

Abstract
Itraconazole (ITZ) is metabolized in vitro to three inhibitory metabolites: hydroxy-ITZ (OH-ITZ), keto-ITZ, and N-desalkyl-ITZ (ND-ITZ). The goal of this study was to determine the contribution of these metabolites to drug-drug interactions caused by ITZ. Six healthy volunteers received 100 mg ITZ orally for seven days and pharmacokinetic analysis was conducted at day 1 and day 7 of the study. The extent of CYP3A4 inhibition by ITZ and its metabolites was predicted using this data. ITZ, OH-ITZ, keto-ITZ and ND-ITZ, were detected in plasma samples of all volunteers. A 3.9-fold decrease in the hepatic intrinsic clearance of a CYP3A4 substrate was predicted using the average unbound steady-state concentrations (Css,ave,u) and liver microsomal inhibition constants for ITZ, OH-ITZ, keto-ITZ, and ND-ITZ. Accounting for circulating metabolites of ITZ significantly improved the in vitro to in vivo extrapolation of CYP3A4 inhibition compared to a consideration of ITZ exposure alone.
INTRODUCTION
Itraconazole (ITZ, Figure 1) is a broad-spectrum triazole antifungal drug used to treat infections caused by dermatophytes, yeasts, dimorphic and demataceous fungi and molds (1). Pharmacokinetics of ITZ is characterized by a large volume of distribution (11 L/kg) and intermediate blood clearance (40 L/h) (1,2). The absorption of ITZ is influenced by food (3) and its bioavailability and half-life are dose-dependant, resulting in a disproportional increase in the area under the plasma concentration vs. time curve (AUC) of ITZ with increasing single oral doses (4,5). Following multiple-dose administration, a 26-60% increase in the elimination half-life (t½) of ITZ and a time-dependent reduction (69-80%) in oral clearance (Cl/F) has been observed (1,4,6). Consistent with concentration, dose and time non-linearity, the elimination of ITZ was found to follow Michaelis-Menten kinetics in vivo with a Michaelis-Menten constant (Km) of 467 nM (7) and it was suggested that ITZ and/or its metabolites saturate intestinal and hepatic CYP3A4 and thus their own CYP3A4 mediated elimination (8).
Figure 1.

Structures of itraconazole (ITZ) and its metabolites hydroxy-itraconazole (OH-ITZ), keto-itraconazole (keto-ITZ) and N-desalkyl-itraconazole (ND-ITZ). * indicates a chiral center.
In agreement with possible saturation of CYP3A4 mediated elimination of ITZ, the drug is a potent inhibitor of CYP3A, causing significant drug-drug interactions when co-administered with other CYP3A substrates (9-13). In vitro to in vivo predictions of drug-drug interactions caused by ITZ have, however, regularly under-predicted the extent of CYP3A4 inhibition observed following ITZ administration (8,14-16). Possible reasons for this under-prediction include active uptake into hepatocytes (14), non-specific binding and circulating inhibitory metabolites (8).
Studies in humans have shown that ITZ undergoes extensive biotransformation, and only 1-2% of the administered dose of ITZ is excreted unchanged in the urine (2,6). The major known ITZ metabolite in humans is hydroxy-itraconazole (OH-ITZ) and it circulates in plasma at equal or higher concentrations than ITZ (2,6,7). In vitro, ITZ is metabolized by CYP3A4 to three products (Figure 1); OH-ITZ, keto-itraconazole (keto-ITZ), and N-desalkyl-itraconazole (ND-ITZ) (8). ND-ITZ has also been described as a urinary and fecal metabolite of ITZ in rats and dogs (17), but circulating plasma concentrations of keto-ITZ and ND-ITZ following ITZ dosing in humans are unknown. OH-ITZ, keto-ITZ and ND-ITZ are all potent inhibitors of CYP3A4 in vitro (8) and could contribute to the overall CYP3A4 inhibition effect observed in vivo if they circulate at sufficiently high concentrations. This study was designed to determine whether keto-ITZ and ND-ITZ are circulating metabolites of ITZ in vivo and can therefore contribute to CYP3A4 inhibition. In addition, the plasma unbound concentrations of ITZ, OH-ITZ, keto-ITZ and ND-ITZ were calculated after single and multiple doses so that their relative contribution to CYP3A4 inhibition could be predicted by in vitro to in vivo extrapolation.
METHODS
Chemicals
ITZ and OH-ITZ were purchased from Janssen Biotech, Research Diagnostics Inc. (Flanders, NJ). Dr. Jan Heeres, Janssen Pharmaceutica N. V. (Beerse, Belgium) generously provided keto-ITZ. ND-ITZ was prepared as previously reported (8,18). The ITZ-d5 (labeled in the terminal two carbons of the pyrazole N-alkyl side chain), used as an internal standard, was purchased from Toronto Research Chemicals Inc. (North York, Ontario, Canada). Acetonitrile was purchased from Fischer Scientific Co. (Fairlawn, NJ), and ammonium acetate from J.T. Baker (Phillipsburg, NJ). Ultrapure water, filtered through a Barnstead Nanopure filter system, was used throughout the study.
Clinical protocol
This protocol was approved by the University of Washington Institutional Review Board (IRB). Six healthy volunteers, two male and four female, with an age range of 19-33 years (mean 25) and all relatively normal weight (body mass index <30), participated in the study after each gave written consent. None of the subjects had significant medical history and none had taken medications (over-the-counter or prescription drugs other than oral contraceptives) for one week prior to and during the drug-dosing period. Potential subjects with known hypersensitivity or adverse reactions to azole antifungals were excluded. Subjects received 100 mg ITZ in oral solution (10 ml of 10 mg/ml) daily in the morning for seven days. Subjects fasted overnight prior to each ITZ dose. On day 1 and day 7 of ITZ dosing, a standard breakfast was served two and one-half hours after the ITZ dose. For the determination of ITZ and metabolite concentrations, blood samples were collected into heparinized tubes before and 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h after ITZ administration via an indwelling venous catheter. After the last ITZ dose (Day-7) additional blood samples were collected at 48 and 72 h. Plasma was isolated from blood samples by centrifugation. After the first and the last ITZ dose, total urine was collected for 24 hours, the volume was measured, and 10 mL aliquots were saved. Plasma and urine samples were stored at -20°C prior to analysis.
