

Abstract
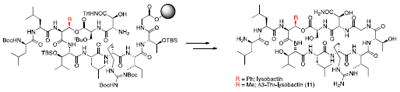
The solid phase synthesis of the cyclic depsipeptide antibiotic lysobactin is described. The natural product was synthesized via a linear approach using mostly Fmoc-strategy solid phase peptide synthesis (SPPS) with a single purification. A lysobactin analog has also been synthesized displaying nanomolar membrane disruption activity not seen with the natural product.
Lysobactin (katanosin B) is an 11 amino acid cyclic depsipeptide antibiotic that was isolated by groups at the Shionogi Research Institute1 and the Squibb Institute of Medical Research.2 The 28-membered macrocycle contains 6 non-proteinogenic amino acids including four that are β-hydroxylated. Lysobactin shows extensive antibacterial activity against a wide range of aerobic and anaerobic Gram-positive bacteria, in some trials displaying 2 to 4-fold greater activity than vancomycin.3 Very strong growth inhibition of methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE) has been shown with minimum inhibitory concentrations ranging from 0.39 to 0.78 μg/mL.2
There have been two syntheses to date of this natural product that employed a modular, solution phase approach. The first employed an approach with minimal use of protecting groups and utilized a crystal structure to guide selection of a site for macrolactamization.4,5 The second employed a more classical approach involving a macrolactamization via activation of a C-terminal Gly residue that allowed epimerization-free cyclization.6 While both of these syntheses provided efficient routes to generate lysobactin, solution phase chemistry greatly restricts the ability to rapidly access structural analogs. Thus, a solid phase approach was desired in order to facilitate our goal of identifying the key structural moieties that are responsible for the excellent antibacterial activity of lysobactin.
As is standard with most solid phase peptide synthesis (SPPS), we chose to synthesize the linear precursor to lysobactin in the C- to N-direction. We selected the same site for macrocyclization that we exploited for our synthesis in solution.6 This enabled epimerization-free cyclization, via a glycine-derived activated ester, and avoided coupling residues of like chirality since these are known to be less efficient than amino acid coupling reactions employing partners having identical absolute configurations.7
All synthetic amino acid building blocks were orthogonally protected to enable trifluoroacetic acid (TFA) promoted global deprotection (Figure 1). Fmoc-allo-Thr(OTBS)-OH (1) was produced starting from Thr as described by Elliott.8 Fmoc-D-Arg(Boc2)-OH (2) was generated by coupling Fmoc-D-Orn-OAllyl with N,N’-bis(tert-butoxycarbonyl)-S-methylisothiourea.9 Fmoc-threo-HyAsn(CONHTrt)-OH (3) was synthesized via our previously established protocol that relied upon a stereospecific oxazoline cyclization to introduce the β-hydroxy stereocenter.10 Fmoc-threo-HyLeu(OTBS)-OH (4) was generated using chemistry first reported by Hamada11 in which RuCl2(binap)-catalyzed hydrogenation of the corresponding β-keto-α-amino acid ester afforded a nearly diastereo- and enantiopure product through dynamic kinetic resolution. threo-Phenylserine was purchased from a commercial source and protected as its corresponding Fmoc derivative (5).
Figure 1.
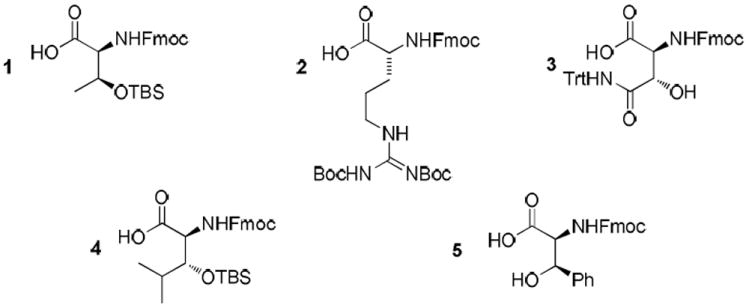
Synthetic Fmoc-protected Building Blocks
The preparation of intermediate 6 began with the C-terminal glycine residue loaded on a 2-chlorotrityl resin (Scheme 1). All peptide couplings were performed using 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT)12 and diisopropylethylamine (DIPEA) in DMF followed by Fmoc deprotection using 20% piperidine in DMF. In an effort to conserve precious materials, only 1.5 equivalents of synthetic amino acids were used during coupling while 5 equivalents were used for commercially available building blocks (excluding Fmoc-threo-phenylserine-OH). Longer reaction times (up to 24 h) were required for couplings using fewer equivalents, but no epimerization was detected as determined by reverse-phase HPLC.
