

Abstract
Advanced contraceptive peptide vaccines suffer from the unavailability of adjuvants capable of enhancing the antibody response with acceptable safety. We sought to overcome this limitation by employing two novel poly(lactic-co-glycolic acid) (PLGA) microsphere formulations to deliver a synthetic human chorionic gonadotropin (hCG) peptide antigen co-synthesized with a T-cell epitope from tetanus toxoid, C-TT2-CTP35: surface-conjugated immunogen to induce phagocytosis; and encapsulated peptide to provide a depot effect, with MgCO3 co-encapsulated in the polymer to neutralize acidity from the biodegrading PLGA polyester. A single immunization of encapsulated peptide in rabbits elicited a stronger antibody response with equivalent duration relative to a positive control — three injections of the peptide administered in a squalene-based water-in-oil emulsion. Surface-conjugated peptide was less effective but enhanced antibody levels at 1/5 the dose, relative to soluble antigen. Most remarkable and unexpected was the finding that co-encapsulation of base was essential to attain the powerful adjuvant effect of the PLGA – MgCO3 system, as the MgCO3-free microspheres were completely ineffective. A promising contraceptive hCG peptide vaccine with acceptable side effects (i.e., local tissue reactions) was achieved by minimizing PLGA and MgCO3 doses, without significantly affecting antibody response.
Keywords: Human chorionic gonadotropin, Poly(lactic-co-glycolic acid) microspheres, Vaccine adjuvant
1. Introduction
For several decades the World Health Organization (WHO) has pursued development of safe, effective and long-acting (e.g., 6 months) nonsteroidal immunocontraceptives to obviate side effects, such as endocrine and other metabolic disturbances, often associated with long-acting steroid hormone preparations [1]. At the forefront of the newly recognized field of immunopharmaceuticals, vaccines against hCG are among the most advanced contraceptive vaccines, utilizing the body's own immune defense system to provide protection against pregnancy [2,3]. A hormone synthesized in appreciable amounts exclusively during pregnancy by the early conceptus or in some cancers, hCG plays an essential role for establishing and maintaining early pregnancy [4,5]. Antibodies against hCG exert antifertility action at the post-fertilization/preimplantation stage without disturbance of the normal ovulation function or hormone secretion. Once the effective antibody level has retreated after several months, immunity can be boosted to continue contraception or discontinued to recover fertility. A successful immunocontraceptive is expected to provide a new and improved method for family planning without the fear of side effects or need of access to high quality services, and lead to a significant public health benefit for millions of families [1].
The C-terminal peptide (CTP) region of the beta chain is a desirable hCG-fragment for immunogen candidates because CTP does not share homology with other hormones, such as human luteinizing hormone [6]. A synthetic peptide antigen, C-TT2-CTP35, consisting of a B-cell epitope from CTP (residues 111-145) [7] and a T-cell epitope from tetanus toxoid (TT) (residues 830-844, designated as TT2) [8,9] has been shown to elicit antibody responses comparable to those induced by the same CTP fragment conjugated to diphtheria toxoid [10]. As a “promiscuous” or universal T-cell epitope, TT2 provides advantage of not being MHC-restricted and thus broadly reactive in multiple haplotypes, as compared with TT or diphtheria toxoid, which often have been used as carriers for hCG vaccines to provide the T-cell helper effect [8,9]. However, strong adjuvant oily vehicles such as a combination of squalene, surface active agents, and/or bacteria surface component analogues were necessary to induce strong immune responses to C-TT2-CTP35 — a recurring problem for poorly immunogenic peptide vaccines [3,10]. Such adjuvants are undesirable because of reported fever, pain, and abscesses accompanying high doses of these chemotactic agents [3].
PLGA polymeric microspheres, by contrast, are among the most studied adjuvants to provide safe and effective single-dose delivery of vaccine antigens, including synthetic peptides, because of their biodegradability, low toxicity, slow-release, and targeting to antigen-presenting cells (APCs) [11-14]. As microparticles of PLGA polyesters degrade to produce lactic and glycolic acids in vivo, encapsulated immunogens are slowly and continuously released resulting in repeated stimulation of lymphocytes, including antigen presenting cells, eliminating the need for multiple immunizations. In addition to antibody production, PLGA microparticles have been observed to induce phagocytosis and elicit cellular immune responses in contrast to aluminum-based adjuvants [15-17]. Despite this progress, until now PLGA microspheres encapsulating hCG peptide antigens have been unsuccessful to achieve strong immune responses when administered in safe (non-oily) aqueous vehicles. Co-adjuvants, squalene-based water-in-oil emulsion containing a normuramyl dipeptide, and mannide monooleate, need to be administrated concurrently (as a mixture or separately) with peptide-containing PLGA microspheres to boost sufficiently high antibody response.
To meet these challenges, we describe two novel microparticle approaches, antigen surface-conjugated to PLGA and antigen encapsulation in PLGA – MgCO3, to improve delivery of peptide antigens. The latter preparation injected in an aqueous vehicle was found to be a powerful adjuvant for the hCG peptide, overcoming the apparent single remaining obstacle in the search for an effective and safe immunocontraceptive. The side effects or local tissue responses of this novel PLGA – MgCO3/hCG vaccine were demonstrated to be minimal and predicted to be acceptable for human use from both macroscopic tissue examination and histopathological evaluation in animal studies.
2. Materials and methods
2.1. Materials
Synthetic hCG peptide antigen, C-TT2-CTP35 (amino acid sequence: CQYIKANSKFIGITEL{TT2}-DDPRFQDSSSSKAPPPSLPSPSRLPGPSDTPILPQ {βhCG 111-145, also CTP35}), was prepared by Peninsula Laboratories (San Carlos, CA). C-MVF-CTP35, which was kindly provided by Dr. P.T.P. Kaumaya of The Ohio State University, was substituted for C-TT2-CTP35 in one instance as described in Results. Poly(D,L-lactide-co-glycolide) 50/50 (end-group capped) with inherent viscosity of 0.19 and 0.24 dl/g in hexafluoroisopropanol solution at 30°C was from Birmingham Polymers (Birmingham, AL) and Alkermes (Cambridge, MA), respectively. Poly(L-lysine) hydrobromide (MW 150–300 kDa), 5,5′ dithio-bis(2-nitrobenzoic acid), L-ornithine hydrochloride, and MgCO3 were from Sigma (St. Louis, MO). N-3-maleimido-butyryloxysulfosuccinimide ester (sulfo-GMBS) was from Pierce (Rockford, IL). All other reagents were of analytical grade or purer and purchased from commercial suppliers.