Determination of itraconazole and metabolite concentrations
For determination of plasma concentrations, all samples were analyzed in duplicate. A volume of 100 μl plasma was used for quantification of analyte concentrations less than 100 nM and 50 μl of plasma was used for concentrations greater than 100 nM. Each plasma sample was analyzed chromatographically for all compounds and the appropriate quantification method was applied based on peak areas. Each sample was prepared as follows: to the appropriate volume of plasma, 10 μl of internal standard (ITZ-d5, 710 nM for 100 μl of plasma and 296 nM for 50 μl of plasma) and 300 μl of acetonitrile were added, the samples were vortexed and centrifuged at 10,000 g for 10 minutes, and the supernatants were transferred to autosampler vials.
ITZ and metabolite plasma concentrations were measured by HPLC-mass spectrometry using an Agilent 1100 MSD system equipped with HP Chemstation data analysis software and operated in the positive ion electrospray mode, with selected ion monitoring as described previously (8). Five μl of each prepared sample supernatant was injected on column. The mobile phase consisted of acetonitrile and aqueous 5 mM ammonium acetate buffer (pH 7.0). Analyte separation was achieved using a Zorbax Eclipse XDB-C8 5 μm column (2.1 mm i.d. × 50 mm; Agilent) equipped with a Phenomenex C8 guard column (2.1 mm i.d × 4 mm) and gradient elution at 0.25 ml/min. The following ions were monitored to generate peak areas for each compound: m/z 705 for ITZ, m/z 721 for OH-ITZ, m/z 719 for keto-ITZ, m/z 649 for ND-ITZ, and m/z 712 for the internal standard (ITZ-d5).
Calibration curves for ITZ, OH-ITZ, keto-ITZ and ND-ITZ were prepared in plasma at concentrations between 1 nM and 100 nM (extracted from 100 μl of plasma) and between 100 nM and 1500 nM (from 50 μl of plasma). The limit of detection for ITZ and its metabolites was 1 nM and the limit of quantification was 5 nM. Quality control samples were included with each analysis. For each analyte, the plasma concentration was 7 nM and 20 nM for the 100 μL extraction, and 300 nM and 1316 nM for a 50 μL extraction. The inter-day variability for all compounds was < 8% at 1316 nM, < 12% at 300 nM, < 10% at 20 nM and < 22% at 7 nM.
The analysis of urine used volumes and a procedure similar to that described above for plasma analysis, that is, concentrations of ITZ and metabolites were determined from 100 μL or 50 μL of urine. A 10 μL volume of internal standard solution and 300 μl of acetonitrile were added, and the mixture was shaken for 20 minutes, centrifuged at 10,000 g for 10 min, and the supernatant was transferred to autosampler vials.
An aliquot of each urine sample was also treated with β-glucuronidase from Helix pomatia (Sigma, St Louis) to measure total conjugated products of ITZ and its metabolites. For β-glucuronidase treatment, the enzyme stock solution was diluted with 100 nM sodium acetate (pH 5) to 200 units/mL. To 1 mL urine samples, 1 mL (200 units) β-glucuronidase solution was added and the mixture was incubated in the dark for 24 hours at 37°C. The reaction was then quenched with additional 200 μL ice-cold acetonitrile, cooled, shaken for 20 min, and centrifuged. The supernatants were transferred to autosampler vials and analyzed using LC-MS, as described above.
Measurement of fraction unbound (fu) in plasma
Due to an apparently high non-specific binding of ITZ and its metabolites to solid surfaces, free drug was lost through adsorption to filters and membranes when employing ultrafiltration and dialysis methods for measurement of plasma free fraction. To overcome this complication, protein binding of ITZ and its metabolites was determined by ultracentrifugation (19). Experiments were conducted using a Beckman TLA-100 bench-top ultracentrifuge with a TLA-100 rotor. Free fraction was calculated in individual plasma samples from the subjects participating in the study. The total concentration range in the analyzed samples was 416 nM – 588 nM and 280 nM – 853 nM for ITZ and OH-ITZ respectively. Keto-ITZ and ND-ITZ did not circulate at sufficiently high concentrations to allow quantification of the unbound ultracentrifugate and thus these metabolites were spiked into the individual samples collected at steady state from the study subjects. Equal amounts of (2R4S2’R)- and (2R4S2’S)-keto-ITZ (the two stereoisomers formed in vivo from ITZ) and ND-ITZ were added to obtain a total concentration of 200 nM and 1000 nM for the two keto-ITZ enantiomers and ND-ITZ, respectively. Samples were vortexed for 30 seconds before aliquoting for analysis. One aliquot (200 μL) was placed into a polycarbonate ultracentrifuge tube (Beckman Coulter # 343775, Palo Alto, CA.) and centrifuged at 445,760 g at 37 °C for 140 min. Another aliquot was incubated for 140 minutes at 37 °C. After ultracentrifugation, a 50 μL aliquot was collected from the clear top layer and from the incubated plasma fraction and both were then prepared for LC-MS analysis. To each plasma and ultracentrifugation supernantant sample, 50 μL of acetonitrile and 10 μL of 710 nM ITZ-d5 (internal standard in acetonitrile) were added. The mixture was vortexed for 30 seconds and the samples were centrifuged at 16,100 g for 10 minutes at room temperature. The supernatants (60 μL) were then transferred to autosampler vials and analyzed by LC-MS, as described above. The plasma free fraction was calculated as the ratio of the free concentration in the ultracentrifugation supernatant and the total concentration in plasma. All free fractions reported are mean values obtained from the six different subject samples.
Ultracentrifugation has been used previously to measure the free fraction of a number of highly bound molecules (19), such as probucol and diazepam, and has been shown to give fu results that are in good agreement with those obtained by other methods. However, to further validate the ultracentrifugation method, we measured the free fractions of midazolam, fluconazole, and (S)-warfarin by the same technique that was applied to ITZ and its metabolites. The free fractions obtained, 1.9% for midazolam, 88% for fluconazole and 1.3% for (S)-warfarin, agreed well with those values reported in the literature using ultrafiltration/dialysis methods (midazolam 1.9 ± 0.6%, fluconazole 89 ± 1% and (S)-warfarin 1.03 ± 0.08%) (19-21).
