

Abstract
A highly regio-, diastereo-, and enantioselective allylic alkylation reaction of 3-monosubstituted oxindoles catalyzed by molybdenum is described. The reaction is affected by the electronic and steric variations of the nucleophile. The use of appropriate N-protecting group is particularly important for achieving high regio- and diastereoselectivity. Products from this reaction, containing vicinal quaternary-tertiary stereogenic centers, are valuable synthetic intermediates and should find utility in alkaloid synthesis.
Keywords: molybdenum, oxindoles, asymmetric allylic alkylation, quaternary center
Introduction
Asymmetric allylic alkylations (AAA) hold much promise for simplifying asymmetric total syntheses and thus new developments represent an important goal.[1] The evolution of the decarboxylation asymmetric allylic alkylation (DAAA) is one such illustration by allowing previously difficult asymmetric alkylations of ketone enolates to be more general.[2] Furthermore, development of systems employing Ir,[3] Cu,[4] and Mo[5] as asymmetric catalysts has significantly expanded the scope of the nucleophile and electrophile. Despite all these advancements, very few allylic alkylation reactions allow the simultaneous stereocontrol at both the nucleophile and the allylic electrophile, especially when a quaternary center is also created in the process. Development of such a reaction is highly desirable, as few methods exist for the asymmetric construction of vicinal quaternary or quaternary-tertiary stereogenic centers that are abundantly present in biologically important molecules. [6] We have reported that asymmetric alkylation of azlactones[5b] and oxalactims[5c] in the presence of a chiral molybdenum catalyst proceeded with excellent stereoselectivity at both the nucleophile and electrophile. However, the extension of this technology into other nucleophiles has been very challenging due to the lack of understanding of the origin of the selectivity.
The present study concerns the use of oxindoles as nucleophiles in the molybdenum reaction. The 3,3′ disubstituted oxindoles are important targets for asymmetric synthesis because of their significance in biological applications. [7] A number of naturally occurring oxindole alkaloids that exhibit interesting medicinal properties contain chiral quaternary centers at the 3 position of the oxindole. Since Julian first demonstrated that the pyrrolidinoindoline core of physostigmine can be prepared by a reductive ring closure of a 3,3′ disubstituted oxindole in 1935, [8] many synthetic efforts towards the oxindole and pyrrolidinoindoline natural products have incorporated oxindoles into their synthetic strategies.[9] However, the asymmetric entry into this important structural motif has been primarily achieved by resolution or diastereoselective methods using chiral auxiliaries or existing chirality. Only very recently have a few enantioselective methods for the synthesis of chiral 3, 3′ disubstituted oxindoles been reported. [10]
The recent success of Mo-catalyzed AAA reactions with azlactones and oxalactims prompted us to investigate whether structurally similar oxindole nucleophiles would also perform well in such reactions. The enolates of azlactones, oxalactims, and oxindoles are all stabilized by aromatic resonance and are therefore more stable than the enolates of the corresponding lactams and lactones. The cyclic nature of these structures not only provides structural rigidity but also alleviates some of the steric difficulties when alkylating. The use of mono-substituted oxindoles as nucleophiles should be more difficult, however, as it involves formation of a quaternary center as opposed to tertiary carbon centers in azlactones and oxalactims due to the fact that one of the substituents is a heteroatom in those latter cases. The following discussion details the development of a Mo-catalyzed regio-, diastereo- and enantioselective allylic alkylation reaction with 3-monosubstituted oxindoles as nucleophiles and our discovery that the high selectivity in the molybdenum reaction can be attained via simple and rational modification of the structure of the nucleophile.
Results and Discussion
Scope of the reaction with 3-aryloxindole as the nucleophile
Given that the steric and electronic properties of the nucleophile can significantly impact the reactivity and selectivity of enolate alkylation reactions, we began our study by looking at two sterically and electronically distinct oxindoles (Table 1). Encouragingly, both 3-methyl (1a) and 3-phenyloxindole (1b) reacted to afford the alkylated products smoothly at room temperature (entry 1 and 2). However, the more stabilized 3-phenyloxindole (1b) gave much better selectivity. Further optimizations of ligand, base and reaction temperature revealed that among the ligands screened, the standard and most easily available bispicolinamide ligand L1 is the best ligand for the reaction with 1b (entry 2–7); the selectivity of the reaction is not sensitive to temperature from 0 °C to 60 °C (entry 9–11); and the use of Na base provided the best regio-, and diastereoselectivity, although the ee is slightly lower than the reaction using Li or K base (entry 8). Thus, the use of ligand L1 (15% mol), Mo(C7H8)(CO)3 (10% mol), and NaOtBu (1.1 equiv) in THF at room temperature or 60 °C (entry 11) became our standard conditions for the alkylation of 3-aryloxindoles.
Table 1.