2.2. Conjugation of C-TT2-CTP35 to PLGA microsphere surface
C-TT2-CTP35 was conjugated via thiol group of cysteine to the surface α-amino groups of PLGA/polylysine microspheres via a bifunctional crosslinker, sulfo-GMBS [18]. The PLGA (0.19 dl/g)/polylysine microspheres were prepared by a solvent evaporation method by utilizing polylysine as an emulsifier in place of commonly used polyvinyl alcohol (PVA) [19]. C-TT2-CTP35 (5–20 mg/ml) in a phosphate buffer solution (pH 7.4–8.0) was reduced by dithiothreitol (DTT) prior to conjugation. The reduced peptide was purified by dialysis (4°C) or size exclusion chromatography. Free thiol groups were determined by 5,5′ dithio-bis (2-nitrobenzoic acid). Sulfo-GMBS was added to microsphere suspension in 0.1 M sodium phosphate buffer (pH 6.6) at 4:1 molar excess, equivalent to 60 nmol/mg microspheres, of lysine residues. The reaction was allowed to proceed for 1–3 h in the dark. Following removal of excess reagent by several centrifugation/washes, suspension aliquots were taken to determine content of maleimido groups introduced by sulfo-GMBS by assaying cysteine consumability. Then, microspheres suspension was mixed with reduced peptide (1.2:1 thiol:maleimido groups) and stirred for 2 h in the dark. Antigen-conjugated microspheres were centrifuged, washed with water and lyophilized in 10% sucrose solution.
2.3. Encapsulation of peptide in PLGA microspheres
According to a double emulsion–solvent evaporation method [20], 100 μl of 70 mg/ml peptide in PBS (pH 7.4) were added to 0.8–1.0 ml of PLGA/CH2Cl2 solution (70% for 0.19 dl/g; 50% for 0.24 dl/g) with or without MgCO3 (< 45 μm). The mixture was homogenized at 15,000 r.p.m. for 1–1.5 min over an ice bath and transferred to 1.6–2 ml of 5% (w/v) PVA solution. The water-in-oil-in-water emulsion was formed by further homogenizing the mixture at 10,000 r.p.m. for 1 min and hardened in 0.5% (w/v) PVA in PBS for 2–3 h. The microspheres were collected by centrifugation, washed with water, and lyophilized.
2.4. Assay of C-TT2-CTP35 peptide
The peptide was quantified by two methods – Coomassie protein assay and amino acid analysis for soluble and conjugated/total peptide, respectively. Coomassie brilliant blue plus protein assay (Pierce, Rockford, IL), a modified Bradford assay, was performed to analyze soluble peptide. An abbreviated amino acid (leucine and/or lysine) analysis was developed for conjugated and total antigen in microspheres, which were completely hydrolyzed (L-ornithine added as internal standard) in 6 N HCl (110°C, 22 h) sealed under light vacuum. After removal of acid, hydrolyzed amino acids were reconstituted in 1 M sodium carbonate solution (pH 9.5), derivatized with o-phthaldialdehyde/2-mercaptoethanol, and separated by RP-HPLC equipped with a Waters™ 474 Scanning Fluorescence Detector (λEx/λEm = 340/455 nm).
2.5. Physicochemical characterization of C-TT2-CTP35-loaded microspheres
The antigen-conjugated and -encapsulated microspheres were characterized regarding peptide loading, particle size, and morphology. Peptide loading in antigen-conjugated microspheres was directly determined by amino acid analysis (see Assay). The antigen-encapsulated microspheres were first dissolved in acetone to extract the peptide. After centrifugation and removal of the polymer solution, the peptide precipitate was reconstituted in PBS solution containing 0.02% Tween 80 (PBST) and incubated at 37°C for at least 1 h. Insoluble materials (e.g., undissolved MgCO3) were removed by centrifugation prior to determining peptide content by Coomassie protein assay. The morphology/size of antigen-associated PLGA microspheres was analyzed with a Hitachi S-3200 N scanning electron microscope (SEM).
2.6. FITC-labeling of C-TT2-CTP35 and laser confocal scanning microscopy (LCSM)
FITC (400 μg) in 100 mM carbonate buffer (pH 9.0) was mixed with 2 ml of 1 mg/ml C-TT2-CTP35 aqueous solution and incubated at 37°C (1 h) in the dark. FITC-labeled peptide, purified by dialysis and lyophilized, was conjugated to PLGA microspheres as described previously (see Conjugation). The control (i.e., peptide-adsorbed) microspheres were prepared by following the same procedures except that the crosslinker was not added. Microspheres washed with 2% (w/v) SDS in PBS to remove physically adsorbed peptides were incubated in PBST at 37°C (14 d) to observe fluorescence retention. The spatial distribution of FITC-labeled peptide in PLGA microspheres was analyzed on a Carl Zeiss LSM 510 laser confocal scanning microscope (Car Zeiss Microimaging, Thornwood, NY) [21]. ProLong® Antifade reagent (Molecular Probes, Eugene, OR) was added to prohibit photobleaching.
2.7. Controlled release and stability of C-TT2-CTP35 in PLGA
After removal of sucrose by washing/centrifugation, peptide-conjugated microspheres (∼ 15 mg) were placed in PBST solution (0.5 ml) and incubated at 37°C under mild agitation. To sample, the buffer was removed at pre-determined time intervals and replaced with new medium. Peptide content in release samples was determined by amino acid analysis. Antigen-encapsulated microparticles (3–4 mg) were similarly incubated in PBST (1 ml) at 37°C. The microspheres were removed; soluble and total peptide in the polymer was determined by Coomassie protein assay and amino acid analysis, respectively. Additionally, structural integrity of peptide extracted from microsphere samples (as in peptide loading measurement) was evaluated by SDS-PAGE using a High Density PhastGel® performed by a PhastSystem (Amersham Pharmacia Biotech AB, Uppsala, Sweden). A modified Coomassie brilliant blue staining method using glutadialdehyde as fixing reagent was developed to stain the low MW peptide bands after separation.
2.8. Immunization of microsphere-associated antigens in rabbits
The immunogenicity of C-TT2-CTP35 antigen conjugated to (200 μg) and/or encapsulated (1.0 mg) in PLGA microspheres was tested in adult, specific pathogen-free New Zealand white rabbits via single-dose i.m. injection of microspheres suspended in PBS. In the initial study, six groups (Group I-VI) of five rabbits were immunized following the immunization scheme as described in Table 1. A synthetic analogue of a surface component of mycobacteria, nor-MDP (25 μg) encapsulated in a separate PLGA microsphere formulation (0.624 mg), was co-administered in several microsphere groups. A single dose of 1.0 mg soluble peptide in PBS plus nor-MDP encapsulated microspheres was used as the negative control (Group VII). Peptide (1.0 mg) in a water-in-oil emulsion, i.e., PBS (containing 25 μg nor-MDP) in squalene:mannide monooleate (4:1) emulsion, served as a positive control (Group VIII) and was boosted at 4 & 10 weeks. Blood samples were collected weekly between 2 and 24 weeks. The serum antibody binding to 125I-labeled hCG was determined by radioimmunoassay (RIA) [22].