Pharmacokinetic analysis
All pharmacokinetic parameters were obtained using the noncompartmental approach and Winnonlin data analysis software (Pharsight Corp. NC). The trough (Cmin) and peak concentrations (Cmax) and times at which peak concentrations were reached (tmax) were obtained directly from the plasma concentration versus time data. The AUC was determined using the linear trapezoidal method and the oral clearance was obtained from the quotient Dose/AUC. The average steady-state concentration (Css,ave) for each compound was calculated from the quotient AUC0-τ /τ where τ equals the dosing interval.
In vitro – in vivo predictions of the extent of hepatic CYP3A4 inhibition by ITZ and its metabolites were made according to Rowland and Matin (22). The effect of an inhibitor on the intrinsic clearance of a substrate is described according to equation 1:
| (1) |
Assuming only hepatic elimination and oral administration of the object drug, the effect of an inhibitor on the AUC of the object can be described using the following equation in which fm equals the fraction metabolized by the inhibited pathway, the inhibition constant (KI) is equal to the concentration of inhibitor at which metabolic turnover is reduced by 50% and [I] is the concentration of inhibitor present at the site of inhibition:
| (2) |
For a drug, such as midazolam or alprazolam, that is extensively metabolized by the inhibited pathway (fm ~1), equation 2 simplifies to:
| (3) |
Following the approach of Wang et al. (23), net inhibition is related to the sum of inhibitory contributions of all circulating inhibitors as described by equation 4:
| (4) |
For ITZ and its metabolites, this can be expressed as:
| (5) |
Similarly, the effect of ITZ and its metabolites on the CYP3A mediated hepatic intrinsic clearance can be expressed as:
| (6) |
The KI -value can be substituted with the IC50 for competitive inhibitors when probe substrate concentration is much less than the Km value, or when the mechanism of inhibition is noncompetitive (24). For the predictions presented in this work an IC50-value of each compound was used in place of the KI. These IC50 values were predicted to be equal to the KI’s because the substrate concentration employed in the inhibition experiments was ≪ Km (8).
To predict the effect of ITZ and its metabolites on hepatic midazolam and alprazolam intrinsic clearance in vivo, four different combinations of inhibitor concentration and IC50 were utilized; unbound IC50 values with unbound steady-state Cmax (Css,max,u) or unbound steady-state Css,ave (Css,ave,u), and total IC50 values with total Css,max or Css,ave.
Statistical analysis
Data are expressed as mean ± SD. Statistical analysis was performed with the Systat 5.04 statistical analysis program (SYSTAT Inc. Evanston, IL). Wilcoxon’s matched pair test was used to evaluate significance of the pharmacokinetic parameters for each compound on day 1 and day 7. The differences in trough concentrations between day 1, day 6, and day 7 were assessed with Friedman’s multiple comparison analysis of variance with Wilcoxon’s matched pair test as a post hoc analysis.
RESULTS
Following administration of 100 mg ITZ in oral solution, three metabolites of ITZ, OH-ITZ, keto-ITZ, and ND-ITZ, were detected in the plasma of all volunteers (figure 2). The mean plasma concentration versus time curves for ITZ, OH-ITZ, keto-ITZ, and ND-ITZ are shown in figure 3 and the average pharmacokinetic parameters obtained from individual subjects are summarized in tables I and II. Cmax of ITZ was observed within 1-2 hours after dosing of the oral solution. The concentrations of OH-ITZ and keto-ITZ peaked simultaneously with ITZ. No accurate Cmax could be determined for ND-ITZ since its concentration versus time profile was fairly flat after 2 hours (figure 3).
Figure 2.
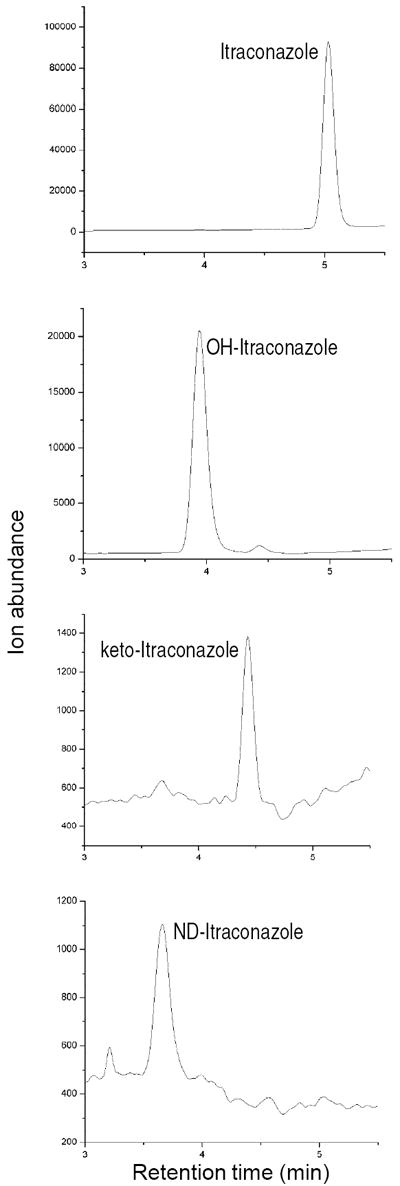
Selected ion chromatograms from a representative subject plasma showing the detection of itraconazole (ITZ), hydroxy-ITZ (OH-ITZ), keto-ITZ and N-desalkyl-ITZ (ND-ITZ) in vivo after administration of 100 mg itraconazole in oral solution.
Figure 3.
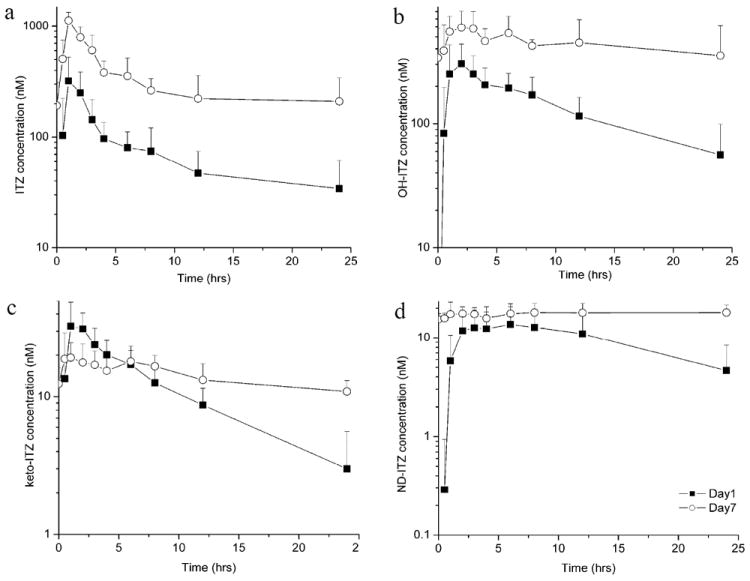
Mean plasma concentration (± SD) versus time curves for itraconazole (a), hydroxy-itraconazole (OH-ITZ) (b), keto-itraconazole (keto-ITZ) (c) and N-desalkyl-itraconazole (ND-ITZ) (d) on day 1 (solid boxes) and day 7 (open circles) of the study in the six volunteers. The symbols are the mean values of the six subjects and the bars show standard deviation. For keto-ITZ only two subjects had quantifiable plasma concentrations at 24 hours after the first dose and for ND-ITZ only one subject had quantifiable plasma concentrations at 0.5 hours after first ITZ dose. For calculation of the mean concentration other plasma concentrations were considered to be zero.