Screening of reaction conditions
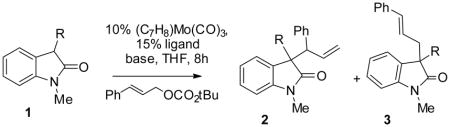
| |||||||
|---|---|---|---|---|---|---|---|
| entry | R | Ligand | Base | Temp | b/l | dr | ee% |
| 1 | Me(2a | L1 | LiOtBu | rt | 2:1 | 1.2:1 | --- |
| 2 | Ph(2b) | L1 | LiOtBu | rt | 4:1 | 8:1 | 95 |
| 3 | Ph | L2 | LiOtBu | rt | 2.5:1 | 5.5:1 | --- |
| 4 | Ph | L3 | LiOtBu | rt | 2.5:1 | 5:1 | --- |
| 5 | Ph | L4 | LiOtBu | rt | 4:1 | 7:1 | --- |
| 6 | Ph | L5 | LiOtBu | rt | 1.2:1 | 3:1 | --- |
| 7 | Ph | L6 | LiOtBu | rt | 5:1 | 4:1 | --- |
| 8 | Ph | L1 | KOtBu | rt | 10:1 | 5:1 | 94 |
| 9 | Ph | L1 | NaOtBu | rt | 16:1 | 9:1 | 92 |
| 10 | Ph | L1 | NaOtBu | 0 °C | 16:1 | 9:1 | 92 |
| 11 | Ph | L1 | NaOtBu | 60 °C | 16:1 | ||
The branch/linear ratio(a:b) and the diastereoselectivity of the branched product (dr) was determined by 1H NMR integration of the crude reaction mixture
With the optimized conditions in hand, we next looked to examine the scope of the reaction with regard to the nucleophile (Table 2). The steric size of the N-protecting group does not seem to be important as methoxymethyl (2a) and benzyl (2b) gave essentially identical results as methyl. The methoxymethyl and benzyl group are valuable as they can be easily removed to afford unprotected oxindoles.[11] We focused our studies on N-methyl compounds, however, as oxindoles bearing methoxymethyl and benzyl groups gave more complicated proton NMR for the precise determination of regio- and diastereoselectivity, and are more difficult to separate on chiral HPLC for the determination of ee.
Table 2.
Scope of the 3-aryloxindoles
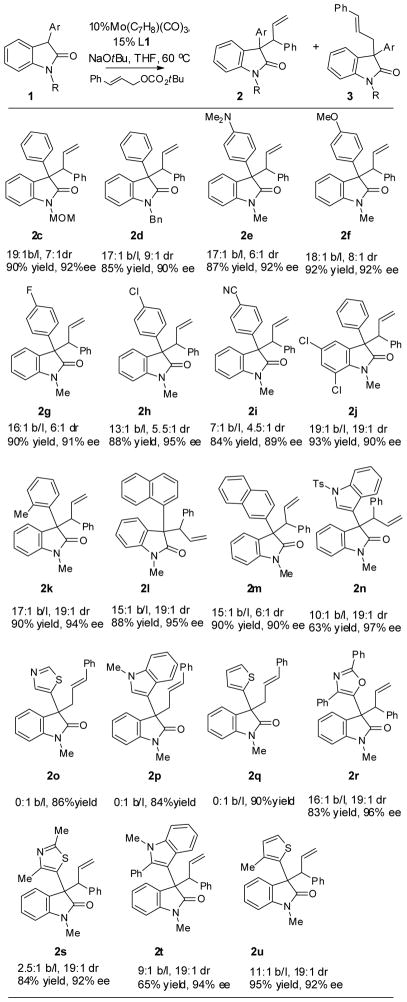
|
The reaction was performed with 0.1 mmol of oxindole 1 and 0.12 mmol of cinnamyl carbonate. The branch/linear ratio(a:b) and the diastereoselectivity of the branched product(dr) was determined by 1H NMR integration of the crude reaction mixture. The ee% was determined by chiral HPLC.
Given that 3-phenyloxindole gave much better selectivity than the more nucleophilic 3-methyoxindole (entry 1 and 2, Table 1), we hypothesized that the selectivity of the reaction is inversely related to the nucleophilicity of the oxindoles. If this hypothesis is correct, oxindoles bearing electron-rich 3-aryl substituents, which are more nucleophilic, should show diminished selectivity in the Mo reaction, compared to the electron-neutral 1b. On the other hand, electron-deficient 3-aryloxindoles should provide enhanced selectivity. Surprisingly, while the selectivity of the reaction changed significantly as we systematically modified the electronics of the 3-aryl substituents on the oxindoles, the trend is not what we had predicted. The regio-, and diastereoselectivity of the reaction decreased as more electron-withdrawing para-substituents were placed on the phenyl ring (2g-2i). Electron-donating groups such as dimethylamino (2e) and methoxy (2f) group, however, had little effects on the selectivity. In all cases, the ee and yield of the reaction showed little sensitivity to the electronic variations.
Steric demand of the nucleophile can have a significant effect on the regioselectivity of the Mo-catalyzed allylic alkylation reaction.[12] In general, small nucleophiles are more selective toward bond formation at the more hindered internal position of the π –allyl, compared to the more bulky ones. Thus, at the outset, we expected that the more compact five-membered heterocycle-substituted oxindoles would give better regioselectivity, favoring the branched product, while oxindoles bearing bulky aryl goups would be less branch-selective. In contrast to this expectation, the bulky 2-tolyl (2k) and 1-naphthyl (2l) substituted oxindoles gave excellent selectivity for the branched products, while smaller thienyl (2q), thiazoyl (2o), and indolyl (2p) substituted ones gave exclusively linear products. Furthermore, installing extra steric bulk on these heterocycles reversed the regioselectivity to give branched products with excellent selectivity (2s, 2t, and 2u). It is also worth noting that bulkier oxindoles also gave better diastereoselectivity (2l vs. 2m, 2k vs. 2b).
New Mechanistic Proposal
The puzzling trend observed in the above electronic and steric studies prompted us to address the question of what is the origin of stereocontrol in the Mo reaction. Previous mechanistic studies have revealed that the Mo reaction is an inner sphere process where the selectivity determining step is the reductive elimination from a π-allylmolybdenum enolate.[13] To evaluate why different nucleophiles behave differently in the Mo reaction, we need to better understand the reductive elimination step.
