

Abstract
A multi-virulence-locus sequence typing (MVLST) scheme was developed for subtyping Listeria monocytogenes, and the results obtained using this scheme were compared to those of pulsed-field gel electrophoresis (PFGE) and the published results of other typing methods, including ribotyping (RT) and multilocus sequence typing (MLST). A set of 28 strains (eight different serotypes and three known genetic lineages) of L. monocytogenes was selected from a strain collection (n > 1,000 strains) to represent the genetic diversity of this species. Internal fragments (ca. 418 to 469 bp) of three virulence genes (prfA, inlB, and inlC) and three virulence-associated genes (dal, lisR, and clpP) were sequenced and analyzed. Multiple DNA sequence alignment identified 10 (prfA), 19 (inlB), 13 (dal), 10 (lisR), 17 (inlC), and 16 (clpP) allelic types and a total of 28 unique sequence types. Comparison of MVLST with automated EcoRI-RT and PFGE with ApaI enzymatic digestion showed that MVLST was able to differentiate strains that were indistinguishable by RT (13 ribotypes; discrimination index = 0.921) or PFGE (22 profiles; discrimination index = 0.970). Comparison of MVLST with housekeeping-gene-based MLST analysis showed that MVLST provided higher discriminatory power for serotype 1/2a and 4b strains than MLST. Cluster analysis based on the intragenic sequences of the selected virulence genes indicated a strain phylogeny closely related to serotypes and genetic lineages. In conclusion, MVLST may improve the discriminatory power of MLST and provide a convenient tool for studying the local epidemiology of L. monocytogenes.
Listeria monocytogenes is a gram-positive, intracellular foodborne pathogen that can contaminate a variety of foods and cause listeriosis, a potentially fatal disease. Although infection by L. monocytogenes is relatively rare, it has the second highest fatality rate (21%) and the highest hospitalization rate (90%) of all foodborne pathogens (4). It was found that L. monocytogenes can grow in a wide variety of potential reservoirs and sources within food-processing plants and contaminate ready-to-eat foods (9). While many different strains of L. monocytogenes have been isolated from processing plant environments and frequently cause costly recalls, only a few virulent strains are known to colonize these environments, contaminate foods, and cause listeriosis (29). Therefore, the ability to differentiate strains of L. monocytogenes is particularly important for tracking transmission of pathogenic strains within food-processing plants and developing more effective intervention strategies to prevent recalls and human illness.
Various fragment-based typing methods have been used to differentiate L. monocytogenes strains at the subspecies or strain level (30). These include (i) PCR-based methods, e.g., randomly amplified polymorphic DNA (19, 32) and repetitive sequence-based PCR (18); (ii) restriction digestion-based methods, e.g., ribotyping (RT) (2, 26) and pulsed-field gel electrophoresis (PFGE) (1, 12, 13); and (iii) some other combined amplification-restriction methods, e.g., amplified fragment length polymorphism (14) and PCR-restriction fragment length polymorphism (17). The methods listed above target nucleotide variations at endonuclease restriction or primer annealing sites, utilize the electrophoretic mobility of digested or amplified DNA fragments in agarose gels, and define L. monocytogenes strains by their unique banding patterns. Although these methods provide better strain differentiation than serotyping and phage typing, their discriminatory abilities are not optimal and sometimes cannot differentiate epidemiologically unrelated strains of L. monocytogenes (13, 30). In addition, experimental protocols of these methods may differ and are difficult to standardize. As a consequence, data comparison among different laboratories is sometimes difficult (13, 30).
To overcome the ambiguities of fragment-based typing methods, multilocus sequence typing (MLST), a DNA sequence-based method, was recently developed by Chan et al. and Maiden et al. (5, 20). MLST targets slowly diversified housekeeping gene sequences to address global epidemiology of pathogenic microorganisms (20). MLST (i) provides unambiguous DNA sequence data that can be easily exchanged and compared via worldwide web databases; (ii) combines PCR and automated DNA sequencing to reduce labor and analysis time; and (iii) provides discriminatory power comparable to or higher than that provided by fragment-based methods (8, 20). Due to limited sequence variation in housekeeping genes (3, 23), however, MLST sometimes lacks the discriminatory power required to address issues of local epidemiology for L. monocytogenes strains, such as how some pathogenic strains colonize and transmit within food-processing plants or whether strains isolated from a localized outbreak or recall are different from those isolated from food-processing plants or contaminated food products.
Certain virulence and virulence-associated genes play very important roles in intracellular survival, cell-to-cell spread, and virulence of L. monocytogenes (6, 7, 10, 15, 21, 22, 28). Due to exposure to frequent environmental changes, e.g., immune system responses, these virulence and virulence-associated genes may evolve more rapidly than housekeeping genes (3, 20). Consequently, these genes may provide a higher degree of nucleotide sequence polymorphism and higher discriminatory power for local epidemiology studies (3, 8, 20). In this study, we selected three virulence genes (prfA, inlB, and inlC) and three virulence-associated genes (dal, lisR, and clpP) of L. monocytogenes (11) for analysis. The goals of this study were (i) to develop a multi-virulence-locus sequence typing (MVLST) scheme for studying the local epidemiology of L. monocytogenes; (ii) to compare the discriminatory power of MVLST with the discriminatory powers of PFGE and of other published subtyping methods, including RT and MLST; and (iii) to generate informative sequence data for studying the virulence of L. monocytogenes.
MATERIALS AND METHODS
Bacterial strains.
A total of 28 strains of L. monocytogenes (Table 1) were selected and obtained from the Listeria strain collection at the Cornell Food Safety Laboratory. These strains represented eight serotypes (primarily listeriosis-associated serotype 1/2a and serotype 4b strains) and three genetic lineages of L. monocytogenes as previously described (31). All strains were previously characterized using serotyping and RT with EcoRI digestion. Bacterial strains were stored in tubes of 15% glycerol at −80°C and grown at 37°C on plates with Trypticase soy agar and yeast extract for the MVLST and PFGE analyses.
TABLE 1.
