | 47.9 | −0.3 | 4.56 |
The z-scores were calculated based on the mean (53.2) and the standard deviation (16.4) of % recovery.
Fig. 4.

Linear regression model between in vitro cell permeability measurements and in silico permeability predictions. Outliers excluded from the regression analysis (8 and 12) are in grey squares.
The physics-based model of permeability is developed upon the idea that “hydrophobicity” is a conformation-based property. As illustrated by our previous work on cyclic and linear peptides,1,6,7,15 permeability could be improved if the permeant could adopt conformations that optimize intramolecular interactions, such as hydrogen bonding, and shield polar functional groups from the lipid environment. In other words, contrary to the conventional expectation, introducing a polar group at a position where it can form a hydrogen bond can in principle improve permeability, or at least not worsen the permeability significantly. For example, the trimethylated 1 has shown higher permeability rates than the unmethylated parent and the per-methylated variant in both the PAMPA and RRCK assays. Another example found in this study is the Thr variant 3, which is the most permeable variant with a hydroxylated substituent. The predicted conformation of 3 suggests that the hydroxyl group could form an intramolecular hydrogen bond with the adjacent backbone carbonyl group (Fig. 5).
Fig. 5.

Predicted membranephilic conformation of 3.
Summary and conclusions
The goal of this study was to evaluate the effect of side chain functionality on the PK properties of a series of compounds based on the same core scaffold. Specifically, we sought to characterize molecules with the structural motif of 1, which spanned a broad range of log D values resulting from polar side chain incorporation. We hypothesized that log D modulation would allow us to define the property space for optimized in vitro clearance and permeability. The ability to introduce polar groups onto an intrinsically orally bioavailable scaffold such as 1 has implications for the design of cyclic peptide-based therapeutics since it can drastically impact structural diversity and property space. Based on in vivo bioavailability data obtained from 5 of the derivatives, there was an obvious trend in the relationship between side chain polarity, permeability and %F. Substitution of Leu with either a positively (Lys) or negatively (Asp) charged residue severely compromised in vitro permeability and ultimately oral bioavailability, whereas the Leu-to-Thr substitution caused only a slight reduction in %F. Surprisingly, removal of the β-methyl to produce the Ser derivative led to a dramatic decrease in both cell permeability and oral bioavailability. This result is consistent with the anecdotal observation that 2° and 3° β-hydroxyl groups are common in lipophilic cyclic peptide natural products (often in the form of non-proteinogenic residues such as β-hydroxyvaline and β-hydroxyleucine), while 1° hydroxyls (e.g., Ser residues) are relatively rare (based on a survey of cyclic peptide natural products in the Antimarin28 database of ~55 000 marine and terrestrial natural products).
Although they were developed independently, the somatostatin analog reported by Kessler and coworkers,30 cyclo-[LPro-LPhe-DTrp(NMe)-LLys(NMe)-LThr-LPhe(NMe)], has the same pattern of N-methylation and shares the same β-hairpin-like backbone conformation as compound 1. The Kessler peptide is similar to our compound 10 in that it contains a single Lys residue, although unlike 10 their peptide had good Caco-2 permeability and fair oral bioavailability (10%) in rat. Its apical-to-basolateral and baso-lateral-to-apical permeability rates were identical in the Caco-2 assay,30 ruling out active transport as a possible mechanism of permeability. When we modeled the Kessler peptide using the above methods (data not shown), we predicted a membrane-associated conformation that is similar to their published NMR structure. However, its low predicted permeability (log Pm = −0.36) was more consistent with the poor permeability and oral bioavailability observed with 10. While the Kessler peptide contains a Thr residue that could form favorable internal hydrogen bonds as illustrated by the Thr variant 3, the peptide also has a cationic Lys residue, which, for 10 and its Nε-Me variant 11, lowers cell permeability dramatically. The Kessler peptide contains three aromatic residues, including a DTrp adjacent to the Lys residue, leading us to speculate that these aromatic residues shield the protonated Lys side chain from the hydrophobic membrane environment through cation–π interactions, contributing to the peptide’s unexpectedly high permeability and oral bioavailability. In future studies we will address this hypothesis by examining the impact of Leu-to-X substitutions on the permeability of 10 where X = Phe, Trp, Tyr, etc.