Scheme 1.
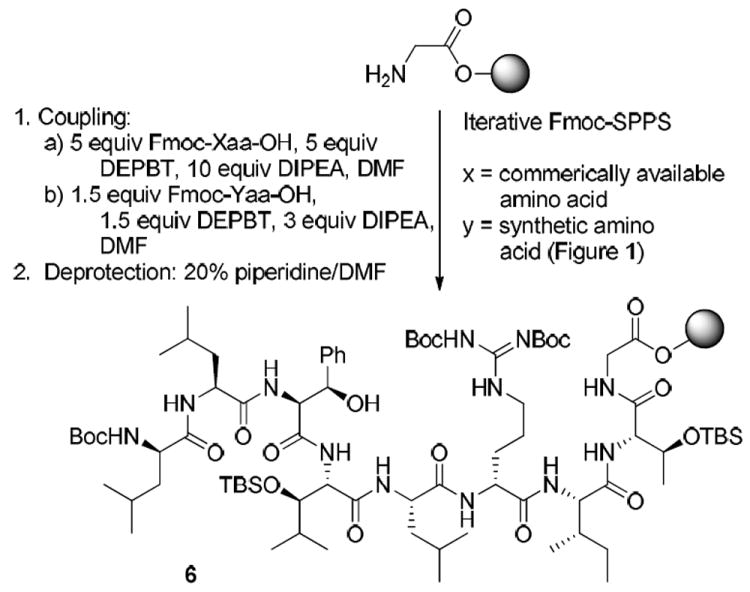
Iterative Fmoc-SPPS of Key Intermediate 6
With linear peptide 6 in hand, our attention was turned to the esterification of the hindered secondary hydroxyl. A variety of coupling reactions employing the use of carbodiimide activating reagents were investigated. It was found that concentration, temperature, dry and distilled reaction solvents, and the choice of α-amino protecting group were critical to both the coupling efficiency and the elimination of by-products arising from epimerization. Initial attempts at coupling Fmoc-Ser(tBu)-OH were plagued by low conversions and unacceptable amounts of epimerization. Furthermore, we found that varying amounts of β-elimination had occurred under standard Fmoc cleavage conditions. To circumvent β-elimination, Alloc-Ser(tBu)-OH was prepared to allow amine deprotection under neutral conditions.13 To our delight, coupling of this building block proceeded with complete conversion and no detectable epimerization when performed at 37 °C (Scheme 2). It is worth noting that these conditions are very similar to those used for macrolactonization of the simplified lysobactin analog synthesized by Bradley.14 Analogous effects of α-amino protecting groups on coupling efficiency have also been reported for the coupling of N-methyl amino acids in Rich’s studies on cyclosporine.15,16
Scheme 2.
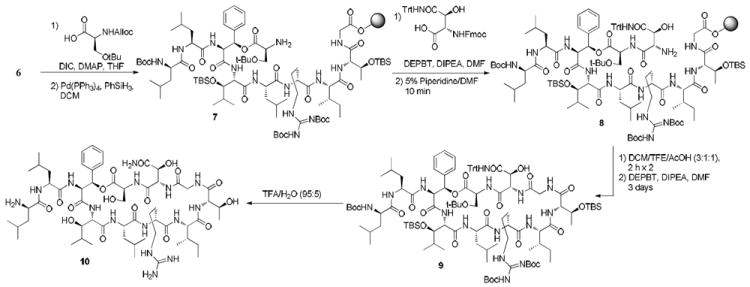
End Game Synthesis
Cleavage of the intermediate allyl carbamate with catalytic Pd(PPh3)4 unfortunately gave a 2:1 mixture of of identical mass while extended reaction times provided a complex mixture of compounds. However, reducing the reaction time to 10 minutes afforded complete deprotection without any appearance of any undesired side product(s). The remaining β-HyAsn (3) was installed under standard conditions (DEPBT, DIPEA, THF) and cleanly deprotected (5% piperidine in DMF) with no evidence of epimerization or β-elimination.