Origins, characteristics and allelic profiles of 28 L. monocytogenes strains analyzed in this study
| Straina | Origina | Serotypea | Lineagea | EcoRI-ribotypea | Apa I-PFGE profile | Allelic profile
|
ST | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| prfA | inlB | dal | lisR | inlC | clpP | |||||||
| FSL J1-101 | Human (sporadic) | 1/2a | II | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| FSL J2-031 | Animal (bovine) | 1/2a | II | 2 | 2 | 1 | 2 | 2 | 1 | 2 | 1 | 2 |
| FSL R2-499 | Human (epidemic) | 1/2a | II | 1 | 1 | 1 | 1 | 1 | 1 | 3 | 1 | 3 |
| FSL J2-003 | Animal (cow) | 1/2a | II | 3 | 3 | 1 | 3 | 3 | 1 | 4 | 1 | 4 |
| FSL J2-017 | Animal (cow) | 1/2a | II | 3 | 4 | 1 | 4 | 2 | 1 | 2 | 2 | 5 |
| FSL C1-117 | Human | 1/2a | II | 2 | 5 | 1 | 5 | 2 | 1 | 5 | 1 | 6 |
| FSL J1-177 | Human (sporadic) | 1/2b | 1 | 4 | 6 | 2 | 6 | 4 | 2 | 6 | 3 | 7 |
| FSL J1-038 | Human | 1/2b | I | 5 | 7 | 2 | 7 | 5 | 2 | 7 | 4 | 8 |
| FSL J2-064 | Animal (cow) | 1/2b | I | 6 | 8 | 3 | 7 | 5 | 2 | 7 | 4 | 9 |
| FSL J1-094 | Human (sporadic) | 1/2c | II | 3 | 9 | 1 | 8 | 6 | 1 | 8 | 1 | 10 |
| FSL J1-022 | Human | 1/2c | II | 3 | 9 | 1 | 8 | 6 | 1 | 9 | 1 | 11 |
| FSL J1-047 | Human | 1/2c | II | 2 | 9 | 1 | 8 | 6 | 1 | 9 | 5 | 12 |
| FSL J1-169 | Human (sporadic) | 3b | I | 6 | 10 | 3 | 7 | 5 | 2 | 10 | 6 | 13 |
| FSL J1-049 | Human (sporadic) | 3c | I | 5 | 11 | 2 | 9 | 5 | 3 | 10 | 4 | 14 |
| FSL J1-168 | Human (sporadic) | 4a | III | 7 | 12 | 4 | 10 | 7 | 4 | 11 | 7 | 15 |
| FSL J1-031 | Human (sporadic) | 4a | III | 8 | 13 | 5 | 11 | 8 | 5 | 12 | 8 | 16 |
| FSL X1-002 | Food | 4a | III | 8 | 13 | 6 | 12 | 9 | 6 | 13 | 8 | 17 |
| FSL J1-119 | Human (epidemic) | 4b | I | 9 | 14 | 7 | 13 | 10 | 3 | 14 | 9 | 18 |
| FSL N1-225 | Human (epidemic) | 4b | I | 10 | 15 | 2 | 13 | 10 | 2 | 10 | 4 | 19 |
| FSL J1-110 | Food (epidemic) | 4b | I | 9 | 14 | 7 | 13 | 10 | 3 | 14 | 4 | 20 |
| FSL J1-116 | Human (epidemic) | 4b | I | 5 | 16 | 2 | 14 | 5 | 3 | 14 | 10 | 21 |
| FSL N3-013 | Food (epidemic) | 4b | I | 5 | 16 | 2 | 14 | 5 | 3 | 14 | 11 | 22 |
| FSL J2-039 | Animal (turkey) | 4b | I | 5 | 17 | 2 | 15 | 5 | 2 | 10 | 12 | 23 |
| FSL J2-045 | Animal (sheep) | 4b | I | 5 | 18 | 2 | 14 | 10 | 2 | 10 | 13 | 24 |
| FSL J1-051 | Human | 4b | I | 6 | 19 | 2 | 16 | 10 | 7 | 7 | 4 | 25 |
| FSL J1-158 | Animal (goat) | 4b | III | 11 | 20 | 8 | 17 | 11 | 8 | 15 | 14 | 26 |
| FSL W1-110 | NAb | 4c | III | 12 | 21 | 9 | 18 | 12 | 9 | 16 | 15 | 27 |
| FSL W1-111 | NAb | 4c | III | 13 | 22 | 10 | 19 | 13 | 10 | 17 | 16 | 28 |
Strain information (origins, serotypes, genetic lineages, and ribotypes) was obtained from the Cornell Food Safety Laboratory.
NA, not applicable.
MVLST.
Intragenic regions of three virulence genes (prfA, inlB, and inlC) and three virulence-associated genes (dal, lisR, and clpP) were selected for the MVLST analysis (Fig. 1). PCR primers (Table 2) were designed using Primer3 software (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi) on the basis of the known sequences of L. monocytogenes strain EGD-e (Lion Bioscience) (11). An approximately 500-bp internal fragment of each gene was amplified to allow accurate sequencing of a ∼450-bp fragment within each gene. Bacterial genomic DNA was extracted using an UltraClean Microbial DNA extraction kit (Mo Bio Laboratories, Solana Beach, Calif.) and was stored at −20°C before use. PCR amplifications were performed using NovaTaqPCR kits (Novagen, Madison, Wis.) with a MasterCycler PCR apparatus (Eppendorf Scientific, Hamburg, Germany). A single PCR program was used for the six virulence-gene amplifications (hot lid, 105°C; initial denaturation at 94°C for 5 min followed by 35 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min, final extension at 72°C for 7 min, and holding at 4°C). A 50-μl reaction system was composed as follows: 40.5 μl of PCR-grade water, 1.0 μl of deoxynucleoside triphosphate mix (final concentration, 0.2 mM each), 1.0 μl of 50 pmol of forward primer/μl, 1.0 μl of 50 pmol of reverse primer/μl, 5 μl of 10× PCR buffer with MgCl2 (final concentration, 1.5 mM), 1 μl of template DNA (∼50 ng/μl), and 0.5 μl of NovaTaqDNA polymerase (2.5 U/μl).