Table 1.
Immunization protocol to evaluate the immune response elicited by i.m. administration of C-TT2-CTP35 vaccine formulations in rabbits.
| Group | Immunogen (Dose) |
PLGA Microspheres administered (mg) |
Co-administered adjuvants (Dose) |
# of immunizations |
|---|---|---|---|---|
| I | SCFa (200 μg) | 382 | None | 1× |
| II | EnFb (1.0 mg) | 143 | None | 1× |
| III | SCFa (200 μg ) + EnFb (1.0 mg) | 525 | None | 1× |
| IV | SCFa (200 μg) | 382 | Nor-MDP/PLGAe (25 μg) | 1× |
| V | EnFb (1.0 mg) | 143 | Nor-MDP/PLGAe (25 μg) | 1× |
| VI | SCFa (200 μg) + EnFb (1.0 mg) | 525 | Nor-MDP/PLGAe (25 μg) | 1× |
| VII | C-TT2-CTP35/PBSc (1.0 mg) | 0.625 | Nor-MDP/PLGAe (25 μg) | 1× |
| VIII | C-TT2-CTP35/w/o emulsiond (1.0 mg) | 0 | Nor-MDPf (25 μg) | 3×g |
Surface-conjugated formulation (SCF): PLGA microsphere formulation with the peptide conjugated on the surface.
Encapsulated formulation (EnF): PLGA microsphere formulation with the peptide encapsulated, co-encapsulated with 3% MgCO3.
The soluble peptide in PBS solution was administered as a negative control.
The peptide was incorporated in a water-in-oil (PBS-in-squalene:mannide monooleate (4:1)) emulsion, as a positive control.
nor-MDP was encapsulated in 0.624 mg PLGA microspheres.
nor-MDP solution was used.
Boosted at 4 and 10 weeks.
A subsequent study was further conducted with four groups of rabbits (n = 4/group) to optimize the microsphere formulation and investigate the effect of varying amount of MgCO3 (MgCO3 : peptide = 0:1, 1:1, 2:1 and 3:1, w/w). Around 65–72 mg of the PLGA/MgCO3 microspheres, corresponding to 1.0 mg of encapsulated C-TT2-CTP35, were administered similarly as described above, and the oil-based peptide (see above) served as the positive control. (Note that nor-MDP was not included in this study.)
2.9. Pathology of rabbit tissue following immunization
The pathology of rabbit tissue at local injection site was examined visually (macroscopically) and by a histopathological microscopic method. A macroscopic scoring system (0–3) was used to evaluate the gross appearance of incised tissues. Normal tissue (score 0) has no visible pathology; yellow fatty/fibrous tissue may appear after complete resolution of inflammation. Score 1 describes minimal pathology showing small (d < 3 mm) hard nodules (encapsulated and resolving sterile abscesses/inflammation). In moderate tissue pathology (score 2), hard (old fibrosis) or soft (more recently encapsulated) nodules are larger (d ∼ 3–10 mm); upon squeezing, pus or injection material may be expressed. Severe tissue pathology (score 3) contains large (d > 10 mm) lesions. Using this scoring system, observations have been made that formulations with a score 0.5 or less in rabbits caused no pain or local reaction in human clinical trials (personal communication with Dr. Dov Michaeli, Aphton Corporation, Larkspur, CA, who devised the above-described scoring system). Both the average and the range of the scores from rabbits in each group were presented. Additionally, histopathological sections of rabbit tissue were also prepared and evaluated under microscope. The extent of myositis, inflammatory leukocyte infiltration, and any other pathological change at microscopic level was assessed.
3. Results and Discussion
3.1. C-TT2-CTP35 antigen conjugated to PLGA microsphere surface
Noticed as early as in 1960s by Gall [23], the importance of the surface of vaccine adjuvants was illustrated by the adjuvancy of many surface active materials including lipids, non-ionic block polymers, water-in-oil and oil-in-water emulsions, and commonly used emulsifiers, such as Span 85, lanolin, methylcellulose and alginate [24-26]. Binding of antigens to, or concentration of antigens and immune-activating opsonins on, a surface appears to be crucial for enhancing immune response to the immunogens by adjuvants [24]. Antigens covalently linked to synthetic non-PLGA microparticle surface have elicited strong CD4+ T-cell responses in mice [27,28]. The presence of antigen on the microsphere surface was suggested to enhance phagocytosis and subsequent induction of immune responses.
To potentially induce phagocytosis, we conjugated C-TT2-CTP35 antigen on biocompatible PLGA microparticles, for which the establishment of moieties conjugatable to antigen onto the PLGA microsphere surface was necessary. This was accomplished by converting polylysine into an efficient aqueous emulsifier via reducing its charge density and raising its α-helix content, which promotes physical entrapment of the polypeptide in PLGA microspheres in a one-step method [19]. Conjugating the thiol group of C-TT2-CTP35 to the surface α-amino groups of polylysine was performed with a water-soluble bifunctional crosslinker, N-3-maleimido-butyryloxysulfosuccinimide ester (sulfo-GMBS) [18].
Antigen-conjugated PLGA microspheres, easily dispersible in aqueous injection vehicles, are displayed in Fig. 1A to be of a size range (1–10 μm) recognizable by antigen presenting cells [16,29]. About 0.8 μg of peptide was coupled to per mg of bulk microspheres which contain only 3 μg polylysine (Fig. 1B). Stable conjugation of CTT2-CTP35 to the microsphere surface was demonstrated qualitatively by laser confocal scanning microscopy (LCSM) with FITC-labeled peptide. Strong fluorescence was observed on the surface of peptide-conjugated microspheres (Fig. 1C), which was not significantly diminished after microsphere incubation for 14 days at 37°C. In control microspheres where crosslinker was not added during conjugation conditions, surface FITC-peptide was undetectable at constant detector gain [30] (data not shown), confirming chemical attachment of surface-associated peptide instead of physical adsorption. Retention of C-TT2-CTP35 from peptide-conjugated microspheres following incubation at 37°C was quantitatively monitored in a phosphate buffer. A steady decline of antigen in peptide-conjugated microspheres was observed, with ∼ 40 % remaining after 2–3 weeks (Fig. 1D), indicating good retention for inducing and maintaining the immune response.