Table I.
Pharmacokinetic parameters of itraconazole and its metabolites after single dose administration of 100 mg oral solution of itraconazole and following multiple dose administration for 7 days.
| Compound and study phase | AUC0→∝ or AUC0→241 (hr*nmol/L) | Css,ave (nM) | Css,ave,u2 (nM) | Cmax (nM) | Cmax,u 2 (nM) |
|---|---|---|---|---|---|
| ITZ (day 1) | 2862 ± 2158 | 362 ± 148 | 13 | ||
| ITZ (day 7) | 7240 ± 3177* | 301 | 11 | 1152 ± 203* | 41 |
| OH-ITZ (day1) | 4237 ± 2175 | 358 ± 118 | 1.6 | ||
| OH-ITZ (day 7) | 9811 ± 5263* | 409 | 1.8 | 608 ± 206* | 2.7 |
| Keto-ITZ (day 1) | 322 ± 96 | 39 ± 11 | 2.1 | ||
| Keto-ITZ (day 7) | 308 ± 81 | 17 | 0.90 | 23 ± 9* | 1.2 |
| ND-ITZ (day 1) | 389 ± 150 | 15 ± 7 | 0.18 | ||
| ND-ITZ (day 7) | 394 ± 133 | 20 | 0.24 | 22 ± 3 | 0.26 |
Values for day 1 are from 0 hrs to infinity and day 7 are for the dosing interval, from 0 hrs to 24 hours.
Unbound values are extrapolated based on in vitro protein binding experiments.
Significantly different (Wilcoxon matched pair test, p<0.05) from the day 1 value.
Table II.
The trough concentrations (nM) of itraconazole (ITZ), hydroxy-itraconaozle (OH-ITZ), keto-itraconazole (keto-ITZ) and N-desalkyl-itraconazole (ND-ITZ).
| ITZ | OH-ITZ | Keto-ITZ | ND-ITZ | |
|---|---|---|---|---|
| Day 1 (unbound) | 34 ± 27* (1.2) | 56 ± 43* (0.25) | 3 ± 3* (0.16) | 5 ± 4* (0.060) |
| Day 6 (unbound) | 193 ± 117 (6.9) | 339 ± 244 (1.5) | 12 ± 4 (0.64) | 16 ± 2 (0.19) |
| Day 7 (unbound) | 210 ± 132 (7.6) | 352 ± 261 (1.6) | 11 ± 2 (0.58) | 18 ± 4 (0.22) |
Significantly different (Wilcoxon matched pair test, p<0.05) from the values obtained on day 7.
The Css,ave values of ITZ and OH-ITZ were approximately 20 times higher than those of keto-ITZ (17 nM) and ND-ITZ (20 nM) following the seventh ITZ dose. The Cmax and Cmin for ITZ and its metabolites are summarized in tables I and II. The significant increase in the trough concentrations of ITZ and each of its metabolites between day 1 and day 7 shows accumulation of all species following multiple dosing. The Cmin of all species measured did not increase significantly (p>0.05) between the trough sample of day 6 and the trough sample of day 7, suggesting an approximation of steady-state conditions.
The urine excretion data are summarized in table III. ITZ, OH-ITZ and ND-ITZ were present predominantly as conjugates and could be detected after treatment of the urine with β-glucuronidase. Keto-ITZ was not detected in urine, but some OH-ITZ was excreted unchanged. At apparent steady state, the sum of the amounts of ITZ, OH-ITZ and ND-ITZ excreted in the urine during the 24-hour dosing interval, free or as conjugates, accounted for less than 2 percent of the administered dose.
Table III.
Amounts of conjugated itraconazole (ITZ), ND-ITZ and the free and conjugated hydroxy-itraconazole (OH-ITZ) excreted into urine during the 24 hour dosing interval on day 1 and day 7 of the study. The β-glucuronidase used has sulfatase activity as well and thus it is possible that some of the cleaved conjugates were sulfates instead of glucuronides.
| ITZ-conjugate (nmol) | ND-ITZ-conjugate (nmol) | OH-ITZ-conjugate (nmol) | OH-ITZ (nmol) | Total OH-ITZ (nmol) | |
|---|---|---|---|---|---|
| Day 1 | 6 ± 6 | 9 ± 6 | 40 ± 17 | 59 ± 44 | 100 ± 49 |
| Day 7 | 19 ± 15 | 4 ± 4 | 204 ± 89 | 62 ± 31 | 266 ± 90 |
Results from protein binding experiments demonstrated a much higher free fraction of ITZ than previously reported (27). The measured free fractions were 3.6% ± 0.3% for ITZ, 0.5% ± 0.2% for OH-ITZ, 5.3% ± 0.7% for keto-ITZ, and 1.2% ± 0.2% for ND-ITZ. Free fraction was independent of the total concentration in the samples and of the other compounds spiked in the same sample within the range tested. Based on the unbound fractions in each individual, Cmax,u, and Css,ave,u were calculated for ITZ and its metabolites (table I). On Day 7, ITZ Css,max,u (41 nM) was more than 15-fold greater than the Css,max,u of OH-ITZ (2.7 nM). In addition, the Css,ave,u of ITZ (11 nM) was approximately 6-fold greater than that of OH-ITZ (1.8 nM), 12-fold greater than keto-ITZ (0.90 nM) and 46-fold greater than that of ND-ITZ (0.24 nM).