FIG. 1.

Genomic locations of the selected virulence loci in L. monocytogenes strain EGD-e. The six loci analyzed are shown in boldface characters.
TABLE 2.
Virulence genes and PCR primers
| Gene | Annotation | Primer | Sequence (5′-3′) |
|---|---|---|---|
| prfA | Listeriolysin positive regulatory protein | prfA-forward | AACGGGATAAAACCAAAACCA |
| prfA-reverse | TGCGATGCCACTTGAATATC | ||
| inlB | Internalin B | inlB-forward | CATGGGAGAGTAACCCAACC |
| inlB-reverse | GCGGTAACCCCTTTGTCATA | ||
| dal | Alanine racemase | dal-forward | GGTTTCTGCGTAGCCATTTT |
| dal-reverse | GGAAGGGGTCAATCCATACA | ||
| lisR | Two-component response regulator | lisR-forward | CGGGGTAGAAGTTTGTCGTC |
| lisR-reverse | ACGCATCACATACCCTGTCC | ||
| inlC | Internalin C | inlC-forward | CGGGAATGCAATTTTTCACTA |
| inlC-reverse | AACCATCTACATAACTCCCACCA | ||
| clpP | Clp protease proteolytic subunit | clpP-forward | CCAACAGTAATTGAACAAACTAGCC |
| clpP-reverse | GATCTGTATCGCGAGCAATG |
Following amplification, PCR mixtures were loaded on a 1.5% UltraClean agarose gel (Mo Bio Laboratories) and separated by electrophoresis at 120 V for 45 min. The DNA bands (ca. 500 bp) were excised from the gel and purified using a QIAquick gel extraction kit (Qiagen Inc., Valencia, Calif.). DNA sequencing was performed with an ABI Prism 3100 DNA sequencer (Applied Biosystems, Inc.) at The Pennsylvania State University Biotechnology Institute. Both forward and reverse PCR primers were used as sequencing primers. DNA sequencing chromatograms were saved as ABI files and SEQ files for analysis.
PFGE.
L. monocytogenes isolates were streaked on plates with Trypticase soy agar and yeast extract and incubated at 37°C for 18 h. Bacterial colonies were suspended in 2 ml of cell suspension Tris-EDTA buffer, and the cell density was adjusted (using a MicroScan turbidity meter [Baxter Diagnostics, Sacramento, Calif.]) to an optical density of 0.80. A 200-μl bacterial suspension was transferred to a 1.5-ml microcentrifuge tube and gently mixed with 60 μl of 10 mg of stock lysozyme solution (Sigma, St. Louis, Mo.)/ml following by a 10-min incubation at 37°C. A preheated (55°C) 300-μl mix of 1.2% SeaKem Gold agarose-1% sodium dodecyl sulfate-0.2 mg of proteinase-K/ml was then added, and the suspension mixture was dispensed into two disposable plug molds (Bio-Rad Laboratories, Hercules, Calif.). Plugs solidified at room temperature were transferred into 50-ml conical tubes with 45 ml of lysis buffer. After 2 h of incubation in a 55°C shaker water bath, the lysis buffer was removed and plugs were washed twice with 45 ml of preheated (55°C) sterile distilled water for 15 min followed by three washes with 45 ml of preheated (55°C) Tris-EDTA buffer in the 55°C shaker water bath.
The 2-mm-thick plug slices were digested with ApaI (New England BioLabs, Beverly, Mass.) (200 U per slice; 5 h at 30°C). The digested slices were then loaded into a 1% SeaKem Gold agarose gel. The PFGE was performed with a CHEF DR II apparatus (Bio-Rad Laboratories) as follows: initial switch time, 4.0 s; final switch time, 40.0 s; run time, 16 h; angle, 120°; gradient, 6.0 V/cm; temperature, 14°C; ramping factor, linear. The gel was stained in ethidium bromide solution for 20 min followed by three distilled water washes. The gel was photographed using a MultiImage gel photographing system (Alpha Innotech Inc., San Leandro, Calif.), and images were saved as TIFF files for analysis.
Data analyses.
For MVLST, multiple sequence alignments were performed using GeneTool software (BioTools, Inc., Edmonton, Canada). Different allelic sequences (with at least a one-nucleotide difference) were assigned arbitrary numbers. For each strain, the combination of 6 alleles defined its allelic profile, and a unique allelic profile was designated as a sequence type (ST). The BLAST 2 sequences program was used to identify genomic locations of the resulting intragenic sequences in strain EGD-e (27). Molecular evolutionary genetics analysis software (version 2.1) (http://www.megasoftware.net) was used to construct the neighbor-joining (N-J) tree of L. monocytogenes strains on the basis of the numbers of nucleotide differences and the results of a bootstrapping test of strain phylogeny. Sequence type analysis and recombinational tests (START) software (version 1.0.5) (http://www.mlst.net) was used to calculate G+C content and perform recombination and selection (dN/dS) tests, and a tree of L. monocytogenes strains was constructed on the basis of their allelic profiles by the unweighted pair group method with arithmetic mean (UPGMA). Sawyer's tests were used to provide statistical evidence of recombinational exchanges of the intragenic sequences analyzed (24).
PFGE patterns with two band differences were compared using the Dice coefficient and Fingerprinting DST Molecular Analyst software (Bio-Rad Laboratories). A dendrogram was constructed on the basis of PFGE banding patterns by the UPGMA.
Discrimination index (D.I.) values were calculated (on the basis of numbers of allelic types [j], numbers of strains belonging to each type [nj], and total numbers of strains analyzed [N]) as previously described (16) with the following equation (higher D.I. values indicate higher discriminatory power):
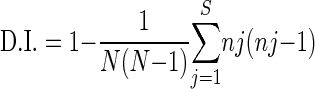 |
Nucleotide sequence accession number.
DNA sequences of the virulence-gene fragments were deposited in GenBank under accession numbers AY259632 through AY259659 (prfA); AY259660 through AY259687 (inlB); AY259688 through AY259715 (dal); AY259716 through AY259743 (lisR); AY259744 through AY259771 (inlC); and AY259772 through AY259799 (clpP).