We were aware of studies of reductive elimination involving Pd enolate complexes and that different bonding modes of Pd enolate can affect the reactivity and selectivity of the reductive elimination. Specifically, Culkin and Hartwig[14, 15] have shown that depending on the particular electronic and steric circumstances, a C-bound or an O-bound Pd enolate isomer may predominate; the C-bound and O-bound isomers react differently; and that the stability and reactivity of the enolates are not controlled by the pKa of the carbonyl compounds.
We consider that the Mo AAA reaction may also involve both or either O-bound and C-bound molybdenum enolate complexes, both of which have been structurally characterized. For example, π-allylmolybdenum complex 4 is an O-bound enolate,[16] but CpMo complex 5 is C-bound. Interestingly, a high-valent molybdenum complex 6 also adopts a C-bound structure as a result of favorable chelation with an α-N atom.[17]
A reaction mechanism involving divergent reaction modes of O-bound and C-bound Mo enolate complexes can rationalize the results from our studies with oxindoles as well as other nucleophiles (Scheme 2). First of all, aromatic stabilization favors O-bound enolate and a favorable “Claisen-like” transition state allows the more substituted allyl terminus to bond to the sp2 carbon of the O-enolate to form the branched product (path A). The generally better regioselectivity observed in the reaction of azlactone than that of oxindole can be ascribed to stronger aromatic stabilization in the case of azlactone. Tuning the electronic and steric property of the oxindole nucleophile, however, should influence the equilibrium ratios of the two enolate isomers and which isomer reacts to give the products. Sterically, a larger aryl group should disfavor the crowded C-bound enolate and favor the O-bound enolate structure, which lead to the normally preferred branched product. On the other hand, the more compact five-membered heterocycle substituted oxindoles should accommodate the C-bound enolate more readily. The steric crowding of a direct reductive elimination to a quaternary sp3 center only allows bonding to the less hindered primary allyl terminus in this case (path B). Electronically, electron-withdrawing 3-aryl substituents should stabilize both enolate complexes and slow down their inter-conversion. Hence, we see the regioselectivity of the reaction deteriorates as more electron-withdrawing groups were introduced.
Scheme 2.

O-bound vs. C-bound Mo enolate
Attempts to isolate and characterize the molybdenum enolate complex by preforming the π-allylmolybdenum and then adding the oxindole nucleophile at low temperature were not successful. Even at −78 °C, we could not spectroscopically detect the presence of any Mo enolate intermediate and only observed the formation of the alkylated product. Interestingly, the regio- and diastereoselectivity of the reaction at −78 °C is much lower than that at room temperature (Table 3), which we attributed to a slower interconversion of the Mo enolate isomers at low temperature that, in turn, lead to unselective reaction via the C-bound molybdenum enolate. This result prompted us to examine whether promoting the equilibration between the enolate isomers would move the reaction toward a Curtin-Hammett type situation and favor reductive elimination via the less hindered O-bound enolate to give the branched product. We reasoned that the use of a more electron-rich ligand should slow down reductive elimination and thus allowing more time for the Mo enolate isomers to equilibrate. Indeed, the use of a bis-methoxy ligand L2 improved the regioselectivity of the reactions with substrate 2i and 2s significantly (Table 4).[18]
Table 3.
Temperature effect on selectivity
| |||||
|---|---|---|---|---|---|
| entry | temp | yield | b/l | dr | ee% |
| 1 | rt | 89% | 10:1 | 8:1 | 92 |
| 2 | −78 | 40% | 1:1 | 2:1 | 91 |
Table 4.
Ligand effects on the regioselectivity.
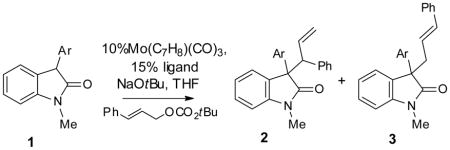
| |||||
|---|---|---|---|---|---|
| entry | Ar | Ligand | b/l | dr | ee % |
| 1 | 4-cyano-phenyl (2l) | L1 | 7:1 | 4.5:1 | 89 |
| 2 | 4-cyano-phenyl (2l) | L2 | 18:1 | 4:1 | 92 |
| 3 | 2,4-dimethyl-5-thiazoyl(2s) | L1 | 2.5:1 | 19:1 | 92 |
| 4 | 2,4-dimethyl-5-thiazoyl(2s) | L2 | 16:1 | 19:1 | 93 |
Scope of the electrophile
The Mo-catalyzed AAA reaction works well with a diverse array of 3-monosubstituted linear allylic electrophiles, giving good to excellent regio-, diastereo-, and enantioselectivity (Table 5). Further, an alkyl substituted allylating agent also gave good selectivity (entry 6).
Table 5.
Scope of the allylic electrophile
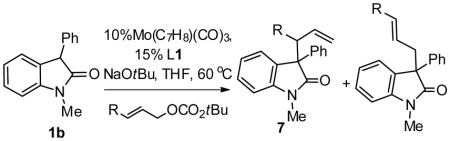
| |||||
|---|---|---|---|---|---|
| entry | R | yield | b/l | dr | ee% |
| 1 | 3,4-Cl2-C6H3 | 87% | 6:1 | 5.5:1 | 89 |
| 2 | 4-OTBS-C6H4 | 90% | 19:1 | 8:1 | 89 |
| 3 | 2-furyl | 92% | 12:1 | 6:1 | 91 |
| 4 | 2-thiophenyl | 88% | 13:1 | 8:1 | 90 |
| 5 | 2NHBocC6H4 | 89% | 19:1 | 19:1 | 93 |
| 6 | 2-(E)-butene | 84% | 6:1 | 6:1 | 91 |
The reaction was performed with 0.1 mmol of oxindole 1b and 0.12 mmol of cinnamyl carbonate. The branch/linear ratio (b/l) and the diastereoselectivity of the branched product(dr) was determined by 1H NMR integration of the crude reaction mixture. The ee% was determined by chiral HPLC.