Based on the single dose data, an ITZ Css,ave (0-24 hours) of 119 nM was expected. However, the observed Css,ave was 301 nM, demonstrating an unexpected accumulation of ITZ between the first and seventh dose. Under classical multiple-dose administration kinetics (assuming constant clearance), the AUC0→τ at steady state is equal to the AUC0→∝ after a single dose (25,26). However, the AUC0→τ for ITZ on day 7 was 2.5 times larger than the AUC0→∝ of ITZ on day 1 (p<0.05), demonstrating either a significant decrease in the systemic ITZ clearance or an increase in its bioavailability following multiple dosing. This was reflected in a 2- to 3-fold decrease in oral clearance of ITZ between day 1 (64 ± 24 L/hr) and day 7 (22 ± 7 L/hr). Similarly, the AUC0→τ for OH-ITZ after the seventh ITZ dose was larger (2.3-fold) than the AUC0→∝ of OH-ITZ following the first dose of ITZ (p<0.05). However, the AUC0→24 on day 7 for keto-ITZ and ND-ITZ was approximately equivalent to the AUC0→∝ of these compounds on day 1. A comparison of the Css,ave,u concentrations of ITZ, OH-ITZ and keto-ITZ to the in vitro unbound Km (Km,u) values towards CYP3A4 can explain in part the observed kinetic non-linearity. For ITZ, the Css,ave,u is 2.8-fold greater than the Km,u. The Css,ave,u/Km,u ratios of OH-ITZ and keto-ITZ were 0.067 and 0.64, respectively. If CYP3A4 is the predominant route of ITZ elimination, this would result in saturation of ITZ clearance and non-linear elimination of ITZ, as was observed.
In agreement with the observed nonlinear elimination kinetics of ITZ, possibly related to CYP3A4 saturation, significant CYP3A4 inhibition was predicted based on the circulating concentrations of ITZ and its metabolites (table IV). The predicted CLint-ratios (CLint/CLint,I) for a CYP3A4 probe are 3.9 and 9.0 based on the Css,ave,u/IC50,u and Css,max,u/IC50,u, respectively, when all inhibitory species are considered. When only ITZ was included, the predicted CLint-ratios were 2.8 and 7.7 based on Css,ave,u/IC50,u and Css,max,u/IC50,u, respectively. As expected for a highly bound drug, the predicted AUC ratio was much greater if total concentration and IC50 values were used rather than the unbound values (table IV).
Table IV.
Predicted in vivo inhibition caused by itraconazole (ITZ) and its metabolites. IC50 values are from reference 8 and determined using human liver microsomes. These IC50 values approximate the KI because the substrate concentration used was much below its Km – value.
| Parameter | ITZ | OH-ITZ | Keto-ITZ | ND-ITZ | CLint/Clint,I* |
|---|---|---|---|---|---|
| Css,ave/IC50 | 10 | 11 | 0.32 | 1.2 | 24 |
| Css,ave,u/IC50,u | 1.8 | 0.39 | 0.13 | 0.60 | 3.9 |
| Css,max/IC50 | 40 | 16 | 0.43 | 1.3 | 59 |
| Css,max,u/IC50,u | 6.7 | 0.59 | 0.17 | 0.59 | 9.0 |
The CLint ratio was predicted according to equation 1. This number will be the same for a prediction of an AUC ratio (AUCi/AUC) for an orally dosed, fully absorbed substrate that is metabolized only by hepatic CYP3A4.
The predicted time course of CYP3A4 inhibition after ITZ dosing is shown in figure 4. The relative roles of ITZ and its metabolites in inhibition of CYP3A4 varied over time and the relative importance of the metabolites in overall CYP3A inhibition depend on the time-window observed after dosing. As shown above, based on average unbound plasma concentrations and IC50,u values, ITZ was the major inhibitor after oral administration. However, the relative role of ND-ITZ increased with time after ITZ dosing (figure IV). After single dose, ND-ITZ contributed approximately 40% of the overall inhibition between 12 and 24 hours after dosing. After multiple ITZ doses, ND-ITZ was predicted to contribute approximately 30-40 % of overall CYP3A4 inhibition 24 hours after last ITZ dose and later.
Figure 4.
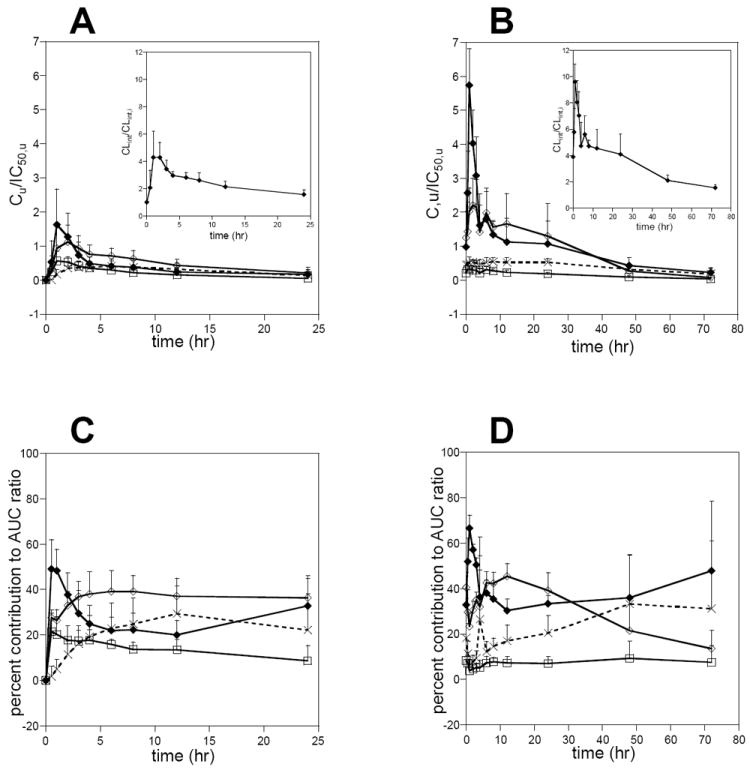
The predicted time-course of CYP3A4 inhibition by ITZ and its metabolites on study day 1 (A) and day 7 (B) and the relative contribution of each inhibitory species to the overall CYP3A4 inhibition on study day 1 (C) and day 7 (D). The insets in A and B depict the time course of the total predicted fold decrease in the intrinsic clearance of a CYP3A4 substrate when ITZ and its metabolites are all used for prediction. Closed diamonds show ITZ, open diamonds OH-ITZ, open boxes keto-ITZ and crosses ND-ITZ.