Figure 1.
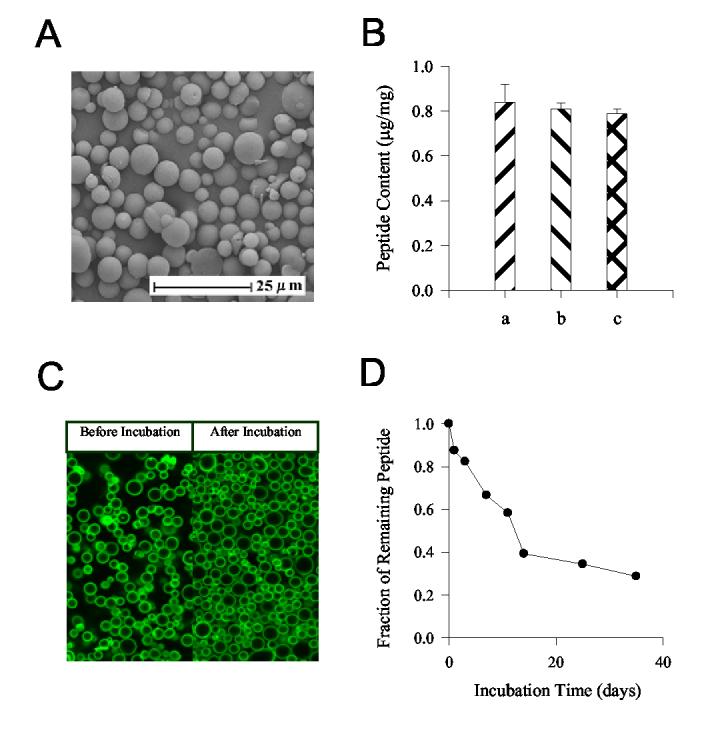
Physicochemical characterization of antigen-conjugated microspheres. (A) Scanning electron micrograph (SEM) of microspheres (5.3 ± 0.1 μm) with surface-conjugated C-TT2-CTP35. (B) The content of C-TT2-CTP35 conjugated to PLGA/polylysine microspheres as determined by amino acid analysis (Mean ± SD, n = 2). The microspheres were washed with purified H2O (a), PBS (b), or 2% (w/v) SDS in PBS solution (c) before acid hydrolysis. (C) The distribution of FITC labeled C-TT2-CTP35 in the peptide-conjugated microspheres. All of the samples were washed with 2% (w/v) SDS in PBS solution to remove the non-covalently adsorbed peptide. The microspheres before and after 14-day incubation at 37°C were observed by LCSM; prior to observation, a ProLong Antifade reagent was added to the microsphere suspension to prevent photo bleaching of fluorescein. Fading of fluorescence during image collection was unnoticeable. (D) The retention of C-TT2-CTP35 peptide following in vitro incubation of antigen-conjugated microspheres in PBST at 37°C, as determined by amino acid analysis of the peptide in release medium.
3.2. Encapsulation and stability of C-TT2-CTP35 antigen in PLGA microspheres
C-TT2-CTP35 was also encapsulated in PLGA microspheres to provide prolonged release of the immunogen and to reduce the need of frequent booster immunizations. The microspheres were prepared with a commonly used double emulsion – solvent evaporation method [31]. Spherical microspheres containing 0.63–0.76% (w/w) immunogen with an average particle size of 3.8 μm were prepared (Fig. 2A). The addition of 3% (w/w) MgCO3 to the microspheres was examined because we have shown this base to be effective in neutralizing the acids (e.g., lactic and glycolic acids) sequestered by the PLGA polyester [32,33], a problem that has frequently been found to damage encapsulated proteins and other acid-labile molecules [14,20]. The encapsulated peptide was slowly and continuously released for up to 1 month in a physiological buffer, regardless of co-encapsulation of MgCO3 (Fig. 2B,C). The stability of the peptide encapsulated in base-containing PLGA microspheres during in vitro release was investigated by comparing the soluble and total peptide remaining in the microspheres following incubation (see Methods). Following 1-month incubation, the majority of the peptide unreleased remained soluble, although slightly lower levels of peptide detected by protein assay relative to amino acid analysis were suggestive of mild aggregation and/or hydrolysis of the peptide (Fig. 2B,C). By contrast, SDS-PAGE revealed largely unaltered peptide bands following erosion of the polymer (Fig. 2D). Thus, initial in vitro analysis indicated a desirable release and retention profile of the antigen during long periods, although minor alterations of the antigen structure could not be ruled out.
Figure 2.
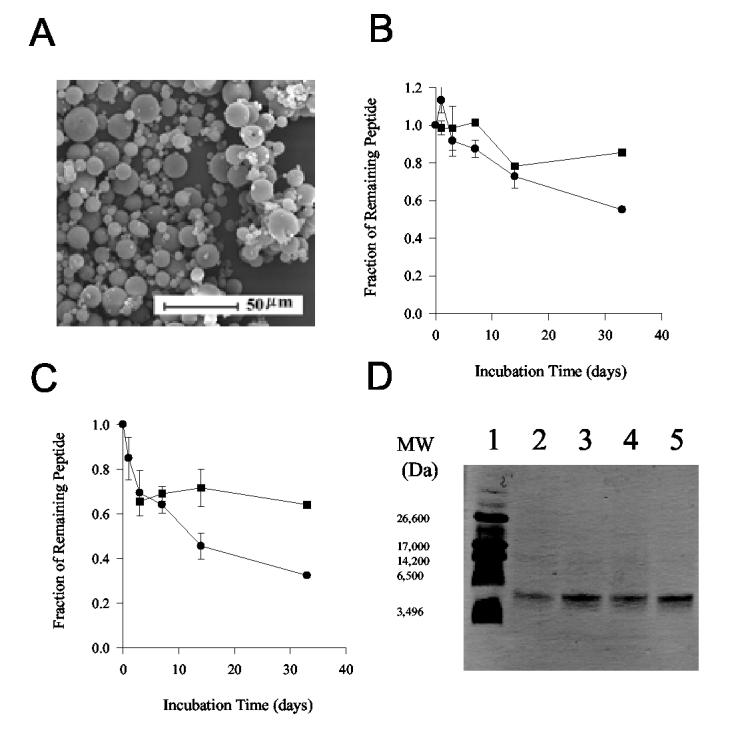
Physicochemical characterization of antigen-encapsulated microspheres. (A) Scanning electron micrograph (SEM) of microspheres containing encapsulated C-TT2-CTP35 peptide. (B, C) The retention kinetics of soluble and total C-TT2-CTP35 in PLGA microspheres, as determined by Coomassie assay (●) and amino acid analysis (■) (Mean ± SD, n = 3). The peptide was encapsulated in the microspheres without (B) or with MgCO3 (C). The release of the antigen can be evaluated by (1 - Fraction of total remaining peptide). (D) SDS-PAGE under reducing condition of C-TT2-CTP35 peptide extracted from PLGA microspheres after 1-day or 33-day incubation at 37°C in PBST. Lane 1: Ultra-low molecular weight markers; Lanes 2 & 3: co-encapsulated with MgCO3, day 33 and 1, respectively; Lanes 4 & 5: without co-encapsulation of MgCO3, day 33 and 1, respectively.