The use of racemic branched allylic electrophiles in the AAA reaction is desirable as the corresponding allylic alcohol is easily prepared by addition of vinyl Grignard to an aldehyde. However, to prepare enantiopure product from racemic starting material requires either a kinetic resolution, where half of the starting material is lost by default, or a dynamic kinetic asymmetric transformation (DYKAT), where equilibration of either the starting material or a reaction intermediate allows the reaction to proceed to completion (Scheme 3).[19] Equilibration of the π –allylmolybdenum complex is known to be sufficiently fast at low concentration of nucleophile to provide synthetically useful DYKAT (k1 ≫ k4).[20] In light of the ligand effect on the regioselectivity of the reaction discussed above, we were intrigued by the possibility that an electron-rich ligand could promote a DYKAT process without modifying the concentration of the nucleophile. Electron-rich ligand stabilizes the η1-allylmetal complex thus facilitating equilibration of π–allylmolybdenum complex. In addition, the rate of nucleophilic attack and reductive elimination should both be slower with electron-rich ligand. The beneficiary effect of the electron-rich ligand was demonstrated by comparing L1 and L2 in reactions with several branched allylic carbonate (Table 6). In all cases, much improved selectivity, especially enantioselectivity, was observed with the more electron-rich ligand L2.
Scheme 3.

Dynamic Kinetic Resolution of branched allylic electrophiles
Table 6.
Mo reaction with branched electrophiles
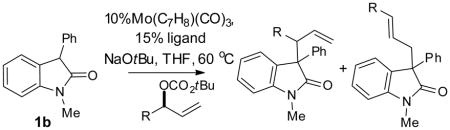
| |||||||
|---|---|---|---|---|---|---|---|
| entry | R | Ligand | yield | b/l | dr | ee% | product |
| 1 | Ph | L1 | 88% | 10:1 | 6:1 | 82 | 2b |
| 2 | Ph | L2 | 85% | 15:1 | 9:1 | 93 | 2b |
| 3 | 3,4Cl2C6H3 | L1 | 90% | 6:1 | 5:1 | 80 | 7a |
| 4 | 3,4Cl2C6H3 | L2 | 87% | 8:1 | 6:1 | 89 | 7a |
| 5 | 4-OTBSC6H4 | L1 | 89% | 12:1 | 6.5:1 | 81 | 7b |
| 6 | 4-OTBSC6H4 | L2 | 89% | 15:1 | 8:1 | 89 | 7b |
The reaction was performed with 0.1 mmol of oxindole 1b and 0.12 mmol of allylic carbonate. The branch/linear ratio (b/l) and the diastereoselectivity of the branched product(dr) was determined by 1H NMR integration of the crude reaction mixture. The ee% was determined by chiral HPLC.
Optimization of 3-alkyloxindoles as nucleophiles
We have discussed earlier that the reaction with the synthetically very useful 1,3-dialkyloxindoles produced a mixture of products with poor selectivity (entry 1, Table 1). On the basis of the assumption that promoting a reaction pathway proceeding through O-bound molybdenum enolate should improve the selectivity, we examined the effect of chelating groups on the nitrogen of the 3-alkyloxindoles with the expectation that the formation of a bis-chelated enolate may disfavor the formation of C-bound enolate (Table 7). We were encouraged that the use of a 2-pyridyl group on the nitrogen improved the regioselectivity of the reaction to a respectable level, although the dr of the reaction remains low (entry 3). Gratifyingly, oxindoles that contain chelating N-carbamoyl groups all gave greatly improved regioselectivity favoring the branched product. Furthermore, the diastereoselectivity was found to correlate inversely with the steric size of the N-carbamoyl group (entry 4–6), and the smallest N-Moc (N-methoxycarbonyl) oxindole provided the best diastereoselectivity of 7:1. This correlation between the steric size of the N-substituent and the diastereoselectivity of the reaction supports the proposed formation of a bis-chelating molybdenum enolate intermediate that brought the substituent on the nitrogen closer to the chiral catalyst. As discussed earlier (Table 2), the reaction with N-alkyl-3-aryloxindole, where only mono-chelation is possible, showed no sensitivity to the steric size of the substituent on the nitrogen. The Moc group is labile under the reaction conditions, and the deprotected amide can act as a catalyst poison.[21] Subsequent optimization revealed that the use of BSA as the stoichiometric base was compatible with the Moc group and afforded consistent conversion with only 2.5% catalyst loading (entry 8).
Table 7.