RESULTS
Allelic nucleotide polymorphism in the select virulence and virulence-associated gene fragments.
Partial coding sequences (CDSs) of the select virulence genes (prfA, inlB, and inlC) and virulence-associated genes (dal, lisR, and clpP) were sequenced and analyzed. These loci are diversely located in the L. monocytogenes genome (at intervals ranging from 253,816 to 681,055 bp) in strain EGD-e (Fig. 1). A total of 2,642 bp (39.35% coverage of the six complete CDSs) were analyzed for each strain. Multiple nucleotide sequence alignment did not exhibit insertion or deletion mutation in the six intragenic fragments analyzed. However, single-nucleotide polymorphism was commonly observed over the entire sequence of these fragments. The number of unique alleles identified ranged from 10 for prfA and lisR to 19 for inlB. The percentages of polymorphic nucleotide sites ranged from 7.036% for prfA to 18.681% for dal. The D.I. of each locus was calculated to allow a comparison of levels of discriminatory power among individual virulence genes (Table 3). The D.I. values of six gene fragments ranged from 0.804 for the prfA gene to 0.966 for the inlB gene. Interestingly, the most polymorphic gene fragment (dal) was not the most discriminatory for the reference strains (D.I. = 0.899) because a high percentage of polymorphic sites occurred in serotype 4a and 4c strains. In contrast, a small portion of inlB (433 bp [22.87% of the complete CDS]) provided 19 alleles (D.I. = 0.966) with 47 polymorphic sites. A subset of three loci, inlB, inlC, and clpP, showed a level of discriminatory power (27 STs; D.I. = 0.997) similar to that seen in the six-locus-based MVLST analysis (28 STs; D.I. = 1.000). This finding may help simplify the MVLST scheme by allowing analysis of the most discriminatory loci.
TABLE 3.
Allelic polymorphisms in the six virulence gene fragments analyzed
| Gene | Size (bp) of fragments analyzed | Coverage of complete CDS (%) | Allelic location (nt) in strain EGDb | No. of alleles | No. of polymorphic sites | % of polymorphic sites | Avg G+C content (%) | D.I. | dN/dS |
|---|---|---|---|---|---|---|---|---|---|
| prfA | 469 | 65.69 | 203701-204169 | 10 | 33 | 7.036 | 34.31 | 0.804 | 0.000 |
| inlB | 433 | 22.87 | 457985-458417 | 19 | 47 | 10.855 | 40.28 | 0.966 | 0.082 |
| dal | 455 | 41.10 | 925132-925586 | 13 | 85 | 18.681 | 40.26 | 0.899 | 0.054 |
| lisR | 448 | 65.79 | 1402760-1403207 | 10 | 54 | 12.054 | 39.11 | 0.823 | 0.000 |
| inlC | 418 | 46.91 | 1860499-1860916 | 17 | 36 | 8.612 | 31.65 | 0.944 | 0.098 |
| clpP | 419 | 70.18 | 2542071-2542489 | 16 | 39 | 9.308 | 39.80 | 0.902 | 0.023 |
| Total | 2642 | 39.35 | NAa | 28 | 294 | 11.128 | 37.57 | 1.000 | NA |
NA, not applicable.
nt, nucleotide.
Although nonsynonymous substitutions were found in all six loci, most of the nucleotide polymorphisms resulted in synonymous substitutions (Table 4 ). Only one amino acid substitution was observed in lisR alleles, while 18 amino acid substitutions were observed in dal alleles. Analysis of prfA, lisR, and clpP genes revealed relatively fewer nonsynonymous substitutions and lower dN/dS ratios than analysis of inlB, inlC and dal. The amino acid sequence homology of these genes may be explained by their important functions for intracellular survival and virulence of L. monocytogenes: The PrfA protein is a key regulatory factor for the differential expression of virulence genes within infected host cells (15). PrfA regulates its own expression, while it positively or negatively regulates the expressions of three other groups of genes (21). This regulation is essential for cell-to-cell spread of L. monocytogenes (21). The lisR gene encodes one of the two-component signal transduction response regulators and is involved in virulence potential and tolerance to antimicrobials (6). The clpP gene codes for one of the caseinolytic proteins (Clps), which is involved in degradation of damaged polypeptides and assists in the rapid adaptive response of intracellular pathogens during the infectious process (10).
TABLE 4.
