

Abstract
Medicinal plants are extensively utilized in traditional and herbal medicines, both in India and around the world due to the presence of diverse low molecular weight natural products such as flavonoids, alkaloids, terpenoids and sterols. Flavonoids which have health benefits for humans are the large class of phenylpropanoid-derived secondary metabolites and are mostly glycosylated by UDP-glycosyltransferases (UGTs). Although large numbers of different UGTs are known from higher plants, very few protein structures have been reported till now. In the present study, the three-dimensional model of flavonoid specific glycosyltransferases (WsFGT) from Withania somnifera was constructed based on the crystal structure of plant UGTs. The resulted model was assessed by various tools and the final refined model revealed GT-B type fold. Further, to understand the sugar donors and acceptors interactions with the active site of WsFGT, docking studies were performed. The amino acids from conserved PSPG box were interacted with sugar donor while His18, Asp110, Trp352 and Asn353 were important for catalytic function. This structural and docking information will be useful to understand the glycosylation mechanism of flavonoid glucosides.
Abbreviations
DOPE - Discrete Optimized Potential Energy, PDB - Protein Data Bank, PSPG - Plant Secondary Product Glycosyltransferase, RMSD - Root Mean Squared Deviation, UDP - Uridine diphosphate, UGT - UDP-glycosyltransferases.
Keywords: Flavonoids, Glycosyltransferase, Homology modeling, Docking, Withania somnifera
Background
Plants contain a very large number of UDP glycosyltransferases (UGTs) that are involved in the glycosylation of natural products [1]. Glycosylation may lead to reduction in the toxicity of endogenous and exogenous toxic substances in plants by adding a sugar moiety to acceptors which modify their properties such as bioactivity, stability, solubility, sub cellular localization and binding properties [2]. UDP-glucose, UDPgalactose, UDP-rhamnose, UDP-xylose and UDP-glucuronic acid are the common activated sugar donors of plant UGTs [3]. Glycosyltransferases (GTs) are the enzymes that synthesize oligosaccharides, polysaccharides and glycoconjugates. GTs have been grouped into 91 families on the basis of sequence similarities. Family-1 has the most number of GTs related to plants [4] and contains a plant secondary product glycosyltransferase (PSPG) box, close to the C-terminal end of the protein. This PSPG box consists of 44 amino acids and is believed to be involved in binding of the activated sugar donors. Within the PSPG-box highly, conserved amino acid residue including the HCGWNS motif are considered to be important for enzymatic function [5]. Despite the fact that many GTs recognize similar donor or acceptor substrates, there is surprisingly limited sequence identity between different families. Until now only two folds have been observed for GTs: fold GT-A, consisting of one α/β/α sandwich domain and characterized by the presence of divalent cation in the binding site while GT-B fold has two such domains [6]. Despite low sequence conservation, the UGTs show highly conserved secondary and tertiary structures. The sugar acceptor and sugar donor substrates of UGTs are accommodated in the cleft formed between the N- and C-terminal domains. Several regions of the primary sequence contribute to the formation of the substrate binding pocket including structurally conserved domains as well as loop regions differing both with respect to their amino acid sequence and sequence length.
Withania somnifera (L.) Dunal (Solanaceae) is used extensively in traditional and herbal medicine, both in India and around the world, primarily due to its antibiotic, antiviral, antiamoebic, antiarthritic and anti-inflammatory properties [7]. It produces diverse low molecular weight natural products such as flavonoids, alkaloids, terpenoids, tannins, resins and sterols through secondary metabolism [5]. Flavonoids and isoflavonoids are major determinants of growth, development and defence in plants. These compounds also possess antioxidant activity, which has potential health benefits for humans and animals. Flavonoids, a large class of phenylpropanoid-derived secondary metabolites, are mostly glycosylated by UGTs with one or more sugar groups [8]. Although large numbers of different UGTs have been reported from higher plants, very few have been crystallized till now due to the difficulties in obtaining crystals. Homology modeling can be useful in prediction of protein structure and to detect potentially important residues [9]. These models have been useful not only for rationalizing experimental data but also for designing directed mutagenesis experiments. In the current study the homology modeling and docking analysis of a flavonoid specific GT from W. somnifera (WsFGT) were performed. The results will provide new insight into molecular interactions of active site residues with substrates for the enzymatic function.