3.3. Immunogenicity of surface-conjugated and/or encapsulated C-TT2-CTP35 antigens in rabbits
The ability of C-TT2-CTP35 antigen associated with PLGA microspheres to induce a strong and long-lasting antibody response was examined in rabbits that were immunized via a single i.m. injection of the microsphere suspension in an aqueous vehicle (i.e., PBS) (see Table 1 for the detailed immunization protocol). Soluble peptide in the buffer vehicle and peptide in a water-in-oil emulsion containing oily components (i.e., squalene and mannide monooelate) were used as negative and positive (boosted at 4 and 10 weeks) controls, respectively. An additional immunostimulant, nor-MDP, was co-administered in several groups to further stimulate the immune system [10].
As shown in Fig. 3A-C, all microsphere-associated antigens (Group I–VI) elicited an immune response quickly with the maximum antibody level observed after 35–56 days, in contrast to the soluble peptide (∼ 77 days). Among these groups, single-dose encapsulated peptide, co-encapsulated with 3% MgCO3, elicited a significantly stronger antibody response (max. mean 1,250 vs. 400 nM, p < 0.1 from statistical student t-test analysis) with an equivalent duration relative to the multiple (three) dose positive control group. The surface-conjugated peptide induced a comparable immunogenicity at 1/5 of the dose, 200 μg, relative to the soluble control group, 1.0 mg, (max. mean 40 vs. 20 nM, p = 0.15 from t-test analysis). As the antigen loading is limited in the surface-conjugated group by the surface area/mass ratio of the polylysine-coated PLGA microspheres, a 200 μg dose was used in this study for the surface-conjugated antigen instead of a standard 1.0 mg dose, because of limitations in the amount of PLGA that could be i.m. injected in the animal. Co-administration of nor-MDP in the microspheres did not further stimulate the antibody response, suggestive of a maximum adjuvant (immuno-stimulating) effect of PLGA – MgCO3 microspheres for the hCG peptide. By contrast, co-immunization of 1.0 mg encapsulated plus 200 μg surface-conjugated antigen induced higher humoral responses than either one administered alone, with an extremely high anti-hCG antibody level (max. mean ∼ 2,800 nM) detected in the rabbit group injected with encapsulated and conjugated peptide plus nor-MDP adjuvant. A prolonged duration of at least six months of high antibody level, comparable to the positive control administered in multiple doses, was observed in these single-dose microsphere vaccines, which is likely due to the prolonged and continuous release of immunogen from the polymer.
Figure 3.

Serum anti-hCG antibody level of rabbits following immunization with PLGA-associated C-TT2-CTP35 antigens (all data were presented as Mean ± S.E.): (A) 200 μg surface-conjugated antigen, alone or with 0.625 mg norMDP/PLGA, or 1.0 mg soluble peptide in PBS plus 0.625 mg nor-MDP/PLGA (negative control). (n = 5). (B) 1.0 mg encapsulated antigen, alone or with 0.625 mg nor-MDP/PLGA, or 1.0 mg peptide in a w/o emulsion, which was boosted at 4 & 10 weeks (positive control). (n = 5). (C) 1.0 mg encapsulated plus 200 μg surface-conjugated antigen, with or without 0.625 mg norMDP/PLGA. (n = 5). (D) 1.0 mg encapsulated antigen in PLGA microspheres co-encapsulated with varying amount of MgCO3 (MgCO3:peptide = 0:1, 1:1, 2:1 and 3:1, w/w), compared with positive control antigens. (n = 4).
3.4. Tissue response of C-TT2-CTP35 antigens in rabbits
Vaccine formulations must be safe with minimal side effects to be administered to a widespread human population. PLGA is well-known as a relatively inert material, one of its advantages as an antigen delivery vehicle [13,14]. Minimal inflammatory reactions and fibrous capsule formation have been reported following implantation of PLGA microspheres, including a series of local events, generally considered as tissue response continuum: injury by injection or implantation, acute inflammation, chronic inflammation, formation of granulation tissue, foreign body reaction and fibrosis [34]. Macroscopic observation may correspondingly reveal lumps and hard tissue, which reflect the fibrous capsules; histopathological evaluation examines microscopic attributes of the tissue reaction (e.g., inflammatory cells).
A qualitative scoring system from 0 (no pathology) to 3 (severe pathology) (see Methods) was used to macroscopically evaluate the pathology of rabbit tissue at sacrifice (24 weeks) following immunization of C-TT2-CTP35 vaccines. Minimal to moderate pathology with appearance of hard nodules was only found to accompany the very high antibody response (score = 0.7 & 1.9 for − & + nor-MDP, respectively) in rabbits administered with combined antigens, as shown in Table 2. Nonetheless, it was observed that lumps were larger at the time of maximum antibody level, about 4–8 weeks. A study of the side effects of the same mass of blank (i.e., not antigen-containing) PLGA microspheres at 4 weeks following administration (score = 0.5 & 0.9 for − & + MgCO3, Table 2) revealed that besides antibody production and co-immunization of nor-MDP, some of the undesirable tissue reaction is also related with the polymer dose and MgCO3.
Table 2.
Tissue reaction at sacrifice following i.m. administration of C-TT2-CTP35 vaccine formulations in rabbits, evaluated by a macroscopic scoring system as described in Section 2.9.
| Groupa | Score of tissue reaction |
Time of sacrifice | |
|---|---|---|---|
| Mean ± S.E. | Range | ||
| I | 0.0 ± 0.0 | 0 – 0 | 24 weeks |
| II | 0.4 ± 0.2 | 0 – 0.5 | 24 weeks |
| III | 0.7 ± 0.4 | 0 – 1 | 24 weeks |
| IV | 0.0 ± 0.0 | 0 – 0 | 24 weeks |
| V | 0.5 ± 0.0 | 0.5 – 0.5 | 24 weeks |
| VI | 1.9 ± 0.2 | 1.5 – 2 | 24 weeks |
| VII | 0.0 ± 0.0 | 0 – 0 | 24 weeks |
| VIII | 0.0 ± 0.0b | 0 – 0 | 24 weeks |
| Blankc | 0.5 ± 0.0 | 0.5 – 0.5 | 4 weeks |
| Blank/MgCO3d | 0.9 ± 0.2 | 0.75 – 1 | 4 weeks |
Groups I-VIII are as described in Table 1.
n = 4. Another group of rabbit (n = 2) sacrificed at antibody peak time, 10 weeks, showed a score of 0.8 ± 0.4 with a range of 0.5 – 1.