DISCUSSION
This study was designed to determine whether keto-ITZ and ND-ITZ, two recently identified new metabolites of ITZ, circulate in vivo, and whether these metabolites of ITZ as well as OH-ITZ could contribute to the inhibition of CYP3A4 in vivo. After ITZ administration, OH-ITZ, keto-ITZ and ND-ITZ were observed in plasma, and they accumulated with multiple dosing. Of the four compounds studied, ITZ and OH-ITZ circulated at comparable concentrations, which were significantly higher than keto-ITZ and ND-ITZ concentrations. Based on unbound plasma concentrations and IC50 values, OH-ITZ and ND-ITZ were predicted to contribute to the inhibition of CYP3A4 in vivo at steady-state.
The single and multiple dose plasma concentrations of ITZ and OH-ITZ obtained in this study agree well with other reports in the literature (2,4,7). Because of the possibility of non-dissociative sequential formation of OH-ITZ, keto-ITZ, and ND-ITZ from ITZ by CYP3A4, it was predicted that if CYP3A4 accounts for a significant portion of ITZ elimination in vivo, keto-ITZ and ND-ITZ would be formed and circulate in vivo regardless of the extent of enzyme saturation by ITZ or OH-ITZ (8). As predicted, keto-ITZ and ND-ITZ were present in all plasma samples collected.
The fu in plasma for ITZ measured in this study (3.6%) was significantly greater than has been previously reported in the itraconazole NDA (0.2%) (27). The fu-values for OH-ITZ, keto-ITZ and ND-ITZ have not been previously reported. A likely explanation for the observed difference in the fu determined for ITZ by ultracentrifugation versus other methods reported previously is the high non-specific binding of ITZ. Dialysis and ultrafiltration methods require the molecule to pass through a membrane interface. Free molecules with high non-specific binding will adsorb to this membrane interface causing loss of detectable free drug. The dialysis method could overcome this problem if equilibrium was achieved but this presumably takes a considerable length of time. Ultracentrifugation avoids the problem of nonspecific loss through a membrane and filter completely.
Calculation of the unbound plasma concentrations of ITZ and its metabolites aids in the interpretation of the dose- and time-dependent changes on the elimination of ITZ. The unbound Cmax of ITZ on day one was 13 nM, a value that is 3- to 10-fold higher than Km,u (3.9 nM) and KI,u (1.3 nM) of ITZ for CYP3A4 measured in vitro (8). This comparison of the Km,u and KI,u values with unbound plasma concentrations suggests that metabolism of ITZ by CYP3A4 should follow saturation (Michaelis-Menten) kinetics in vivo. If a large fraction of ITZ is eliminated via CYP3A4 metabolism, the overall elimination should be non-linear. Consistent with this prediction, an approximately 2.5-fold increase in the AUC of ITZ and OH-ITZ was observed between the first dose (0→∝) administration and day 7 (0→τ) in this study, corresponding to a significant decrease in oral clearance of ITZ, most likely a result of saturation of intestinal and hepatic CYP3A4 leading to decreased systemic clearance and increased oral bioavailability. Barone et al. (7) found the Km of ITZ in humans in vivo to be 467 nM (total concentration), which would correspond to a free concentration of 17 nM (based on fu = 3.6%). This unbound value is similar to the in vitro Km,u of ITZ for CYP3A4 (4 nM). Based on in vitro experiments, OH-ITZ, keto-ITZ and ND-ITZ are formed from ITZ only via CYP3A4 (8), but mass balance for ITZ following IV administration has not been determined. Therefore, it is not known what fraction of the ITZ dose is eliminated via the pathway that begins with OH-ITZ formation.
Saturation of CYP3A4 by ITZ and its metabolites in vivo is in agreement with the clinically observed interactions between ITZ and other CYP3A substrates. Based on pharmacokinetic theory, saturable elimination kinetics are expected when the clearance of a drug is dependent on a single enzyme, and the drug causes inhibitory interactions with other drugs cleared by that same enzyme (i.e. CYP3A4). Based on the Css,u/IC50,u values for ITZ, OH-ITZ, keto-ITZ and ND-ITZ (1.8, 0.39, 0.13 and 0.60 respectively), ITZ alone is predicted to partially saturate CYP3A4, but the metabolites, and ND-ITZ in particular, should also contribute to inhibition of CYP3A4 mediated metabolism. Interestingly, ND-ITZ appears to have a much longer half-life than ITZ, OH-ITZ and keto-ITZ suggesting that ND-ITZ could be responsible for the clinically observed persistent inhibition of CYP3A4-mediated clearance of immunosuppressants co-administered with ITZ (29,30).
The prediction of clinically important drug-drug interactions caused by ITZ using the unbound concentrations and metabolite data described here agrees well with available clinical observations, and provides a significant improvement over previous in vitro-to-in vivo predictions. If the predicted intrinsic clearance value obtained here (3.9-fold) is compared to the clearance ratio (CL/CLI =3.2) obtained after iv administration of midazolam before and after four days of ITZ (200 mg/day) treatment (10), a slight overprediction is obtained. The discrepancy is most likely due to fact that midazolam is an intermediate extraction ratio drug and thus hepatic blood flow partially limits its systemic clearance dampening the effect of the inhibitor. The average steady state ITZ concentration measured in this study was similar to the ITZ concentration measured in the interaction study (10). Alprazolam is a low extraction ratio CYP3A4 substrate that is not subject to significant intestinal metabolism (28). Thus we compared our predicted AUC ratio (equation 4) to the observed effect of four days of ITZ treatment (200 mg/day) on alprazolam oral clearance (28). Again, the observed in vivo effect on AUC was in relatively good agreement with our predicted data (observed ratio=2.5, predicted ratio=3.9). The slight overprediction is possibly a result of the contribution from other enzymes besides CYP3A to alprazolam clearance. It should also be noted, that the dosing of ITZ was different in the two interaction studies and in our study, which might significantly affect metabolite plasma concentrations and subsequently CYP3A inhibitory effect and future studies that measure simultaneously both the concentration of the inhibitory metabolites and the inhibitory effect are needed.