Methodology
Sequence Alignment:
Protein sequence of WsFGT (NCBI Ac. No. FJ560880) was selected. BLAST algorithm against protein data bank (PDB) was used to carry out the sequence homology searches. The sequence and 3D structure of template proteins were extracted from the PDB database [10]. Multiple sequence alignments of the target and template sequences were carried out using ClustalW program [11] with default parameters.
Homology modeling of WsFGT:
Secondary structure was predicted via the PSIpred v 3.0 servers [12]. Based on high-resolution crystal structures of homologous proteins, 3D models of WsFGT were built using the homology modeling software MODELLER 9v9 [13] on windows operating environment. Loop refinement tool of MODELLER was used to refine the loop conformation of the model. Further, this model was improved by Chimera [14]. Amber parameters were used for standard residues, and Amber's Antechamber module was used to assign parameters to nonstandard residues. The model having lowest DOPE scores was selected for validation.
Model Validation:
Stereochemical quality of the polypeptide backbone and side chains was evaluated using Ramachandran plot obtained from PROCHECK [15]. Amino acid environment was evaluated using ERRAT plot [16] which assesses the distribution of different types of atoms with respect to one another in the protein model. ProSAII program was utilized for structure validation. It uses knowledge based potentials of mean force to evaluate model accuracy and it shows local model quality by plotting energies as a function of amino acid sequence position [17]. Pair wise structural superimposition of modelled WsFGT was done with templates using Chimera match maker program.
Molecular Docking:
Structure Data File (SDF) of ligand molecules were downloaded from the pubchem (http://pubchem.ncbi.nlm.nih.gov) and were converted to PDB format using Pymol software. The WsFGT structure and substrate ligands were prepared using AutoDock Tools v.1.5.2 software [18]. Molecular docking was performed using Autodock vina 1.1.2 software [19]. The lowest binding energy conformation in the first cluster was considered as the most favorable docking pose. Hydrogen bonds, bond lengths and close contacts between enzyme active site and substrate atoms were analyzed.
Discussion
Model Construction:
The secondary structure prediction run at the PSIpred server showed that the WsFGT consists of 45% α-helix (15 helices; 179 residues), 5% extended-beta (13 strands; 52 residues) and 50% random coil (29 coils; 228 residues) configuration (Figure S1). Homology modeling relies on establishing an evolutionary relationship between the sequence of a protein of interest and other members of the protein family whose structures have been solved experimentally by X-ray crystallography or NMR. WsFGT amino acid sequence alignment with available protein sequences showed a significant percentage of identity with Medicago truncatula UDP-glucuronosyl/UDPglucosyltransferase (2PQ6: 31% identity) (Figures 1a & 1b), Arabidopsis thaliana hydroquinone glucosyltransferase (2VG8: 29%), Vitis vinifera UDP-glucose flavonoid 3-O glycosyltransferase (2C1Z: 29%), Medicago truncatula triterpene UDP-glucosyltransferase (2ACW: 30%) and Medicago truncatula flavonoid 3-O-glucosyltransferase (3HBJ: 27%). These proteins were selected as templates for the modeling. In general, sequence identities of 30% are enough to construct the 3D model of target proteins through homology modeling. Due to low sequence identity SALIGN was employed to construct multiple structure alignments of templates. The 3D models of WsFGT were generated by aligning the target sequence with this multiple structures-based alignment.
Figure 1.