Cleavage from the 2-Cl-Trt resin with acetic acid/trifluoroethanol/dichloromethane (1:1:3) yielded the linear peptide in high purity (>90% by HPLC). No additional peptide was collected after a second treatment of the resin under the same conditions. The crude peptide was cyclized using the conditions (DEPBT, DIPEA, DMF) from our previous synthesis of lysobactin.5 The cyclized product was submitted to global deprotection conditions17 (TFA/Et3SiH/DCM; 90/5/5) that removed all protecting groups save for a single TBDMS ether. Treatment with neat TFA for an additional 9-12 h was required for full deprotection. Deprotection conditions used in our previous synthesis (TFA/H2O; 95/5) removed all protecting groups reproducibly in only 4 h; however, there was no significant increase in yield when compared to the best results under anhydrous conditions.
Reverse-phase preparatory HPLC purification yielded lysobactin in 8.4% overall yield. Co-injection with an authentic sample showed a single, symmetrical peak under all HPLC conditions. Furthermore, 1H NMR, 13C NMR, optical rotation, and high resolution ESI-TOF mass spectral analysis correlated with synthetic lysobactin.18
With the ability to rapidly generate structurally diverse lysobactin analogs in place, we chose to begin studies focused on a potentially important cation-π interaction bridging the macrocycle. As seen in the crystal structure of lysobactin reported by von Nussbaum, the guanidine moiety of D-Arg lies in close proximity to the phenyl substituent of phenylserine.3 If this structural attribute is critical for the biological activity of lysobactin, we hypothesized that the removal of this interaction would result in increased conformational flexibility and diminished antibacterial activity.
Our first target for synthesis was lysobactin analog 11 for which our solid phase strategy only required incorporation of of Fmoc-Thr-OH in place of threo-phenylserine. The newly introduced methyl group would eliminate the possibility of any cation-π interaction.
Upon completion of the solid-phase synthesis of 11, comparison of its circular dichroism spectrum with that obtained from lysobactin, revealed a distinct conformational change/relaxation.19 Minimum inhibitory concentrations for lysobactin and 11 were determined against Bacillus subtilis. The MIC values were 0.06 μg/mL and 2 μg/mL for lysobactin and 11, respectively. Although the analog (11) shows a 32-fold decrease in activity, the MIC still remains at a respectable level meaning that either the cation-π interaction is not critical for antibacterial activity or that 11 may be achieving its antibacterial activity via an additional/different mechanism.19,20
A bacterial cell membrane disruption assay was performed in order to compare the activity of 11 with the natural product as lysobactin has previously shown limited amounts of membrane permeabilization at concentrations above its MIC.1 To this end, exponentially growing wild-type B. subtilis cells were treated with varying concentrations of antibiotic in the presence of propidium iodide (PI). PI is a normally membrane impermeable dye, which becomes up to 40 times more fluorescent upon intercalation into membrane enclosed DNA. Thus, cells with compromised membranes show a time dependent enhancement of PI fluorescence compared to cells with intact membranes (Figure 3).21,22 In agreement with previous reports, lysobactin showed poor PI influx until a concentration of 64 to 128 times the MIC is reached (Figure 3 and Supporting Information, Figure 2). Surprisingly, 11 caused a rapid PI influx even at concentrations 4 times lower than its MIC. This difference in rate of PI influx relative to the respective MIC values for lysobactin and 11, suggests that the antibacterial activity for the lysobactin analog (11) may be the result of membrane permeabilization.
Figure 3.
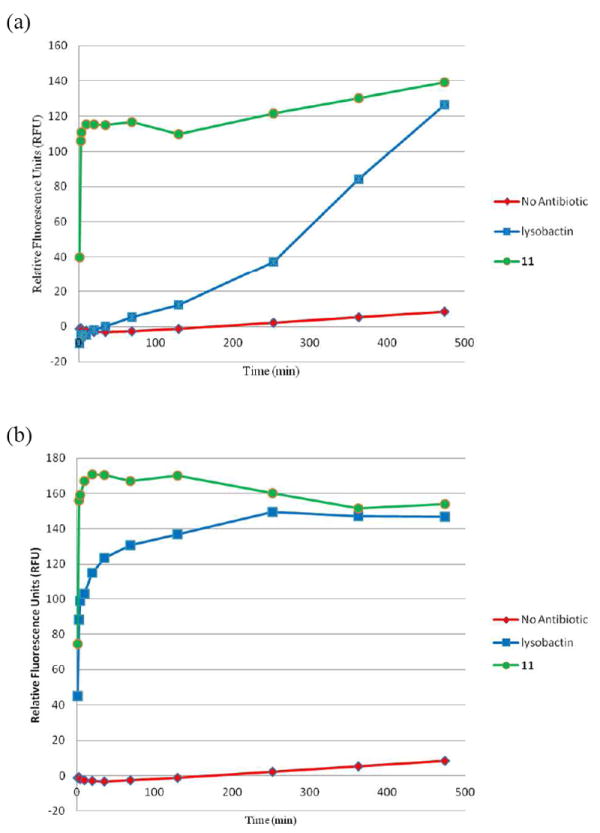
Propidium iodide bacterial membrane disruption assay. Cell culture of B. subtilis PY79 treated with depsipeptide concentrations of: (a) 1 μg/mL (b) 8 μg/mL
This notion was further supported by a killing kinetics experiment with B. subtilis, showing that cell death after treatment with 11 occurred more rapidly than after treatment with lysobactin, a characteristic result that is consistent with known membrane disrupting antibiotics such as nisin.23 At identical concentrations, lysobactin showed a gradual decline to ~50% cell survival while the analog displayed an exponential decrease in viability with levels of cell survival falling below 20% within 1 h (Supporting Information, Figure 4).