Nonsynonymous substitution sites in each of the six gene fragments
| Strain | Substitution by gene, location, and 50% consensus amino acid identity
|
|||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
prfA
|
inlB
|
dal
|
||||||||||||||||||||||
| 136 | 140 | 145 | 50 | 52 | 66 | 69 | 75 | 101 | 118 | 124 | 125 | 142 | 143 | 38 | 43 | 48 | 49 | 68 | 69 | 73 | 76 | 77 | 78 | |
| K | D | T | N | S | M | A | A | L | R | G | K/E | S | L | E | -b | A | P | T | E/D | R | T | T | I | |
| J1-101 | . | . | . | . | N | V | V | . | F | K | . | . | . | . | . | N | . | T | . | . | . | A | . | S |
| J2-031 | . | . | . | . | N | V | . | T | . | . | . | . | . | . | . | N | . | T | . | . | . | A | . | S |
| R2-499 | . | . | . | . | N | V | V | . | F | K | . | . | . | . | . | N | . | T | . | . | . | A | . | S |
| J2-003 | . | . | . | . | N | V | . | T | . | . | . | . | . | . | . | H | . | . | V | . | Q | . | . | . |
| J2-017 | . | . | . | . | N | V | . | T | . | . | . | . | . | . | . | N | . | T | . | . | . | A | . | S |
| C1-117 | . | . | . | . | N | V | . | . | . | . | . | . | . | . | . | N | . | T | . | . | . | A | . | S |
| J1-177 | . | . | . | D | . | . | V | . | . | . | . | . | . | . | . | D | . | . | . | D | . | . | . | . |
| J1-038 | . | . | . | D | . | . | V | . | . | . | . | . | . | . | . | D | . | . | . | D | . | . | . | . |
| J2-064 | . | . | . | D | . | . | V | . | . | . | . | . | . | . | . | D | . | . | . | D | . | . | . | . |
| J1-094 | . | . | . | . | N | V | . | . | . | . | . | . | . | . | . | N | . | T | . | . | . | A | . | S |
| J1-022 | . | . | . | . | N | V | . | . | . | . | . | . | . | . | . | N | . | T | . | . | . | A | . | S |
| J1-047 | . | . | . | . | N | V | . | . | . | . | . | . | . | . | . | N | . | T | . | . | . | A | . | S |
| J1-169 | . | . | . | D | . | . | V | . | . | . | . | . | . | . | . | D | . | . | . | D | . | . | . | . |
| J1-049 | . | . | . | D | . | . | V | . | . | . | . | . | . | . | . | D | . | . | . | D | . | . | . | . |
| J1-168 | . | . | A | . | . | . | . | . | . | . | S | E | . | . | . | D | . | . | V | . | Q | . | . | . |
| J1-031 | E | . | . | . | . | . | . | . | . | . | S | E | . | . | . | D | V | . | . | D | . | . | . | . |
| X1-002 | . | . | A | . | . | . | . | . | . | . | S | E | . | . | . | D | . | . | V | . | Q | . | . | . |
| J1-119 | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | E | . | . | . | D | . | . | . | . |
| N1-225 | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | E | . | . | . | D | . | . | . | . |
| J1-110 | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | E | . | . | . | D | . | . | . | . |
| J1-116 | . | . | . | . | . | I | . | . | . | . | S | E | . | . | . | D | . | . | . | D | . | . | . | . |
| N3-013 | . | . | . | . | . | I | . | . | . | . | S | E | . | . | . | D | . | . | . | D | . | . | . | . |
| J2-039 | . | . | . | D | . | . | . | . | . | . | . | E | . | . | . | D | . | . | . | D | . | . | . | . |
| J2-045 | . | . | . | . | . | I | . | . | . | . | S | E | . | . | . | E | . | . | . | D | . | . | . | . |
| J1-051 | . | . | . | D | . | . | . | . | . | . | . | E | . | . | . | E | . | . | . | D | . | . | . | . |
| J1-158 | . | N | A | . | . | . | . | . | . | . | . | E | P | S | A | N | . | . | V | . | Q | . | M | . |
| W1-110 | . | N | A | . | . | . | . | . | . | . | . | E | . | . | . | E | . | . | V | . | Q | . | . | . |
| W1-111 | I | N | A | . | . | . | . | . | . | . | . | E | . | . | . | N | . | . | V | . | Q | . | . | . |
Locations of the variable amino acid sites are shown by the numbers above in vertical format. The amino acids with no less than 50% consensus are shown in boldface characters. Amino acids that are the same as the 50% consensus amino acids at each polymorphic site are indicated by periods.
-, no 50% consensus amino acid was identified.
Discriminatory power of RT, PFGE, and MVLST.
The 28 strains were divided into 13 ribotypes by automated EcoRI-RT (strain ribotypes were obtained from the Cornell Food Safety Laboratory), 23 types by ApaI-PFGE, and 28 STs by MVLST (Table 1). The D.I. for each method was calculated as previously described (16), and D.I. values were compared as indicators of discriminatory power. In this study, PFGE (D.I. = 0.970) and MVLST (D.I. = 1.000) provided satisfactory discriminatory power for the 28 L. monocytogenes strains. RT (D.I. = 0.921) could not differentiate epidemiologically unrelated serotype 1/2a and 1/2c strains (e.g., strains J2-003 [1/2a] and J1-094 [1/2c]) or strains within serovar 4b (e.g., strains J1-116 and J2-039) (Table 1). ApaI-PFGE analysis successfully differentiated strains from different serovars; however, it could not differentiate some epidemiologically unrelated strains within the same serovars (e.g., strains J1-101 [serotype 4b; isolated from a human sporadic case in 1985] and R2-499 [serotype 4b; isolated from a human epidemic outbreak in 2000]). MVLST analysis provided the highest discriminatory power and resolved all the RT- or PFGE-indistinguishable strains by at least one allelic difference.
Cluster analyses of PFGE and MVLST.
The PFGE dendrogram was constructed by the UPGMA on the basis of the banding patterns of ApaI-digested genomic DNA fragments (Fig. 2). The 28 strains were divided into two major clusters (>40% similarity). Cluster I included strains of serotypes 1/2b, 1/2c, 3b, 3c, 4a, 4b, and 4c; cluster II included serotype 1/2a strains and one serotype 4b strain. The N-J method with 1,000 bootstrapping replications was used to construct a phylogenetic tree for MVLST analysis on the basis of the number of nucleotide differences in the six virulence gene fragments (Fig. 3). Three major clusters were identified from N-J analysis. Cluster A mainly included serotype 4b (n = 8), 1/2b (n = 3), 3b (n = 1), and 3c (n = 1) strains. Cluster B included serotype 1/2a (n = 6) and 1/2c (n = 3) strains. Cluster C included serotype 4a (n = 3), 4b (n = 1), and 4c (n = 2) strains. Interestingly, the N-J clusters were highly congruent with genetic lineages as previously described by Wiedmann et al. (31) for investigations using RT and PCR-restriction fragment length polymorphism methods. All genetic lineage I strains (n = 13) were grouped in cluster A, all genetic lineage II strains (n = 9) were in cluster B, and all genetic lineage III strains (n = 6) were in cluster C. Within the major N-J clusters, subgroups were observed and correlated with serovars; e.g., in cluster B, all serotype 1/2c strains were in one subgroup and all serotype 1/2a strains were in another subgroup. Both trees indicated that some serotypes of strains, including serotype 1/2b and 4b strains, serotype 3b and 3c strains, serotype 4a and 4c strains, and strains of serotype 1/2a, exhibited close genetic relatedness.
FIG. 2.

(A) PFGE dendrogram constructed by the UPGMA. (B) Computer-normalized PFGE banding patterns with ApaI enzymatic digestion.