Screening of chelating groups
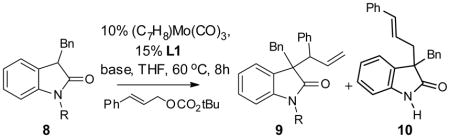
| ||||||
|---|---|---|---|---|---|---|
| entry | R | Base | yield | b/l | dr | ee % |
| 1 | Me (8a) | NaOtBu (1 eq) | 92% | 2:1 | 1.2:1 | 92 |
| 2 | OAc(8b) | NaOtBu (1 eq) | ||||
| 3 | 2-pyridyl | NaOtBu (1 eq) | 89% | 14:1 | 1:1 | |
| 4 | CO2tBu | NaOtBu (1 eq) | 85% | 12:1 | 3:1 | 98 |
| 5 | CO2Et(8e) | NaOtBu (1 eq) | 86% | 15:1 | 5:1 | 99 |
| 6 | CO2Me(8f) | NaOtBu (1 eq) | 60% | 15:1 | 7:1 | 98 |
| 7 | CO2Me(8f) | NaOtBu (0.1 eq) BSA (1.2 eq) |
88% | 15:1 | 7:1 | 98 |
| 8* | CO2Me(8f) | NaOtBu (0.1 eq) BSA (1.2 eq) |
83% | 15:1 | 7:1 | 98 |
performed with 2.5% (C7H8)Mo(CO)3 and 3.75% L1
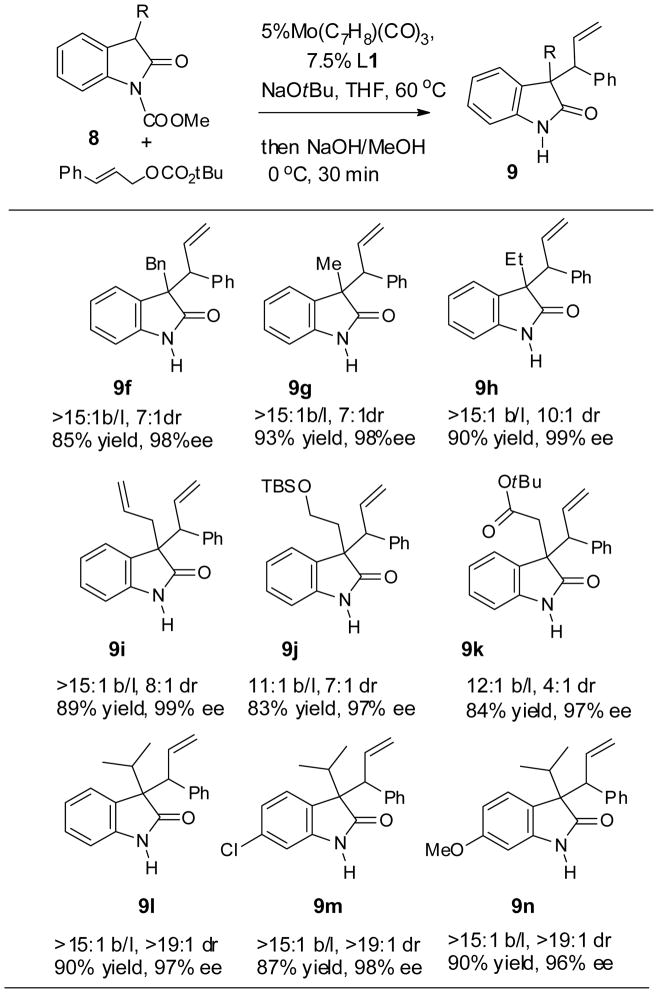
Having identified the key parameters of the catalytic system, the scope of the molybdenum reaction was next explored, in which a variety of 3-alkyloxindoles were efficiently and selectively alkylated (Table 8). Substitution on the oxindole aromatic ring does not affect the reaction (9l–9n). Functional groups such as allyl (9i), TBS ether (9j) and ester groups (9k) at the 3 positions of the oxindoles are tolerated. Notably, increasing the size of the 3-alkyl group increased the diastereoselectivity significantly, while maintaining excellent levels of regio- and enantioselectivity (9g vs. 9h vs. 9l). The largest N-isopropyl substituted oxindoles (9l–9n), which are difficult substrates in several other asymmetric oxindole syntheses, gave the branched product as essentially a single enantiomer.[22] For the convenience of handling and purification, the N-Moc group was in-situ removed with 1 eq. of NaOH in methanol upon completion of the alkylation reaction.
Table 8.
Scope of the 3-alkyloxindoles
|
The reaction was performed with 0.2 mmol of oxindole 8 and 0.24 mmol of cinnamyl carbonate. The branch/linear ratio (b/l) and the diastereoselectivity of the branched product(dr) was determined by 1H NMR integration of the crude reaction mixture. The ee% was determined by chiral HPLC.
The observation that the combination of a small N-carbamoyl group and a large 3-alkyl group provided the best diastereoselectivity is consistent with our analysis that the reaction proceeds through a bis-chelating O-bound molybdenum enolate intermediate, as shown in Scheme 4. Path A is favored as it avoids the steric interaction between the 3-alkyl group and the ligand. Reductive elimination from intermediate A via a “Claisen-like” transition state provides the branched product. However, as the size of the carbamoyl group increases, the steric congestion between the N-carbamoyl group and the ligand disfavors path A and leads to deterioration of the diastereoselectivity. The proposed transition state is in agreement with the stereochemical outcome of the reaction that was established by X-ray crystallographic analysis of compound 9g.
Scheme 4.

Model for stereoselectivity and X-ray structure of 9g
Conclusions
In conclusion, we have developed a molybdenum-catalyzed allylic alkylation reaction with oxindoles containing either an aryl or an alkyl substituent the 3 position of the oxindole that proceeds with high regio-, diastereo-, and enantioselectivity. The products of the reaction are highly useful intermediates for the preparation of oxindole and indoline natural products containing adjacent quaternary-tertiary stereocenters that are difficult to access via other methods. In the case of 3-aryloxindoles, the selectivity of the reaction is sensitive towards the steric and electronic variation of the aryl group. However, the use of an electron-rich ligand can improve the regioselectivity. In the case of 3-alkyloxinoles, the ability to influence regio- and diastereoselectivity of the molybdenum reaction via chelation control is a significant finding. Furthermore, a predictive model involving the intermediacy of C vs. O enolates has been developed and should have broad applicability with other classes of nucleophiles. In particular, we are in the process of identifying other nucleophiles that are capable of chelation control with the molybdenum catalyst.