In B. subtilis, lysobactin began to show significant levels of membrane disruption at concentrations approaching its MIC values for some Gram-negative bacteria (i.e., ~8 μg/mL, Figure 3(b)).1 We reasoned that the ability to permeabilize bacterial membranes might, in part, contribute to its moderate activity against Gram-negative bacteria. However, repeating the membrane disruption assay with a Gram-negative organism (E. coli) showed no significant PI influx for 11 even at the highest concentration concentration tested (64 μg/mL), whereas lysobactin only caused moderate levels of PI influx at high concentrations (32-64 μg/mL) (Supporting Information, Figure 3).
In summary, we have completed a highly efficient solid phase total synthesis of the cyclic depsipeptide antibiotic lysobactin with only a single purification of the final product. Using this route, we have synthesized Δ3-Thr-lysobactin (11) that lacks the functionality required for the cation-π interaction that has been observed in the parent natural product. Additional experiments revealed that B. subtilis cells (a Gram-positive organism) treated with 11 showed significant membrane permeabilization at concentrations four times lower than its MIC suggesting that membrane permeabilization may contibute significantly to its observed antibacterial activity, a notion also supported by killing kinetics experiments. Interestingly, in E. coli (a Gram-negative organism), only lysobactin showed any evidence of membrane permeabilization and that was at concentrations significantly greater (32-64 μg/mL) than its reported MIC value.1
Additional analogues will reveal the key structural attributes that are responsible for the biological activity of lysobactin and enable studies directed toward determining conformation of the natural product when bound to its cellular target. Results of these studies will be reported in due course.
Supplementary Material
Figure 2.
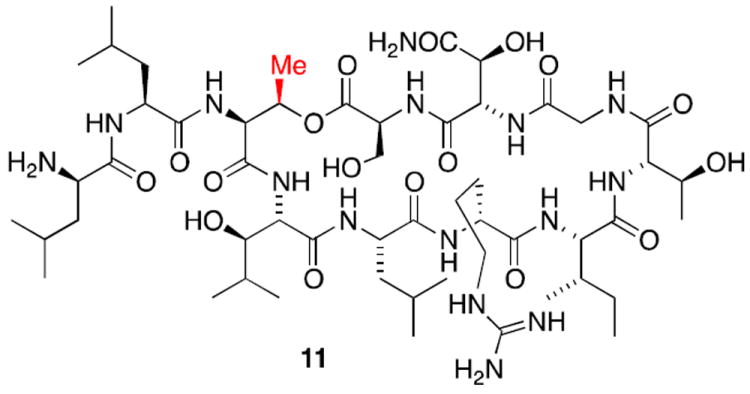
Δ3-Thr-lysobactin
Acknowledgments
Financial support provided by the National Institutes of Health (AI 059327) and Indiana University is gratefully acknowledged.