Blank microspheres. Neither antigen nor MgCO3 was encapsulated; n = 2.
Blank microspheres with 3% MgCO3; n = 2.
3.5. C-TT2-CTP35/PLGA–MgCO3 vaccine composition with minimal side effects
A promising contraceptive hCG peptide vaccine with acceptable side effects (i.e., local tissue reactions) was achieved by minimizing PLGA and MgCO3 doses, without significantly affecting antibody response.
Henceforth we sought to find a C-TT2-CTP35 microsphere vaccine capable of inducing a sufficiently high antibody level with minimal toxicity by: 1) reducing the PLGA dose, and 2) minimizing the MgCO3 amount required for antibody production. Encapsulation of C-TT2-CTP35 peptide at higher peptide loading (∼ 1.5% loading with ∼ 85% recovery) in PLGA (i.v. = 0.24 dl/g) enabled the polymer dose to be lowered to about 70 mg as compared to 143 mg in the previous antigen-encapsulated formula. A series of peptide-encapsulated PLGA microsphere formulations were prepared co-encapsulating various amount of base, which was added at a ratio of MgCO3:peptide = 0:1, 1:1, 2:1 and 3:1, wt/wt. The morphology, shape and size of the particles were not influenced by co-encapsulation of MgCO3 (data not shown).
Remarkably, as shown in Fig. 3D, co-encapsulation of MgCO3 was essential for eliciting anti-hCG antibody response as base-free microspheres were completely ineffective. By contrast, MgCO3-containing formulations induced strong antibody response comparable to the positive control, with no significant difference among the formulations encapsulating different amounts of MgCO3. As learned from stability analysis that MgCO3 appears not to significantly increase antigen stability or release, the exceptional adjuvancy of co-encapsulated MgCO3 reflect alternative adjuvant mechanisms. An insoluble metal salt, similar to well-used inorganic aluminum salts which are among the first discovered and commonly used vaccine adjuvants [35], MgCO3 may independently act as an additional adjuvant besides PLGA microspheres. The greater non-specific inflammatory responses at local injection sites in MgCO3-containing microspheres may invoke higher antibody production. The slow erosion of PLGA slowly releasing antigen and some undissolved MgCO3 also may lead to prolonged exposure of antigen associated with MgCO3 for further stimulation of the immune system [11].
The macroscopic tissue reactions examined at 24 weeks were minimal and acceptable (scores < 0.5) for all these formulations. The microscopic histopathological evaluation of antigen*-encapsulated PLGA microspheres containing 2:1 MgCO3:peptide was further examined at antibody peak time (8 weeks) when the tissue reactions were found most severe. Among five rabbits, varying degree of myositis and inflammatory leukocytic infiltratration were observed (Fig. 4B,C) as compared to the control rabbit (Fig. 4A). While in some rabbits multiple foci of polymorphonuclear cells were the predominating inflammatory cell types (Fig. 4B), the others exhibited obvious infiltration by lymphoplasmacytic cells (Fig. 4C). Overall, the histopathological evaluation revealed that in rabbits, no discomfort or intolerable inflammatory response is likely to be induced by the selected vaccine formulation if it was administered to humans. (*C-MVF {a T-cell epitope sequence from measles protein F}-CTP35 antigen was used in place of C-TT2-CTP35; the antibody profile following immunization in rabbits was found to be comparable to C-TT2-CTP35 antigen in a separate study, with max. mean antibody level of 572 nM).
Figure 4.

Histopathological evaluation of the tissue of control rabbit (A) and rabbits immunized with C-MVF-CTP35 in PLGA microspheres (B,C). The antigen was co-encapsulated with magnesium carbonate (2:1 MgCO3:peptide, wt/wt) in PLGA. All rabbits were sacrificed at antibody peak time (8 weeks) thus the pathology examined herein reflects the peak of local tissue reaction within the duration of immunization (24 weeks).
3.6. Potential use of PLGA/MgCO3 for peptide vaccine delivery
Historically, vaccines have been developed to protect humans and domestic animals against infectious diseases. Most of these vaccines utilized live attenuated pathogens, whole inactivated organisms, and inactivated toxins as immunogens, which were recognized by the body as foreign agents and were strongly immunogenic when administered intramuscularly or applied to mucosal surfaces. As these microorganisms display multiple sites recognized by T and B lymphocytes as well as antigen presenting cells, the impurities or other components of organisms also serve as adjuvant or immunostimulant to induce the cellular or humoral immune response needed to prevent infection [36]. Traditional vaccines, although still currently being used, are often associated with toxicity and severe side effects by reverting to a virulent form or by causing excessive inflammation and thus posing biohazardous potential [37,38]. Also, since the aqueous delivery vehicles offered no depot effect, multiple immunizations were often required to initiate and maintain effective immunity.
During the past few decades, extensive progress has been made in developing new vaccines and making improvements in old ones [38-40]. New vaccines often target specific regions of infectious organisms encompassing only one or a few epitopes and still others, target “self” antigens that are not normally immunogenic [41,42]. Most of these new approaches employ recombinant protein subunit, synthetic peptide or DNA antigens, which is safer and more efficient than the traditional vaccines. In spite of their advantages of defined physical and chemical characteristics, these new generation vaccines are often poorly immunogenic when used alone [38] and some form of immunostimulant or adjuvant as a component of the vaccine formulation is critical. Aluminum salts, the only adjuvant approved for human use by US FDA, have been used with limited success in a few vaccines [36,38]. Other adjuvants under development, although effective towards enhancing the immune response, are often limited by safety concerns, such as tissue damage at the site of injection and later granulomatous reactions, pyrogenicity, arthritis, and other side effects [40]. On the other hand, improvements in traditional vaccines are needed in order to reduce the number of immunizations required to establish an effective immunity. An ideal adjuvant or delivery system must provide the desirable actions (i.e., effective and long-lasting) and, at the same time, be completely safe from systemic toxicity or adverse reactions at the site of injection [37,43].