Experimental Section
All reactions were carried out in oven-dried flasks or pyrex test tubes under a positive pressure of argon. Anhydrous solvents were obtained from elution through alumina column, except for THF, which was distilled from Na-benzophenone ketyl. All reagents were purchased commercially and used without further purification unless stated otherwise. TLC was performed on precoated glass plates (Merck), flash chromatography with silica gel 60, 230–400 mesh. Enantiomeric excesses were determined by HPLC on Tsp Spectra series P100/UV100 apparatus with a chiral stationary phase (see details where applies). Melting points (uncorrected) were determined in open capillary tubes using a Thomas-Hoover apparatus. 1H-NMR (0 ppm as internal standard) and 13C-NMR (77 ppm as internal standard) spectra were recorded on Varian Mercury 400 (400 MHz for 1H-NMR) and on Unity Inova 500 (500 MHz for 1H-NMR). IR spectra (cm−1) were obtained with a Perkin-Elmer FT-IR Paragon 500 spectrometer, either using neat sample on NaCl pad (for oils and liquids), or preparing a KBr pellet.
General procedure for Mo catalyzed AAA reactions with 3-aryloxindoles
1-methyl-3-phenyl-3-(1-phenylallyl)indolin-2-one (2b)
To an oven-dried test tube equipped with a magnetic stir bar was added ligand L1 (4.8 mg, 0.015 mmol) and (C7H8)Mo(CO)3 (2.7 mg, 0.01 mmol). The test tube was sealed with a rubber septum and THF (freshly distilled and degassed, 0.3 mL) was added via syringe. The test tube was then placed in a pre-heated oil bath at 60 °C for 5 min until the solution turns deep purple. The reaction was then allowed to cool to rt. A second oven-dried test tube containing 0.1 mmol of the oxindole was transferred to the dry box and to it sodium tert-butoxide (0.11mmol, 11mg) was added. The test tube was sealed, taken out of the dry box, and to it was added degassed THF (0.7 mL) at room temperature. The resulting solution was stirred for 5 min before the solution of the active catalyst solution (0.3 mL) was added via cannula, followed by the addition of cinnamyl tert-butyl carbonate (0.12 mmol) via syringe. The reaction was then stirred at 60 °C under Ar for 2 h at which point TLC indicated complete consumption of the starting oxindole. The reaction was then concentrated and column purification (10% EtOAc-pentane) afforded the desired product as a mixture of regio- and diastereomers. (31 mg, 92%, 16:1 b/l, 9:1 dr, 92% ee) Chiral HPLC, AD-column, flow rate 0.7 mL/min, 90:10 heptane / i-PrOH, tA (minor) = 8.387 min, tB (major) = 9.556 min. IR (film) λmax/cm−1: 3058, 2926, 1708, 1610, 1493, 1470, 1373, 1349, 1255, 1131, 1089, 1025, 923, 757, 689. 1H NMR (500 MHz, CDCl3) major diastereomer: δ 7.61-7.58 (m, 2H), 7.34-7.27 (m, 2H), 7.26-7.21 (m, 2H), 7.08-6.98 (m, 5H), 6.89-6.87 (m, 2H), 6.69(d, J=7.5, 1H), 6.06 (ddd, J= 9.5, 10, 17, 1H), 5.18 (dm, J = 17, 1H), 5.08 (dd, J = 1.5, 10, 1H), 4.48(d, J= 9.5, 1H), 3.09 (s, 3H). 13C NMR (125 MHz, CDCl3) major diastereomer: δ 177.1, 143.5, 139.0, 138.0, 135.8, 129.5, 129.3, 128.2, 128.1, 128.0, 127.3, 127.2, 126.7, 126.4, 121.7, 118.5, 107.9, 60.7, 57.2, 26.1 HRMS—EI (m/z): calcd for C24H21NO: 339.1623; found: 339.1629.