Footnotes
Supporting Information Available. Experimental details and spectral data for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Shoji J, Hinoo H, Matsumoto K, Hattori T, Yoshida T, Matsuura S, Kondo E. J Antibiot. 1988;41:713–718. doi: 10.7164/antibiotics.41.713. [DOI] [PubMed] [Google Scholar]
- 2.O’Sullivan J, McCullough JE, Tymiak AA, Kirsch DR, Trejo WH, Principe PA. J Antibiot. 1988:1740–1744. doi: 10.7164/antibiotics.41.1740. [DOI] [PubMed] [Google Scholar]
- 3.Bonner DP, O’Sullivan J, Tanaka SK, Clark JM, Whitney RR. J Antibiot. 1988:1745–1751. doi: 10.7164/antibiotics.41.1745. [DOI] [PubMed] [Google Scholar]
- 4.von Nussbaum F, Anlauf S, Benet-Buchholz J, Häbich D, Köbberling J, Musza L, Telser J, Rübsamen-Waigmann H, Brunner NA. Angew Chem Int Ed. 2007:2039–2042. doi: 10.1002/anie.200604232. [DOI] [PubMed] [Google Scholar]
- 5.(a) von Nussbaum F, Anlauf S, Koebberling J, Telser J, Haebich D. Procedure for synthesis of cyclic depsipeptides such as Lysobactin via peptide fragment assembly and intramolecular cyclization. Aicuris G.m.b.H. & Co. K.-G; Germany: 2007. DE102006018250A1. [Google Scholar]; (b) von Nussbaum F, Brunner N, Endermann R, Fuerstner C, Hartmann E, Paulsen H, Ragot J, Schiffer G, Schuhmacher J, Svenstrup N, Telser J, Anlauf S, Bruening M-A. Synthesis of lysobactin derivs. for use as antibacterial agents in the treatment or prevention of disease. Bayer Healthcare AG; 2006. DE102004053407A1. [Google Scholar]
- 6.Guzman-Martinez A, VanNieuwenhze MS. J Am Chem Soc. 2007;129:6017–6021. doi: 10.1021/ja067648h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Rich DH, Bhatnagar P, Mathiaparanam P, Grant JA, Tam JP. J Org Chem. 1978;43:296–302. doi: 10.1021/jo00396a027. [DOI] [PubMed] [Google Scholar]; (b) Brady SF, Varga SL, Freidinger RM, Schwenk DA, Mendlowski M, Holly FW, Veber DF. J Org Chem. 1979;44:3101–3105. [Google Scholar]; (c) Birch D, Hill RR, Jeffs GE, North M. Chem Commun. 1999:941–942. [Google Scholar]
- 8.Elliott DF. J Chem Soc. 1949:589–594. [Google Scholar]
- 9.Markowski PJ. J Pept Sci. 2005;11:60–64. doi: 10.1002/psc.585. [DOI] [PubMed] [Google Scholar]
- 10.Guzman-Martinez A, VanNieuwenhze MS. Synlett. 2007;10:1513–1516. doi: 10.1055/s-2007-982544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makino K, Okamoto N, Hara O, Hamada Y. Tetrahedron: Asymmetry. 2001;12:1757–1762. [Google Scholar]
- 12.(a) Li H, Jiang X, Ye Y-h, Fan C-X, Romoff T, Goodman M. Org Lett. 1999;1(1):91–93. doi: 10.1021/ol990573k. [DOI] [PubMed] [Google Scholar]; (b) Ye Y-H, Li H, Jiang X. Biopolymers. 2005;80:172–178. doi: 10.1002/bip.20201. [DOI] [PubMed] [Google Scholar]
- 13.Albericio F, Thieriet N, Alsina J, Giralt E, Guibe F. Tetrahedron Lett. 1997;38:7275–7278. [Google Scholar]
- 14.Egner JE, Bradley M. Tetrahedron. 1997;53:14021–14030. [Google Scholar]
- 15.Tung RD, Rich DH. J Am Chem Soc. 1985;107:4342–4343. [Google Scholar]
- 16.Colucci WJ, Tung RD, Petri JA, Rich DH. J Org Chem. 1990;55:2895–2903. [Google Scholar]
- 17.Initial deprotection conditions omitted water with the intent of avoiding any undesired hydrolytic ring-opening reactions
- 18.Lysobactin derivative 11 was produced in 5.4% overall yield
- 19.See Supporting Information
- 20.Lysobactin is an inhibitor of the transglycosyation reaction in the bacterial cell wall biosynthetic pathway: Maki H, Miura K, Yamano Y. Antimicrob Agents Chemother. 2001;45(6):1823–1827. doi: 10.1128/AAC.45.6.1823-1827.2001.
- 21.Bhakdi S, Martin E. Infect Immun. 1991;59:2955–2962. doi: 10.1128/iai.59.9.2955-2962.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Virto R, Manas P, Alvarez I, Condon S, Raso J. Appl Environ Microb. 2005;71:5022–5028. doi: 10.1128/AEM.71.9.5022-5028.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruhr E, Sahl HG. Antimicrob Agents Chemother. 1985;27:841–845. doi: 10.1128/aac.27.5.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.