The PLGA – MgCO3 microsphere vaccine delivery system described in this report addresses each of the problems outlined above for old and new vaccines. First, the system provided a high level of humoral immunity against a small peptide antigen that is virtually non-immunogenic when administered alone in saline or PBS. Second, there is no evidence that any of the components of the system induce systemic toxicity and we have shown that tissue reactions at the injection site are minimal at effective doses. Third, this system initiates and sustains an effective level of immunity for several months from a single immunization.
In closing, we have demonstrated the novel, and unanticipated, finding that co-encapsulation of MgCO3 in PLGA microspheres is essential to provide a powerful adjuvant effect for a contraceptive hCG-based peptide antigen in rabbits with good safety. Initial analysis suggests that MgCO3 does not significantly improve stability and release characteristics of this particular antigen, but instead enhances antibody production via some alternative adjuvant mechanism. The use of these novel microspheres may, in the future, be useful to augment immunity against numerous other peptide vaccines suffering from poor antibody responses, including those directed against HIV.
Acknowledgment
We thank John Powell and Nancy Carney for conducting the numerous antibody assays reported here, and Dr. P.T.P. Kaumaya for peptide synthesis and helpful discussions. This work was supported by the Special Programme of Research, Development, and Research Training in Human Reproduction, World Health Organization, and NIH HL 68345.
Abbreviations
- PLGA
poly(lactic-co-glycolic acid)
- hCG
human chorionic gonadotropin
- CTP
C-terminal peptide
- PBS
phosphate buffered saline
- PBST
phosphate buffered saline containing 0.02% Tween 80
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Annual Technical Report, A.T. Department of Reproductive Health and Research (RHR) World Health Organization; 2003. [Google Scholar]
- 2.Talwar GP. Fertility regulating and immunotherapeutic vaccines reaching human trials stage. Hum. Reprod. Update. 1997;3(4):301–310. doi: 10.1093/humupd/3.4.301. [DOI] [PubMed] [Google Scholar]
- 3.Stevens VC. Progress in the development of human chorionic gonadotropin antifertility vaccines. Am. J. Reprod. Immunol. 1996;35(3):148–155. doi: 10.1111/j.1600-0897.1996.tb00024.x. [DOI] [PubMed] [Google Scholar]
- 4.Fishel SB, Edwards RG, Evans CJ. Human chorionic gonadotropin secreted by preimplantation embryos cultured in vitro. Science. 1984;223(4638):816–818. doi: 10.1126/science.6546453. [DOI] [PubMed] [Google Scholar]
- 5.Hearn JP, Gidley-Baird AA, Hodges JK, Summers PM, Webley GE. Embryonic signals during the peri-implantation period in primates. J. Reprod. Fertil. Suppl. 1988;36:49–58. [PubMed] [Google Scholar]
- 6.Thanavala YM, Hay FC, Stevens VC. Affinity, cross-reactivity and biological effectiveness of rabbit antibodies against a synthetic 37 amino acid C-terminal peptide of human chorionic gonadotrophin. Clin. Exp. Immunol. 1980;39(1):112–118. [PMC free article] [PubMed] [Google Scholar]
- 7.Stevens VC, Cinader B, Powell JE, Lee AC, Koh SW. Preparation and formulation of a human chorionic gonadotropin antifertility vaccine: selection of a peptide immunogen. Am. J. Reprod. Immunol. 1981;1(1981):307–314. [Google Scholar]
- 8.Kaumaya PT, Kobs-Conrad S, Seo YH, et al. Peptide vaccines incorporating a ‘promiscuous’ T-cell epitope bypass certain haplotype restricted immune responses and provide broad spectrum immunogenicity. J. Mol. Recognit. 1993;6(2):81–94. doi: 10.1002/jmr.300060206. [DOI] [PubMed] [Google Scholar]
- 9.Panina-Bordignon P, Tan A, Termijtelen A, Demotz S, Corradin G, Lanzavecchia A. Universally immunogenic T cell epitopes: promiscuous binding to human MHC class II and promiscuous recognition by T cells. Eur. J. Immunol. 1989;19(12):2237–2242. doi: 10.1002/eji.1830191209. [DOI] [PubMed] [Google Scholar]
- 10.Stevens VC, Jones WR. Vaccines to prevent pregnancy. In: Levine MM, Woodrow GC, Kaper JB, Cobon GS, editors. New Generation Vaccines. Marcel Dekker, Inc.; New York: 1997. pp. 1131–1143. [Google Scholar]
- 11.Langer R, Cleland JL, Hanes J. New advances in microsphere-based single-dose vaccines. Adv. Drug Deliv. Rev. 1997;28(1):97–119. doi: 10.1016/s0169-409x(97)00053-7. [DOI] [PubMed] [Google Scholar]
- 12.Cleland JL. Design and production of single-immunization vaccines using polylactide polyglycolide microsphere systems. Pharm. Biotechnol. 1995;6:439–462. doi: 10.1007/978-1-4615-1823-5_18. [DOI] [PubMed] [Google Scholar]
- 13.Johansen P, Men Y, Merkle HP, Gander B. Revisiting PLA/PLGA microspheres: an analysis of their potential in parenteral vaccination. Eur. J. Pharm. Biopharm. 2000;50(1):129–146. doi: 10.1016/s0939-6411(00)00079-5. [DOI] [PubMed] [Google Scholar]
- 14.Schwendeman SP. Recent advances in the stabilization of proteins encapsulated in injectable PLGA delivery systems. Crit. Rev. Ther. Drug Carrier Syst. 2002;19(1):73–98. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.20. [DOI] [PubMed] [Google Scholar]
- 15.Newman KD, Elamanchili P, Kwon GS, Samuel J. Uptake of poly(D,Llactic-co-glycolic acid) microspheres by antigen-presenting cells in vivo. J. Biomed. Mater. Res. 2002;60(3):480–486. doi: 10.1002/jbm.10019. [DOI] [PubMed] [Google Scholar]
- 16.Tabata Y, Ikada Y. Effect of the size and surface charge of polymer microspheres on their phagocytosis by macrophage. Biomaterials. 1988;9(4):356–362. doi: 10.1016/0142-9612(88)90033-6. [DOI] [PubMed] [Google Scholar]