Several factors complicate the quantitative assessment of the contribution of ITZ metabolites to in vivo CYP3A4 inhibition. First, the total hepatic concentration of ITZ can greatly exceed the total plasma concentration (11- to 14-fold) and active uptake for ITZ has been proposed based on rat data (15). It is possible that the metabolites are also actively taken up into the hepatocytes and therefore, unbound ITZ and metabolite concentrations in the hepatocytes may exceed that found in plasma. Second, the net inhibitory effect from ITZ administration may be non-classical, complicated by the possibility of non-dissociative sequential metabolism of ITZ to keto-ITZ and ND-ITZ. ITZ itself has high affinity and apparent slow dissociation rate from CYP3A4 (31), and the concentration/abundance of the CYP3A4-metabolite complex may be under-predicted from unbound plasma and hepatocyte concentrations. Assessment of active uptake of ITZ and its metabolites into hepatocytes and the role of sequential metabolism in CYP3A4 inhibition requires further study.
Stereochemical issues further complicate interpretation of the in vivo and in vitro kinetic data for ITZ metabolism. ITZ is administered as a mixture of four stereoisomers, of which all inhibit CYP3A4 but only two are metabolized by CYP3A4 to OH-ITZ, keto-ITZ and ND-ITZ, resulting in different circulating concentrations of ITZ stereoisomers (32,33). The stereoselective metabolism of ITZ leads to the formation of three OH-ITZ, two keto-ITZ and a single ND-ITZ stereoisomer in comparison to mixtures of eight, four, and two stereoisomers, respectively, used for in vitro inhibition studies. The inhibitory potency of the individual stereoisomers of the metabolites is currently unknown but it is possible that there are significant differences in their ability to inhibit CYP3A4 in vitro that will in turn alter the in vitro to in vivo predictions presented here.
Finally, this is the first study to report evidence for glucuronidation of ITZ or its metabolites. One earlier study suggested that ITZ is subject to enterohepatic recycling (4) but did not identify the metabolite(s). N-Glucuronidation of the new triazole antifugal posaconazole by UGT1A4 was recently reported and several glucuronides of this azole were detected in urine (34,35). Posaconazole is structurally similar to OH-ITZ and thus it seems possible that OH-ITZ may undergo similar glucuronidation. Moreover, the low recovery of the OH-ITZ glucuronides in urine may be due to the preferential excretion of these glucuronides into bile, which is the main elimination route of the posaconazole glucuronides. It should be noted that the β-glucuronidase used in this study also has sulfatase activity and therefore the detected conjugates cannot be assigned as glucuronides with certainity. It is possible that these conjugates or other unidentified metabolites of ITZ inhibit CYP3A4 mediated metabolism in vivo.
In conclusion, sequentially formed ITZ metabolites OH-ITZ, keto-ITZ and ND-ITZ were observed to circulate in vivo at appreciable levels and were predicted to contribute to in vivo CYP3A4 inhibition observed after ITZ dosing. Of the metabolites identified, ND-ITZ was predicted to play the most significant role in CYP3A4 inhibition. Use of the unbound average steady state concentrations of ITZ and its metabolites provided significantly improved in vitro to in vivo prediction of CYP3A4 inhibition by ITZ. These observations show that inhibitory metabolites need to be taken into account when in vivo drug-drug interactions are predicted for novel compounds and when extent of interaction is evaluated for known compounds.
Acknowledgments
This study was supported in part by National Institutes of Health Grants PO1 GM32165, K24DA00417 and P30 ES07033. A portion of this work was conducted through the Clinical Research Center Facility at the University of Washington and supported by the National Institutes of Health Grant M01-RR-00037.
References
- 1.Haria M, Bryson HM, Goa KL. Itraconazole. A reappraisal of its pharmacological properties and therapeutic use in the management of superficial fungal infections. Drugs. 1996;51:585–620. doi: 10.2165/00003495-199651040-00006. [DOI] [PubMed] [Google Scholar]
- 2.Poirier JM, Cheymol G. Optimisation of itraconazole therapy using target drug concentrations. Clin Pharmacokinet. 1998;35:461–473. doi: 10.2165/00003088-199835060-00004. [DOI] [PubMed] [Google Scholar]
- 3.Barone JA, et al. Enhanced bioavailability of itraconazole in hydroxypropyl-beta-cyclodextrin solution versus capsules in healthy volunteers. Antimicrob Agents Chemother. 1998;42:1862–1865. doi: 10.1128/aac.42.7.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardin TC, et al. Pharmacokinetics of itraconazole following oral administration to normal volunteers. Antimicrob Agents Chemother. 1988;32:1310–1313. doi: 10.1128/aac.32.9.1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schäfer-Körting M. Pharmacokinetic optimisation of oral antifungal therapy. Clin Pharmacokinet. 1993;25:329–341. doi: 10.2165/00003088-199325040-00006. [DOI] [PubMed] [Google Scholar]
- 6.Heykants J, et al. The clinical pharmacokinetics of itraconazole: an overview. Mycoses. 1989;32(Suppl 1):67–87. doi: 10.1111/j.1439-0507.1989.tb02296.x. [DOI] [PubMed] [Google Scholar]
- 7.Barone JA, et al. Food interaction and steady-state pharmacokinetics of itraconazole capsules in healthy male volunteers. Antimicrob Agents Chemother. 1993;37:778–784. doi: 10.1128/aac.37.4.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Isoherranen N, Kunze KL, Allen KE, Nelson WL, Thummel KE. Role of itraconazole metabolites in CYP3A inhibition. Drug Metab Dispos. 2004;32:1121–1131. doi: 10.1124/dmd.104.000315. [DOI] [PubMed] [Google Scholar]