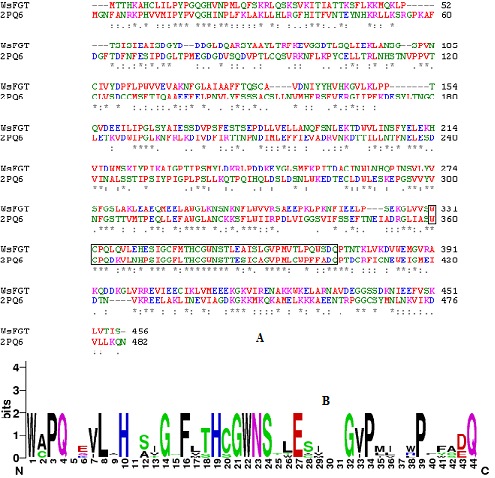
(A) Sequence alignment of WsFGT from W. somnifera with template (PDB code: 2PQ6) using ClustalW. The conserved regions are indicated by ‘*’ and box indicates PSPG motif. (B) Conserved PSPG box created using WebLogo (http://weblogo.berkeley.edu/logo.cgi).
Structure Validation:
We selected 5 out of 50 models with lower DOPE score. In order to select the best model, the structural validity of the models was performed by PROCHECK, ERRAT and ProSAII programs. The best model was subjected to loop refinement at region where ERRAT plot show more than 95% error value and disallowed amino acid region showed by PROCHECK. To improve and verify the stability of the loop model, an energy minimization procedure was applied to the WsFGT model. Ramachandran plot provided by the program PROCHECK, assured very good confidence for the predicted protein with 91.6% residues in most allowed region, 8.4% in additional allowed region, 0.0% in generously allowed region and 0.0% in disallowed region (Figure 2), Table 1 (see supplementary material). ERRAT plot gave an overall quality factor of 74.330 to the modeled structure which was very much satisfactory. The ProSAII program (Protein Structure Analysis) is an established tool, which is frequently employed in structure prediction, refinement and validation of experimental protein structures. It generates Z score of model, which is a measure of compatibility between its sequence and structure. The model Z score should be comparable to the Z scores obtained from the template [20, 21]. ProSAII analysis showed that protein folding energy of our modeled structure was in good agreement with that of the template. It showed a Z-Score of -10.28, implying no significant deviation from the templates 2ACW (-12.75), 2C1Z (-10.96), 2PQ6 (-10.07), 2VG8 (-11.73) and 3HBJ (-11.44) (Figures S2A & S2B). Thus, validation results suggested that the predicted model was a reliable 3D structure of WsFGT. The final model structure of WsFGT and superimposition with template (2PQ6) was displayed in (Figures 3a & 3b). Similarly, a homology model of a F7GAT UGT88D7 from Perilla frutescents was generated using VvGT1 structure as a template [22]. Further, Root Mean Squared Deviation (RMSD) and TM-score were used to quantitatively compare the 3D-structures of the models Table 2 (see supplementary material) which revealed high similarity in topology and overall fold of the model with templates. Overall, these analyses confirmed the stability of WsFGT structure and GT-B fold type. Till now, all structures of plant UGTs (M. truncatula UGT71G1, UGT85H2 and UGT78G1; V. vinifera VvGT1 and A. thaliana UGT72B1) which have been reported exhibit GT-B fold.
Figure 2.
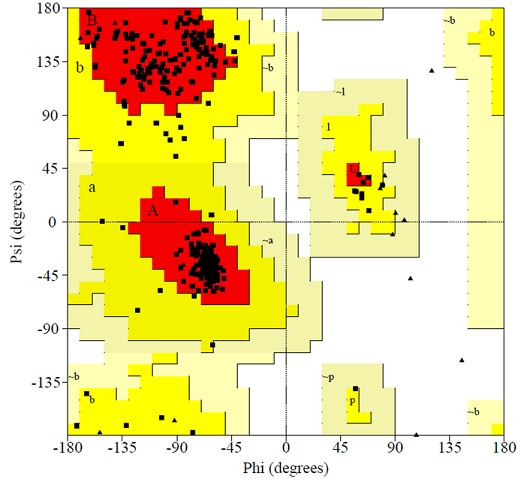
Residue profile of WsFGT in Ramachandran plot.