FIG. 3.

N-J tree of 28 L. monocytogenes strains; the tree was constructed on the basis of the number of nucleotide differences in the six virulence-gene fragments analyzed. Bootstrap values (1,000 replications) are shown at the interior branches.
Evidence of recombination and selection in the six virulence loci.
Sawyer's test was used to provide statistical evidence of recombinational exchanges within the aligned virulence gene sequences by determining the sum of the squares of the condensed fragment lengths (SSCF) and the maximum condensed fragment (24). Sawyer's tests with 10,000 trials did not reveal detectable cases of intragenic recombination for prfA (SSCF P = 0.3477), lisR (SSCF P = 1.000), inlC (SSCF P = 0.5015), or clpP (SSCF P = 1.0000); however, intragenic recombination was indicated with inlB (SSCF P = 0.0007) and dal (SSCF P = 0.0000).
DISCUSSION
Comparison of RT, PFGE, and MVLST.
RT and PFGE are presently the most commonly used methods for subtyping L. monocytogenes (25, 30). RT typically targets conserved ribosomal DNA regions and is less discriminatory than PFGE; therefore, its utility for local epidemiological purposes is limited (2, 30). PFGE targets DNA variations at multiple endonuclease restriction sites across the bacterial genome and generally provides satisfactory discriminatory power for investigations of L. monocytogenes. However, PFGE is less automated than RT and requires greater experimental skill. In addition, the interpretations of the PFGE banding patterns differ between researchers (13, 30). This study demonstrated that the discriminatory power of MVLST analysis was higher than that of EcoRI-RT and comparable to that of PFGE. Interestingly, dendrograms of PFGE and MVLST indicated different genetic relatedness of the 28 strains, which was probably a reflection of recombination in some selected genes (e.g., inlB and dal).
Comparison of MLST and MVLST.
A total of 14 of the strains investigated in this study were previously analyzed by sequencing the complete CDSs of three housekeeping genes, sigB (780 bp; stress responsive alternative sigma factor B), prs (957 bp; phosphoribosyl synthetase), and recA (1,047 bp; recombinase A) (3). For these 14 strains, the percentage of nucleotide polymorphisms in these genes ranged from 4.9% for prs (nine allelic types) to ∼10% for sigB (nine allelic types) and recA (seven allelic types). These housekeeping genes were found to be less discriminatory than some virulence genes, namely, actA (14 allelic types) and inlA (13 allelic types) (3). In addition, housekeeping gene-based sequence analysis could not differentiate between some L. monocytogenes serotypes (e.g., serotypes 1/2b and 4b and serotypes 1/2a and 1/2c) and between some epidemiologically unrelated strains within the same serovars (e.g., serotype 1/2a strains J2-017 and C1-117 and serotype 1/2c strains J1-022 and J1-047) (3). However, in the MVLST analysis, strains of different serotypes were differentiated by individual virulence genes, e.g., inlB. Epidemiologically unrelated strains within serovars were clearly differentiated by analysis of a combination of 3 to 6 loci.
In another recent study, partial CDSs of nine housekeeping genes in L. monocytogenes were analyzed by MLST (23). Most of these gene fragments showed relatively lower nucleotide polymorphism levels, e.g., abcZ (6.1%), bglA (5.0%), cat (7.8%), dapE (8.3%), ldh (4.3%), lhkA (3.5%), pgm (4.4%), and sod (2.9%). In addition, MLST was unable to differentiate epidemiologically unrelated strains, especially for the disease-related serotype 4b strains. Results of MVLST supported the finding of MLST that strains of serotypes 1/2a and 4b were genetically more similar than strains of other serotypes (23). By simultaneously targeting three to six virulence genes, the present MVLST analysis provided a clear differentiation of strains within these serotypes.
Applications in epidemiological studies.
As PCR amplification and DNA sequencing become increasingly automated and commercially available, MVLST may provide a more convenient tool for studying epidemiology of L. monocytogenes than fragment-based typing methods. World Wide Web access to gene sequences for different pathogens may further facilitate the dissemination and comparison of sequences or STs, making MVLST a powerful tool for studying the local epidemiology of foodborne pathogens. Highly discriminatory MVLST analysis may assist governmental agencies and food processors in identifying critical control points and establishing effective intervention strategies to prevent contamination of ready-to-eat foods by pathogenic L. monocytogenes strains (25, 29). MVLST analysis targets rapidly evolving virulence genes and therefore may not be suitable for population genetics of bacterial pathogens. More extensive MVLST analyses conducted on the basis of the investigation of a larger number of strains (n > 100) may provide critical validation of the present study and informative clues for the study of the virulence genes and pathogenesis of L. monocytogenes. By the selection of appropriate virulence loci, the MVLST scheme may also be used to subtype other bacterial genera or species to improve the discriminatory power of MLST-based analyses.
Table 4a.