- 9.Neuvonen PJ, Kantola T, Kivisto KT. Simvastatin but not pravastatin is very susceptible to interaction with the CYP3A4 inhibitor itraconazole. Clin Pharmacol Ther. 1998;63:332–341. doi: 10.1016/S0009-9236(98)90165-5. [DOI] [PubMed] [Google Scholar]
- 10.Olkkola KT, Ahonen J, Neuvonen PJ. The effect of systemic antimycotics, itraconazole and fluconazole, on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Anesthesia and Analgesia. 1996;82:511–516. doi: 10.1097/00000539-199603000-00015. [DOI] [PubMed] [Google Scholar]
- 11.Backman JT, Kivisto KT, Olkkola KT, Neuvonen PJ. The area under the plasma concentration-time curve for oral midazolam is 400-fold larger during treatment with itraconazole than with rifampicin. Eur J Clin Pharmacol. 1998;54:53–58. doi: 10.1007/s002280050420. [DOI] [PubMed] [Google Scholar]
- 12.Florea NR, Capitano B, Nightingale CH, Hull D, Leitz GJ, Nicolau DP. Beneficial pharmacokinetic interaction between cyclosporine and itraconazole in renal transplant recipients. Transplant Proc. 2003;35:2873–2877. doi: 10.1016/j.transproceed.2003.10.058. [DOI] [PubMed] [Google Scholar]
- 13.Mahnke CB, et al. Tacrolimus dosage requirements after initiation of azole antifungal therapy in pediatric thoracic organ transplantation. Pediatr Transplant. 2003;7:474–478. doi: 10.1046/j.1397-3142.2003.00103.x. [DOI] [PubMed] [Google Scholar]
- 14.Yamano K, Yamamoto K, Kotaki H, Sawada Y, Iga T. Quantitative prediction of metabolic inhibition of midazolamby itraconazole and ketoconazole in rats: Implication of concentrative uptake of inhibitors into liver. Drug Metab Dispos. 1999;27:395–402. [PubMed] [Google Scholar]
- 15.Yamano K, et al. Prediction of midazolam-CYP3A inhibitors interaction in the human liver from in vivo/in vitro absorption, distribution, and metabolism data. Drug Metab Dispos. 2001;29:443–452. [PubMed] [Google Scholar]
- 16.Galetin A, Ito K, Hallifax D, Houston JB. CYP3A4 Substrate Selection and Substitutions in the Prediction of Potential Drug-Drug Interactions. J Pharmacol Exp Ther. 2005;314:180–190. doi: 10.1124/jpet.104.082826. [DOI] [PubMed] [Google Scholar]
- 17.Heykants J, et al. The pharmacokinetics of itraconazole in animals and man: an overview. In: Fromtling RA, editor. Recent Trends in the Discovery, Development, and Evaluation of Antifungal Agents. J.R. Prous Science Publishers; Barcelona: 1987. [Google Scholar]
- 18.Heeres J, Backx LJ, Van Cutsem J. Antimycotic azoles. 7. Synthesis and antifungal properties of a series of novel triazol-3-ones. J Med Chem. 1984;27:894–900. doi: 10.1021/jm00373a015. [DOI] [PubMed] [Google Scholar]
- 19.Nakai D, Kumamoto K, Sakikawa C, Kosaka T, Tokui T. Evaluation of the Protein Binding Ratio of Drugs by A Micro-Scale Ultracentrifugation Method. J Pharm Sci. 2004;93(4):847–854. doi: 10.1002/jps.20012. [DOI] [PubMed] [Google Scholar]
- 20.Brunton LL, Lazo JS, Parker KL. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 11. McGraw-Hill; New York: 2006. [Google Scholar]
- 21.Trouvin JH, et al. Pharmacokinetics of midazolam in anaesthetized cirrhotic patients. Br J Anaesth. 1988;60:762–7. doi: 10.1093/bja/60.7.762. [DOI] [PubMed] [Google Scholar]
- 22.Rowland M, Matin SB. Kinetics of Drug-Drug Interactions. J Pharm Bio. 1973;1(6):553–568. [Google Scholar]
- 23.Wang Y-H, Jones DR, Hall SD. Prediction of Cytochrome P450 3A Inhibition by Verapamil Enantiomers and Their Metabolites. Drug Metab Dispos. 2004;32:259–266. doi: 10.1124/dmd.32.2.259. [DOI] [PubMed] [Google Scholar]
- 24.Webb JL. Enzyme and metabolic inhibitors. Academic Press; New York: 1963. [Google Scholar]
- 25.Rowland M, Tozer TN. Clinical Pharmacokinetics: Concepts and Applications. 3. Lippincott Williams & Wilkins; Philadelphia: 1995. [Google Scholar]
- 26.Gibaldi M, Perrier D. Pharmacokinetics. 2. Marcel Dekker Inc.; New York: 1982. [Google Scholar]
- 27.Meuldermans W, Heykants J. The plasma protein binding of itraconazole and its distribution in blood. Janssen Pharmaceutica Preclinical Research Report. 1986 R51211/33. [Google Scholar]
- 28.Yasui N, et al. Effect of itraconazole on the single oral dose pharmacokinetics and pharmacodynamics of alprazolam. Psychopharmacology. 1998;139:269–273. doi: 10.1007/s002130050715. [DOI] [PubMed] [Google Scholar]
- 29.Banerjee R, Leaver N, Lyster H, Banner NR. Coadministration of itraconazole and tacrolimus after thoracic organ transplantation. Transplant Proc. 2001;33:1600–1602. doi: 10.1016/s0041-1345(00)02608-7. [DOI] [PubMed] [Google Scholar]
- 30.Kwan JT, Foxall PJ, Davidson DG, Bending MR, Eisinger AJ. Interaction of cyclosporin and itraconazole. Lancet. 1987;2:282. doi: 10.1016/s0140-6736(87)90873-7. [DOI] [PubMed] [Google Scholar]
- 31.Pearson JT, et al. Surface plasmon resonance analysis of antifungal azoles binding to CYP3A4 with kinetic resolution of multiple binding orientations. Biochemistry. 2006;45(20):6341–6353. doi: 10.1021/bi0600042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Breadmore MC, Thormann W. Capillary electrophoresis evidence for the stereoselective metabollism of itraconazole in man. Electrophoresis. 2003;24:2588–2597. doi: 10.1002/elps.200305494. [DOI] [PubMed] [Google Scholar]
- 33.Kunze KL, Nelson WL, Kharasch ED, Thummel KE, Isoherranen N. Stereochemical aspects of itraconazole metabolism in vitro and in vivo. Drug Metab Dispos. 2006;34(4):583–90. doi: 10.1124/dmd.105.008508. [DOI] [PubMed] [Google Scholar]
- 34.Ghosal A, et al. Identification of human UDP-glucuronosyltransferase enzyme(s) responsible for the glucuronidation of posaconazole (Noxafil) Drug Metab Dispos. 2004;32:267–71. doi: 10.1124/dmd.32.2.267. [DOI] [PubMed] [Google Scholar]
- 35.Krieter P, et al. Disposition of posaconazole following single-dose oral administration in healthy subjects. Antimicrob Agents Chemother. 2004;48:3543–51. doi: 10.1128/AAC.48.9.3543-3551.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]