General procedure for Mo catalyzed AAA reactions with 3-alkyloxindoles
3-methyl-3-(1-phenylallyl)indolin-2-one(9g)
To an oven-dried test tube equipped with a magnetic stir bar was added Ligand L1 (4.8 mg, 0.015 mmol) and (C7H8)Mo(CO)3 (2.7 mg, 0.01 mmol). The test tube was sealed with a rubber septum and THF (freshly distilled and degassed, 0.3 mL) was added via syringe. The test tube was then placed in a pre-heated oil bath at 60 °C for 5 min until the solution turns deep purple. The test tube was then taken out of the oil bath and let cool to rt. A second test tube containing oxindole 8g (40 mg, 0.2 mmol) was dissolved in 1 mL dry THF and cooled to 0 °C. A freshly prepared solution of sodium tert-butoxide (4 mg, 0.02 mmol) in 0.3 mL THF was added dropwise. The resulting solution was warmed to rt and BSA (0.24 mmol) was added via syringe. The resulting solution stirred for 5 min before the solution of the active catalyst solution was added via cannula, followed by the addition of cinnamyl tert-butyl carbonate (0.24 mmol) via syringe. The resulting solution was stirred at 60 °C under Ar for 6 h until TLC indicates complete consumption of the starting oxindole. The reaction mixture was then cooled to 0 °C, and a solution of NaOH (0.25 mL, 1M in MeOH) was added dropwise. The resulting solution was stirred for 30 min and the reaction was then quenched with H2O (10 mL). The mixture was then extracted with EtOAc (3×10 mL), dried over sodium sulfate, concentrated, and purified through silica gel (15% EtOAc-pentane) to give 9g as a clear oil (50 mg, 93% yield, 98% ee, 15:1 b/l, 7:1 dr). Chiral HPLC, OD-column, flow rate 1.0 mL/min, 97:3 heptane / i-PrOH, tA (minor) = 13.346 min, tB (major) = 15.047 min. IR (film) λmax/cm−1: 3210, 3081, 3061, , 3030, 2975, 2887, 1707, 1619, 1471, 1452, 1334, 1230, 1202, 1123, 994, 917, 753, 726, 701; 1H NMR (500 MHz, CDCl3): δ 8.50(s, 1H), 7.25-7.14 (m,6H), 7.04-6.97(m, 2H), 6.84(d, J = 7.5, 1H), 6.19-6.07 (m, 1H), 5.21 (dm, J =17, 1H), 5.08 (dm, J = 10, 1H), 3.73(d, J = 10, 1H), 1.38(s, 3H; 13C NMR (125 MHz, CDCl3) δ 182.3, 140.5, 139.3, 135.7, 132.5, 129.2, 127.8, 126.8, 124.6, 121.8, 117.9, 109.5, 57.0, 52.5, 22.2. HRMS(MH+): calcd for C18H18NO: 264.1388; found: 264.1379
Supplementary Material
Scheme 1.

Molybdenum enolate
Acknowledgments
We thank the National Institutes of Health (NIH-13598) for their generous support of our program. Y. Z. thanks Amgen for a graduate fellowship.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.For recent reviews on asymmetric allylic alkylation reactions, see: Trost BM. J Org Chem. 2004;69:5813. doi: 10.1021/jo0491004.Trost BM. Chem Phar Bull. 2002;38:1. doi: 10.1248/cpb.50.1.
- 2.Stoltz BM, Mohr JT. Chem Asian J. 2007;2:1476. doi: 10.1002/asia.200700183.And references therein. Trost BM, Xu J. J Am Chem Soc. 2005;127:2846. doi: 10.1021/ja043472c.Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044. doi: 10.1021/ja044812x.
- 3.a) Takeuchi R. Synthesis. 2006:3349. [Google Scholar]; (b) Bartels B, Helmchen G. Chem Comm. 1999:741–742. [Google Scholar]; (c) Ohmura T, Hartwig JF. J Am Chem Soc. 2002;120:15164. doi: 10.1021/ja028614m. [DOI] [PubMed] [Google Scholar]; (d) Tissot-Croset K, Polet D, Alexakis A. Angew Chem Int Ed. 2004;43:2426. doi: 10.1002/anie.200353744. [DOI] [PubMed] [Google Scholar]
- 4.a) Falciola CA, Alexakis A. Eur J Org Chem. 2008;22:3755. [Google Scholar]; b) Luchaco-Cullis C, Murphy KE, Hoveyda AH. Angew Chem Int Ed. 2001;40:1456. doi: 10.1002/1521-3773(20010417)40:8<1456::AID-ANIE1456>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 5.a) Trost BM, Hachiya I. J Am Chem Soc. 1998;120:1104–1105. [Google Scholar]; b) Trost BM, Dogra K. J Am Chem Soc. 2002;124:14320. doi: 10.1021/ja020290e. [DOI] [PubMed] [Google Scholar]; c) Trost BM, Dogra K, Franzini M. J Am Chem Soc. 2004;126:1944. doi: 10.1021/ja031539a. [DOI] [PubMed] [Google Scholar]; d) Trost BM, Zhang Y. J Am Chem Soc. 2007;129:14548. doi: 10.1021/ja0755717. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Belda O, Moberg C. Acc Chem Res. 2004;37:159. doi: 10.1021/ar030239v. [DOI] [PubMed] [Google Scholar]
- 6.For recent examples of asymmetric synthesis of natural products containing vicinal quaternary or quaternary-tertiary stereogenic centers, see: Overman LE, Paone DV, Stearns BA. J Am Chem Soc. 1999;121:7702.Trost BM, Tang W. J Am Chem Soc. 2002;124:14542. doi: 10.1021/ja0283394.Boezio AA, Jarvo ER, Lawrence BM, Jacobsen EN. Angew Chem Int Ed Engl. 2005;44:6046. doi: 10.1002/anie.200502178.Boeckman RK, Shao P, Wrobleski ST, Boehmler DJ, Heintzelman GR, Barbosa AJ. J Am Chem Soc. 2006;128:10572. doi: 10.1021/ja0581346.Enquist JA, Stoltz BM. Nature. 2008;453:1228. doi: 10.1038/nature07046.
- 7.For some representative biologically important targets containing oxindole structures, see: Greig NH, Pei X-F, Soncrant TT, Ingram DK, Brossi A. Med Res Rev. 1995;15:3–31. doi: 10.1002/med.2610150103.Trost BM, Cramer N, Bernsmann H. J Am Chem Soc. 2007;129:3086–3087. doi: 10.1021/ja070142u.Lin H, Danishefsky SJ. Angew Chem Int Ed Engl. 2003;42:36. doi: 10.1002/anie.200390048.Wearing XZ, Cook JM. Org Lett. 2002;24:4237. doi: 10.1021/ol020170b.