| Substitution by gene, location, and 50% consensus amino acid identity
| ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
dal
|
lisR
|
inlC
|
clpP
|
|||||||||||||||||||||||
| 79 | 80 | 82 | 109 | 120 | 127 | 130 | 144 | 3 | 6 | 7 | 12 | 15 | 16 | 20 | 25 | 29 | 39 | 46 | 58 | 68 | 74 | 123 | 19 | 20 | 30 | 90 |
| A | K | N | A | N | T | S | I | T | N | Q | S | K | D | L | V | R | A | V | I | I | K | R | D | N | L | N |
| T | N | H | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | K | . | . | . | . |
| T | N | H | . | K | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | K | . | . | . | . |
| T | N | H | . | . | . | . | . | . | . | . | . | . | . | F | . | . | . | L | . | . | . | K | . | . | . | . |
| . | S | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | K | . | . | . | . |
| T | N | H | . | K | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | K | . | . | . | . |
| T | N | H | . | K | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | K | . | . | . | . |
| . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . |
| . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | M | . | . | . | . | . | . | . | . | . | . | . |
| . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | M | . | . | . | . | . | . | . | . | . | . | . |
| T | N | H | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | K | . | . | . | . |
| T | N | H | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | K | . | . | . | . |
| T | N | H | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | K | . | . | . | . |
| . | . | . | . | . | . | . | . | . | . | . | . | N | . | . | M | . | . | . | . | . | . | . | . | . | F | . |
| . | . | . | . | . | . | . | . | . | . | . | . | N | . | . | M | . | . | . | . | . | . | . | . | . | . | . |
| . | S | . | . | . | . | . | . | . | . | . | . | . | . | . | . | K | . | . | . | . | . | . | . | . | . | S |
| T | . | . | . | . | . | . | . | . | . | . | G | . | . | . | . | K | . | . | . | . | . | . | . | . | . | S |
| . | S | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | . | . | . | . | . | . | S |
| . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . |
| . | . | . | . | . | . | . | . | . | . | . | . | N | . | . | M | . | . | . | . | . | . | . | . | . | . | . |
| . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . |
| . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | K | . | . |
| . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . |
| . | . | . | . | . | . | . | . | . | . | . | . | N | . | . | M | . | . | . | . | . | . | . | . | . | . | . |
| . | . | . | . | . | . | . | . | . | . | . | . | N | . | . | M | . | . | . | . | . | . | . | E | . | . | . |
| . | . | . | . | . | . | . | . | . | P | . | . | . | . | . | M | . | . | . | . | . | . | . | . | . | . | . |
| . | S | S | . | . | V | P | T | . | . | K | G | . | . | . | . | K | . | . | V | L | . | . | . | . | . | S |
| . | S | . | T | . | . | . | . | . | . | . | . | . | N | . | . | K | T | . | . | . | R | . | . | . | . | S |
| . | S | S | . | . | V | . | . | . | H | . | G | . | . | . | . | K | . | . | V | L | R | . | . | . | . | S |
Acknowledgments
This study was supported by a U.S. Department of Agriculture Special Milk Safety grant to The Pennsylvania State University.
We thank Martin Wiedmann for providing the bacterial strains, Narashima Hegde for providing assistance with PFGE analysis, and Chitrita DebRoy for critical review of the manuscript. We also are grateful to Masatoshi Nei for his valuable suggestions for phylogenetic analysis.
REFERENCES
- 1.Brosch, R., M. Brett, B. Catimel, J. B. Luchansky, B. Ojeniyi, and J. Rocourt. 1996. Genomic fingerprinting of 80 strains from the WHO. multicenter international typing study of Listeria monocytogenes via pulsed-field gel electrophoresis (PFGE). Int. J. Food Microbiol. 32:343-355. [DOI] [PubMed] [Google Scholar]
- 2.Bruce, J. L., R. J. Hubner, E. M. Cole, C. I. McDowell, and J. A. Webster. 1995. Sets of EcoRI fragments containing ribosomal RNA sequences are conserved among different strains of Listeria monocytogenes. Proc. Natl. Acad. Sci. USA 92:5229-5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai, S., D. Y. Kabuki, A. Y. Kuaye, T. G. Cargioli, M. S. Chung, R. Nielsen, and M. Wiedmann. 2002. Rational design of DNA sequence-based strategies for subtyping Listeria monocytogenes. J. Clin. Microbiol. 40:3319-3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. 2001. Draft assessment of the relative risk to public health from food-borne Listeria monocytogenes among selected categories of ready-to-eat foods. [Online.] http://vm.cfsan.fda.gov/∼dms/lmrisk.html. Accessed 1 March 2003.
- 5.Chan, M., M. C. J. Maiden, and B. G. Spratt. 2001. Database-driven multi locus sequence typing (MLST) of bacterial pathogens. Bioinformatics 17:1077-1083. [DOI] [PubMed] [Google Scholar]
- 6.Cotter, P. D., N. Emerson, C. G. M. Gahan, and C. Hill. 1999. Identification and disruption of lisRK, a genetic locus encoding a two-component signal transduction system involved in stress tolerance and virulence in Listeria monocytogenes. J. Bacteriol. 181:6840-6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelbrecht, F., S. K. Chun, C. Ochs, J. Hess, F. Lottspeich, W. Goebel, and Z. Sokolovic. 1996. A new PrfA-regulated gene of Listeria monocytogenes encoding a small, secreted protein which belongs to the family of internalins. Mol. Microbiol. 21:823-837. [DOI] [PubMed] [Google Scholar]
- 8.Enright, M. C., and B. G. Spratt. 1999. Multilocus sequence typing. Trends Microbiol. 7:482-487. [DOI] [PubMed] [Google Scholar]
- 9.Fenlon, D. R. 1999. Listeria monocytogenes in the natural environment, p. 21-39. In E. T. Ryser and E. H. Marth (ed.), Listeria, listeriosis, and food safety, 2nd ed. Marcel Dekker Inc., New York, N.Y.