It may be worth noting that, in general, charged and polar, neutral residues (e.g., Gln, Asn) are rare in N-methylated cyclic peptide natural products.28 Crystal structures and low-dielectric solution structures of these compounds are generally characterized by networks of intramolecular hydrogen bonds, which we hypothesize correspond to their membrane-associated conformations. However, their conformations can change dramatically when bound to their respective biological targets, at least in the few cyclic peptides for which both free and target-bound structures have been reported (e.g., CSA,19–21 luzopeptin,22–25 and YM-25489026,27). In their low-dielectric structures, most of the polar backbone groups in these cyclic peptides are sequestered in intramolecular hydrogen bonds. However, in their bound conformations, the backbones rearrange, forming hydrogen bonds with their targets. Thus, in the de novo design of orally bioavailable cyclic peptides that interact with specific targets, polar contacts may be better achieved through exposure of backbone C=O and amide groups from energetically accessible, high-dielectric conformations, rather than through the display of side chain functional groups on a conformationally defined scaffold. Recent work from the Kessler group has uncovered a relationship between N-methylation and permeability in cyclic peptides that, unlike 1, are not passively permeable in cell-free membrane model systems.29 In these and similar cases, backbone conformation will need to take other modes of transport into account, such as the paracellular route and active uptake by transporters.
Nevertheless, we believe that it is possible to design compounds inspired from 1 which show improved clearance, permeability and bioavailability potential. For example, substitution of Leu with non-natural amino acids (as 15) yielded promising log D values of <3, with a corresponding improvement in in vitro CL and in vitro permeability values relative to compounds containing polar natural amino acids (e.g. 10). This illustrates the significance and utility of novel and diverse unnatural amino acids, as these synthons would enable the systematic interrogation of molecular properties beyond log D, for example pKa.
Supplementary Material
Acknowledgments
We thank Angela C. Doran for technical support of intestinal tissue collection and brush border membrane vesicle preparation. MPJ is a consultant to Schrodinger LLC. Development of the computational model was supported by NIH grant R01-GM086602 to MPJ.
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures for the synthesis of compounds in Tables 1 and 2, including NMR spectra and LCMS traces of pure compounds, experimental details of in vitro metabolism studies, and experimental details of the in vivo pharmacokinetic studies.
Contributor Information
Alan M. Mathiowetz, Email: alan.m.mathiowetz@pfizer.com.
Matthew P. Jacobson, Email: Matt.Jacobson@ucsf.edu.
R. Scott Lokey, Email: slokey@ucsc.edu.
References
- 1.White TR, Renzelman CM, Rand AC, Rezai T, McEwen CM, Gelev VM, Turner RA, Linington RG, Leung SSF, Kalgutkar AS, Bauman JN, Zhang Y, Liras S, Price DA, Mathiowetz AM, Jacobson MP, Lokey RS. Nat Chem Biol. 2011;7:810. doi: 10.1038/nchembio.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di L, Umland JP, Zhang H, Zhang X, Gebhard DF, Lai Y, Federico JJ, 3rd, Davidson RE, Smith R, Reyner EL, Lee C, Feng B, Rotter C, Varma MV, Kempshell S, Fenner K, El-Kattan AF, Lison TE, Troutman MD. J Pharm Sci. 2011;100:4974. doi: 10.1002/jps.22674. [DOI] [PubMed] [Google Scholar]
- 3.Hop CECA, Cole MJ, Davidson RE, Duignan DB, Federico J, Janiszewski JS, Jenkins K, Krueger S, Lebowitz R, Liston TE, Mitchell W, Snyder M, Steyn SJ, Soglia JR, Taylor C, Troutman MD, Umland J, West M, Whalen KM, Zelesky V, Zhao SX. Curr Drug Metab. 2008;9:847. doi: 10.2174/138920008786485092. [DOI] [PubMed] [Google Scholar]
- 4.For the Fa calculation, gut wall metabolism was assumed to be minimal (i.e., Fg = 1) based on the observed low CLp, and Fh was estimated to be 0.93 based on the equation Fh = 1 − hepatic extraction ratio (obtained by dividing the CLp of 4.5 mL min−1 kg−1 by the rat hepatic blood flow of 70 mL min−1 kg−1).