- 8.Julian PL, Pikl J. J Am Chem Soc. 1935;57:755–757. [Google Scholar]
- 9.Lebsack AD, Link JT, Overman LE, Steans BA. J Am Chem Soc. 2002;124:9008–9009. doi: 10.1021/ja0267425. [DOI] [PubMed] [Google Scholar]
- 10.a) Trost BM, Frederiksen MU. Angew Chem, Int Ed. 2005;44:308. doi: 10.1002/anie.200460335. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Zhang Y. J Am Chem Soc. 2006;128:4590. doi: 10.1021/ja060560j. [DOI] [PubMed] [Google Scholar]; (c) Shaw SA, Aleman P, Vedeis E. J Am Chem Soc. 2003;125:13368. doi: 10.1021/ja037223k. [DOI] [PubMed] [Google Scholar]; (d) Hills ID, Fu GC. Angew Chem, Int Ed. 2003;42:3921. doi: 10.1002/anie.200351666. [DOI] [PubMed] [Google Scholar]; (e) Hamashima Y, Suzuki T, Takano H, Shimura Y, Sedeoka M. J Am Chem Soc. 2005;127:10164. doi: 10.1021/ja0513077. [DOI] [PubMed] [Google Scholar]; (f) Ishimaru T, Shibata N, Horikawa T, Yasuda N, Nakamura S, Toru T, Shiro M. Angew Chem, Int Ed. 2008;47:4157. doi: 10.1002/anie.200800717. [DOI] [PubMed] [Google Scholar]; (g) Ishimaru T, Shibata N, Nagai J, Nakamura S, Toru T, Kanemasa S. J Am Chem Soc. 2006;128:16488. doi: 10.1021/ja0668825. [DOI] [PubMed] [Google Scholar]; (b) Sano D, Nagata K, Itoh T. Org Lett. 2008;10:1593. doi: 10.1021/ol800260r. [DOI] [PubMed] [Google Scholar]; (h) Ogawa S, Shibata N, Inagaki J, Nakamura S, Toru T, Shiro M. Angew Chem, Int Ed. 2007;46:8666. doi: 10.1002/anie.200703317. [DOI] [PubMed] [Google Scholar]; (i) Tian X, Jiang K, Peng J, Du W, Chen Y-C. Org Lett. 2008;10:3583. doi: 10.1021/ol801351j. [DOI] [PubMed] [Google Scholar]; (j) Linton EC, Kozlowski MC. J Am Chem Soc. 2008;130:16162. doi: 10.1021/ja807026z. [DOI] [PubMed] [Google Scholar]; (k) Taylor AM, Altman RA, Buchwald SL. J Am Chem Soc. 2009;131:9900. doi: 10.1021/ja903880q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukuyama T, Gang L. J Am Chem Soc. 1996;118:7426–7427. [Google Scholar]
- 12.Trost BM, Lautens M. J Am Chem Soc. 1983;105:3343. [Google Scholar]
- 13.(a) Lloyd-Jones GC, Krska SW, Hughes DL, Gouriou L, Bonnet VD, Jack K, Sun Y, Reamer RA. J Am Chem Soc. 2004;126:702. doi: 10.1021/ja0376339. [DOI] [PubMed] [Google Scholar]; (b) Hughes DL, Lloyd-Jones GC, Krska SW, Gouriou L, Bonnet VD, Jack K, Sun Y, Mathre DJ, Reamer RA. Proc Natl Acad Sci USA. 2004;101:5363. doi: 10.1073/pnas.0306918101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Culkin DA, Hartwig JF. Organometallics. 2004;23:3398. [Google Scholar]
- 15.Reductive elimination from a η3-palladium enolate complex has also been proposed. See: Altman RA, Hyde AM, Huang X, Buchwald SL. J Am Chem Soc. 2008;130:9613. doi: 10.1021/ja803179s.
- 16.Morales D, Clemente MEN, Perez J, Riera L, Riera V. Organometallics. 2003;22:4124. [Google Scholar]
- 17.Cameron PA, Britovsek GJP, Gibson VC, Williams DJ, White AJP. Chem Commun. 1998:737. [Google Scholar]
- 18.Moberg has reported the use of L2 in molybdenum catalyzed allylic alkylation of malonates: Belda O, Moberg C. Synthesis. 2002;1601
- 19.Trost BM, Fandrick DR. Aldrichimica Acta. 2007;40:59. [Google Scholar]
- 20.Hughes DL, Palucki M, Yasuda N, Reamer RA, Reider PJ. J Org Chem. 2002;67:2762. doi: 10.1021/jo015871l. [DOI] [PubMed] [Google Scholar]
- 21.Suarez-Castillo O, Sanchez-Zavala M, Melendez-Rodriguez M, Castelan-Duarte LE, Morales-Rios MS, Joseph-Nathan P. Tetrahedron. 2006;62:3040. [Google Scholar]
- 22.a) Tian X, Jiang K, Peng J, Du W, Chen Y-C. Org Lett. 2008;10:3583. doi: 10.1021/ol801351j. [DOI] [PubMed] [Google Scholar]; b) Shibata N, Suzuki E, Asahi T, Shiro M. J Am Chem Soc. 2001;123:7001. doi: 10.1021/ja010789t. [DOI] [PubMed] [Google Scholar]; c) Huang A, Kodanko JJ, Overman LE. J Am Chem Soc. 126:14043. doi: 10.1021/ja046690e. [DOI] [PubMed] [Google Scholar]; d) Ishimaru T, Shibata N, Nagai J, Nakamura S, Toru T, Kanemasa S. J Am Chem Soc. 2006;128:16488. doi: 10.1021/ja0668825. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.