- 10.Gaillot, O., E. Pellegrini, S. Bregenholt, S. Nair, and P. Berche. 2000. The ClpP serine protease is essential for the intracellular parasitism and virulence of Listeria monocytogenes. Mol. Microbiol. 35:1286-1294. [DOI] [PubMed] [Google Scholar]
- 11.Glaser, P., L. Frangeul, C. Buchrieser, C. Rusniok, A. Amend, F. Baquero, P. Berche, H. Bloecker, P. Brandt, T. Chakraborty, A. Charbit, F. Chetouani, E. Couvé, A. de Daruvar, P. Dehoux, E. Domann, G. Domínguez-Bernal, E. Duchaud, L. Durant, O. Dussurget, K.-D. Entian, H. Fsihi, F. Garcia-Del Portillo, P. Garrido, L. Gautier, W. Goebel, N. Gómez-López, T. Hain, J. Hauf, D. Jackson, L.-M. Jones, U. Kaerst, J. Kreft, M. Kuhn, F. Kunst, G. Kurapkat, E. Madueño, A. Maitournam, J. Mata Vicente, E. Ng, H. Nedjari, G. Nordsiek, S. Novella, B. de Pablos, J.-C. Pérez-Diaz, R. Purcell, B. Remmel, M. Rose, T. Schlueter, N. Simoes, A. Tierrez, J.-A. Vázquez-Boland, H. Voss, J. Wehland, and P. Cossart. 2001. Comparative genomics of Listeria species. Science 294:849-852. [DOI] [PubMed] [Google Scholar]
- 12.Graves, L. M., and B. Swaminathan. 2001. PulseNet standardized protocol for subtyping Listeria monocytogenes by macrorestriction and pulsed-field gel electrophoresis. Int. J. Food Microbiol. 65:55-62. [DOI] [PubMed] [Google Scholar]
- 13.Gravesen, A. T. Jacobsen, P. L. Moller, F. Hansen, A. G. Larse, and S. Knochel. 2000. Genotyping of Listeria monocytogenes: comparison of RAPD, ITS, and PFGE. Int. J. Food Microbiol. 57:43-51. [Google Scholar]
- 14.Guerra, M. M., F. Bernardo, and J. McLauchlin. 2002. Amplified fragment length polymorphism (AFLP) analysis of Listeria monocytogenes. Syst. Appl. Microbiol. 25:456-461. [DOI] [PubMed] [Google Scholar]
- 15.Herler, M., A. Bubert, M. Goetz, Y. Vega, J. A. Vazquez-Boland, and W. Goebel. 2001. Positive selection of mutations leading to loss or reduction of transcriptional activity of PrfA, the central regulator of Listeria monocytogenes virulence. J. Bacteriol. 183:5562-5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunter, P. R., and M. A. Gaston. 1988. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J. Clin. Microbiol. 26:2465-2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeffers, G. T., J. L. Bruce, P. L. McDonough, J. Scarlett, K. J. Boor, and M. Wiedmann. 2001. Comparative genetic characterization of Listeria monocytogenes isolates from human and animal listeriosis cases. Microbiology 147:1095-1104. [DOI] [PubMed] [Google Scholar]
- 18.Jersek, B., P. Gilot, M. Gubina, N. Klun, J. Mehle, E. Tcherneva, N. Rijpens, and L. Herman. 1999. Typing of Listeria monocytogenes strains by repetitive element sequence-based PCR. J. Clin. Microbiol. 37:103-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawrence, L. M., J. Harvey, and A. Gilmour. 1993. Development of a random amplification of polymorphic DNA typing method for Listeria monocytogenes. Appl. Environ. Microbiol. 59:3117-3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maiden, M. C. J., J. A. Bygraves, E. Feil, G. Morelli, J. E. Russell, R. Urwin, Q. Zhang, J. Zhou, K. Zurth, D. A. Caugant, I. M. Feavers, M. Achtman, and B. G. Spratt. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA 95:3140-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milohanic, E., P. Glaser, J. Y. Coppee, L. Frangeul, Y. Vega, J. A. Vazquez-Boland, F. Kunst, P. Cossart, and C. Buchrieser. 2003. Transcriptome analysis of Listeria monocytogenes identifies three groups of genes differently regulated by PrfA. Mol. Microbiol. 47:1613-1625. [DOI] [PubMed] [Google Scholar]
- 22.Parida, S. K., E. Domann, M. Rohde, S. Muller, A. Darji, T. Hain, J. Wehland, and T. Chakraborty. 1998. Internalin B is essential for adhesion and mediates the invasion of Listeria monocytogenes into human endothelial cells. Mol. Microbiol. 28:81-93. [DOI] [PubMed] [Google Scholar]
- 23.Salcedo, C., L. Arreaza, B. Alcala, L. De La Fuente, and J. A. Vazquez. 2003. Development of a multilocus sequence typing method for analysis of Listeria monocytogenes clones. J. Clin. Microbiol. 41:757-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sawyer, S. 1989. Statistical tests for detecting gene conversion. Mol. Biol. Evol. 6:526-538. [DOI] [PubMed] [Google Scholar]
- 25.Sveum, W. H., L. J. Moberg, R. A. Rude, and J. F. Frank. 1992. Microbiological monitoring of the food processing environment, p. 51-74. In C. Vanderzant and D. F. Splittstoesser (ed.), Compendium of methods for the microbiological examination of foods, 3rd ed. American Public Health Association, Washington, D.C.
- 26.Swaminathan, B., S. B. Hunter, P. M. Desmarchelier, P. Gerner-Smidt, L. M. Graves, S. Harlander, Hubner, R. Jacquet, C. Pedersen, B. Reineccius, K. Ridley, A. Saunders, N. A., and J. A. Webster. 1996. WHO-sponsored international collaborative study to evaluate methods for subtyping Listeria monocytogenes: restriction fragment length polymorphism (RFLP) analysis using ribotyping and Southern hybridization with two probes derived from L. monocytogenes chromosome. Int. J. Food Microbiol. 32:263-278. [DOI] [PubMed] [Google Scholar]
- 27.Tatusova, T. A., and T. L. Madden. 1999. Blast 2 sequences-a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 174:247-250. [DOI] [PubMed] [Google Scholar]
- 28.Thompson, R. J., H. G. A. Bouwer, D. A. Portnoy, and F. R. Frankel. 1998. Pathogenicity and immunogenicity of a Listeria monocytogenes strain that requires d-alanine for growth. Infect. Immun. 66:3552-3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tompkin, R. B. 2002. Control of Listeria monocytogenes in the food-processing environment. J. Food Prot. 65:709-725. [DOI] [PubMed] [Google Scholar]
- 30.Wiedmann, M. 2002. Molecular subtyping methods for Listeria monocytogenes. J. Assoc. Off. Anal. Chem. Int. 85:524-531. [PubMed] [Google Scholar]
- 31.Wiedmann, M., J. L. Bruce, C. Keating, A. E. Johnson, P. L. McDonough, and C. A. Batt. 1997. Ribotypes and virulence gene polymorphisms suggest three distinct Listeria monocytogenes lineages with differences in pathogenic potential. Infect. Immun. 65:2707-2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida, T., M. Takeuchi, M. Sato, and K. Hirai. 1999. Typing Listeria monocytogenes by random amplified polymorphic DNA (RAPD) fingerprinting. J. Vet. Med. Sci. 61:850-860. [DOI] [PubMed] [Google Scholar]