- 5.Sharma P, Varma MVS, Chawla HPS, Panchagnula R. Il Farmaco. 2005;60:884–893. doi: 10.1016/j.farmac.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 6.Rezai T, Yu B, Millhauser GL, Jacobson MP, Lokey RS. J Am Chem Soc. 2006;128:2510. doi: 10.1021/ja0563455. [DOI] [PubMed] [Google Scholar]
- 7.Rezai T, Bock JE, Zhou MV, Kalyanaraman C, Lokey RS, Jacobson MP. J Am Chem Soc. 2006;128:14073. doi: 10.1021/ja063076p. [DOI] [PubMed] [Google Scholar]
- 8.Leung SSF, Mijalkovic J, Borrelli K, Jacobson MP. J Chem Inf Model. 2012;52:1621. doi: 10.1021/ci200583t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diamond JM, Katz Y. J Membr Biol. 1974;17:121. doi: 10.1007/BF01870176. [DOI] [PubMed] [Google Scholar]
- 10.Xiang TX, Anderson BD. Biophys J. 1994;66:561. doi: 10.1016/s0006-3495(94)80833-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiang TX, Anderson BD. J Membr Biol. 1994;140:111. doi: 10.1007/BF00232899. [DOI] [PubMed] [Google Scholar]
- 12.Xiang TX, Anderson BD. J Membr Biol. 1995;148:157. doi: 10.1007/BF00207271. [DOI] [PubMed] [Google Scholar]
- 13.Seelig A. J Mol Neurosci. 2007;33:32. doi: 10.1007/s12031-007-0055-y. [DOI] [PubMed] [Google Scholar]
- 14.Kalyanaraman C, Jacobson MP. J Comput-Aided Mol Des. 2007;21:675. doi: 10.1007/s10822-007-9141-z. [DOI] [PubMed] [Google Scholar]
- 15.Rafi SB, Hearn BR, Vedantham P, Jacobson MP, Renslo AR. J Med Chem. 2012;55:3163. doi: 10.1021/jm201634q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Epik, version 2.2. Schrodinger, L.L.C; New York, NY: 2011. [Google Scholar]
- 17.Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, Friesner RA. Proteins: Struct, Funct, Bioinf. 2004;55:351. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 18.Jorgensen WL, Maxwell DS, TiradoRives J. J Am Chem Soc. 1996;118:11225. [Google Scholar]
- 19.Loosli HR, Kessler H, Oschkinat H, Weber HP, Petcher TJ, Widmer A. Helv Chim Acta. 1985;68:682–704. [Google Scholar]
- 20.Kessler H, Kock M, Wein T, Gehrke M. Helv Chim Acta. 1990;73:1818–1832. [Google Scholar]
- 21.Jin L, Harrison SC. Proc Natl Acad Sci U S A. 2002;99:13522–13526. doi: 10.1073/pnas.212504399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnold E, Clardy J. J Am Chem Soc. 1981;103:1243–1244. [Google Scholar]
- 23.Searle MS, Hall JG, Wakelin PG. Biochem J. 1988;256:271–278. doi: 10.1042/bj2560271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Searle MS, Hall JG, Denny WA, Wakelin LP. Biochemistry. 1988;27:4340–4349. doi: 10.1021/bi00412a022. [DOI] [PubMed] [Google Scholar]
- 25.Zhang XL, Patel DJ. Biochemistry. 1991;30:4026–4041. doi: 10.1021/bi00230a030. [DOI] [PubMed] [Google Scholar]
- 26.Miyamae A, Fujioka M, Koda S, Morimoto Y. J Chem Soc, Perkin Trans. 1989;1 [Google Scholar]
- 27.Nishimura A, Kitano K, Takasaki J, Taniguchi M, Mizuno N, Tago K, Hakoshima T, Itoh H. Proc Natl Acad Sci U S A. 2010;107:13666–13671. doi: 10.1073/pnas.1003553107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.http://www.chem.canterbury.ac.nz/marinlit/marinlit.shtml.
- 29.Beck JG, Chatterjee J, Laufer B, Kiran MU, Frank AO, Neubauer S, Ovadia O, Greenberg S, Gilon C, Hoffman A, Kessler H. J Am Chem Soc. 2012 doi: 10.1021/ja303200d. [DOI] [PubMed] [Google Scholar]
- 30.Biron E, Chatterjee J, Ovadia O, Langenegger D, Brueggen J, Hoyer D, Schmid HA, Jelinek R, Gilon C, Hoffman A, Kessler H. Angew Chem, Int Ed. 2008;47:2595–2599. doi: 10.1002/anie.200705797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.