- 17.Men Y, Tamber H, Audran R, Gander B, Corradin G. Induction of a cytotoxic T lymphocyte response by immunization with a malaria specific CTL peptide entrapped in biodegradable polymer microspheres. Vaccine. 1997;15(12-13):1405–1412. doi: 10.1016/s0264-410x(97)00047-9. [DOI] [PubMed] [Google Scholar]
- 18.Lee AC, Powell JE, Tregear GW, Niall HD, Stevens VC. A method for preparing beta-hCG COOH peptide-carrier conjugates of predictable composition. Mol. Immunol. 1980;17(6):749–756. doi: 10.1016/0161-5890(80)90145-5. [DOI] [PubMed] [Google Scholar]
- 19.Cui C, Schwendeman SP. Surface entrapment of polylysine in biodegradable poly(DL-lactide-co-glycolide) microparticles. Macromolecules. 2001;34(24):8426–8433. [Google Scholar]
- 20.Zhu G, Mallery SR, Schwendeman SP. Stabilization of proteins encapsulated in injectable poly (lactide- co-glycolide) Nat. Biotechnol. 2000;18(1):52–57. doi: 10.1038/71916. [DOI] [PubMed] [Google Scholar]
- 21.Kang J, Schwendeman SP. Determination of diffusion coefficient of a small hydrophobic probe in poly(lactide-co-glycolide) microparticles by laser scanning confocal microscopy. Macromolecules. 2003;36(2003):1324–1330. [Google Scholar]
- 22.Powell JE, Lee AC, Tregear GW, Niall HD, Stevens VC. Characteristics of antibodies raised to carboxy-terminal peptides of hCG beta subunit. J. Reprod. Immunol. 1980;2(1980):1–13. [Google Scholar]
- 23.Gall D. The adjuvant activity of aliphatic nitrogenous bases. Immunol. 1966;11(4):369–386. [PMC free article] [PubMed] [Google Scholar]
- 24.Woodard LF. Surface chemistry and classification of vaccine adjuvants and vehicles. Adv. Biotechnol. Processes (Bacterial Vaccines) 1990;13(1990):281–306. [PubMed] [Google Scholar]
- 25.Cox JC, Coulter AR. Adjuvants - a classification and review of their modes of action. Vaccine. 1997;15(3):248–256. doi: 10.1016/s0264-410x(96)00183-1. [DOI] [PubMed] [Google Scholar]
- 26.Hunter R, Strickland F, Kezdy F. The adjuvant activity of nonionic block polymer surfactants I. The role of hydrophil-lipophile balance. J. Immunol. 1981;127(3):1244–1250. [PubMed] [Google Scholar]
- 27.Sedlik C, Perraut R, Bonnemains B, Leclerc C. Antigens linked to synthetic microspheres induce immune responses in primates in the absence of adjuvant. Immunobiology. 1996;195(1):105–118. doi: 10.1016/S0171-2985(96)80009-X. [DOI] [PubMed] [Google Scholar]
- 28.Gengoux C, Leclerc C. In vivo induction of CD4+ T cell responses by antigens covalently linked to synthetic microspheres does not require adjuvant. Int. Immunol. 1995;7(1):45–53. doi: 10.1093/intimm/7.1.45. [DOI] [PubMed] [Google Scholar]
- 29.Eldridge JH, Staas JK, Meulbroek JA, Tice TR, Gilley RM. Biodegradable and biocompatible poly(DL-lactide-co-glycolide) microspheres as an adjuvant for staphylococcal enterotoxin B toxoid which enhances the level of toxin-neutralizing antibodies. Infect. Immun. 1991;59(9):2978–2986. doi: 10.1128/iai.59.9.2978-2986.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui C. Thesis in Pharmaceutics. Univ. Michigan; Ann Arbor, MI: 2003. Ph.D. [Google Scholar]
- 31.Thies C. Formation of degradable drug-loaded microparticles by in-liquid drying processes. In: Donbrow M, editor. Microcapsules and nanoparticles in medicine and pharmacy. CRS Press; Boca Raton, FL: 1992. pp. 47–71. [Google Scholar]
- 32.Fu K, Pack DW, Klibanov AM, Langer R. Visual evidence of acidic environment within degrading poly(lactic-co-glycolic acid) (PLGA) microspheres. Pharm. Res. 2000;17(1):100–106. doi: 10.1023/a:1007582911958. [DOI] [PubMed] [Google Scholar]
- 33.Shenderova A, Burke TG, Schwendeman SP. The acidic microclimate in poly(lactide-co-glycolide) microspheres stabilizes camptothecins. Pharm. Res. 1999;16(2):241–248. doi: 10.1023/a:1018876308346. [DOI] [PubMed] [Google Scholar]
- 34.Shive MS, Anderson JM. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997;28(1):5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 35.Glenny AT, Pope CG, Waddington H, Wallace V. The antigenic value of toxoid precipitated by potassium alum. J. Pathol. Bacteriol. 1926;29(1926):38–45. [Google Scholar]
- 36.Gupta RK, Siber GR. Adjuvants for human vaccines - current status, problems, and future prospects. Vaccine. 1995;13(14):1263–1276. doi: 10.1016/0264-410x(95)00011-o. [DOI] [PubMed] [Google Scholar]
- 37.Zauner W, Lingnau K, Mattner F, von Gabain A, Buschle M. Defined synthetic vaccines. Biol. Chem. 2001;382(4):581–595. doi: 10.1515/BC.2001.071. [DOI] [PubMed] [Google Scholar]
- 38.Sheikh NA, al-Shamisi M, Morrow WJ. Delivery systems for molecular vaccination. Curr. Opin. Mol. Ther. 2000;2(1):37–54. [PubMed] [Google Scholar]
- 39.Singh M, O'Hagan D. Advances in vaccine adjuvants. Nat. Biotechnol. 1999;17(11):1075–1081. doi: 10.1038/15058. [DOI] [PubMed] [Google Scholar]
- 40.Rabinovich NR, McInnes P, Klein DL, Hall BF. Vaccine technologies: view to the future. Science. 1994;265(5177):1401–1404. doi: 10.1126/science.7521064. [DOI] [PubMed] [Google Scholar]
- 41.Raychaudhuri S, Rock KL. Fully mobilizing host defense: building better vaccines. Nat. Biotechnol. 1998;16(11):1025–1031. doi: 10.1038/3469. [DOI] [PubMed] [Google Scholar]
- 42.Mesa C, Fernandez LE. Challenges facing adjuvants for cancer immunotherapy. Immunol. Cell Biol. 2004;82(6):644–650. doi: 10.1111/j.0818-9641.2004.01279.x. [DOI] [PubMed] [Google Scholar]
- 43.Ben-Yedidia T, Arnon R. Design of peptide and polypeptide vaccines. Curr. Opin. Biotechnol. 1997;8(4):442–448. doi: 10.1016/s0958-1669(97)80066-3. [DOI] [PubMed] [Google Scholar]