Figure 3.
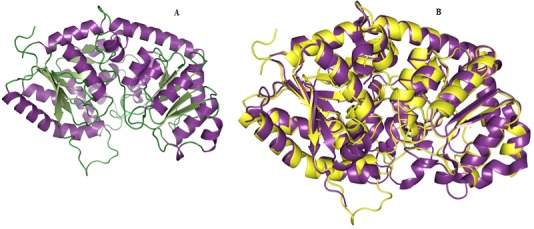
(A) Homology model of WsFGT represented in ribbon diagram. (B) Superimposition of 3D structure of WsFGT (yellow) and M. truncatula UDP-glucuronosyl/UDP-glucosyltransferase (2PQ6: violet purple).
Molecular Interactions:
WsFGT docking studies were carried out with diadzein, apigenin, luteolin, naringenin, genistein and kaempferol as acceptors and UDP-glucose as sugar donor. Substrate binding pocket was made up from 13 residues (Tyr13, His18, Asn20, Gln133, Lys245, Ser278, Gln334, His349, Trp352, Asn353, Ser354, Glu357 and Trp371). Among these, 6 residues were polar uncharged, 3 residues each were positively charged and aromatic, with 1 negatively charged residue. These residues interacted with substrate molecules forming hydrogen bonds. UDP-glucose was almost completely buried in a long, narrow channel mainly the C-terminal domain of the enzyme. The enzyme interacted with UDP mainly through residues in the UGT signature PSPG motif (His18, Asn20, Gln133, Lys245, Ser278, Gln334, His349, Asn353, Ser354, Glu357 and Trp371) in WsFGT structure. The interaction energy of UDP-glucose with WsFGT complex was -10.9 kcal/mol and the hydrogen bond distances were observed within the range of 2.1 Å to 3.4 Å.
Comparative analysis of WsFGT-substrate conformations:
A very narrow, deep pocket was observed adjacent to the UDP binding site, mainly consisting of residues from the N-terminal domain, which would appear to be the binding pocket for acceptor molecules. Docking conformations of all the flavonoid group of substrates (i.e. flavonols- kaempferol, flavononesnaringenin; iso flavones- diadzein and genistein; flavonesapigenin and luteolin) with WsFGT complex revealed the lowest interaction energy of -9.3 kcal/mol with luteolin. The active residues for luteolin: Tyr13, His18, Gln133, His349, Trp352 and Asn353 with seven hydrogen bonds. All the hydrogen bond distances between WsFGT and luteolin complex were observed within the range of 2.1 Å to 3.1 Å. WsFGT docking conformations with naringenin, apigenin, kaempferol, diadzein and genistein were shown with interaction energies of -9.2, -9.2, -9.2, -8.4 and -8.4 respectively, as shown in Table 3 (see supplementary material) and in (Figures 4A-F). Together, these analyses identified Trp352 and Asn353 as critical amino acids due to their involvement in interactions with 5 different substrates. His-18 was identified as a key residue for enzyme activity. Similarly in all UGT structures, a highly conserved histidine (His22 in UGT71G1 and His20 in VvGT1) was observed in the active site. This may act as a general base and catalytic residue for enzyme activity to abstract a proton from the acceptor substrate [23]. Asp110 was another conserved residue in WsFGT (Asp121 in UGT71G1 and Asp119 in VvGT1) which formed a hydrogen bond with His18. This interaction may stabilize the histidine and balances its charge after deprotonating the acceptor [23]. For flavonoid group of substrates, molecular docking showed that the 7-hydroxyl group of flavonoids can be docked into the active site of the enzyme with higher interaction energies than the other hydroxyl groups. The docking study also showed that the interaction energy for iso-flavones were slightly lower compared with other flavonoids group. These studies provided clear evidence of the importance of these amino acid residues in the active site of WsFGT. Chemical synthesis of glycosides is often difficult and plant UGTs can be exploited for enzymatic synthesis. The data presented may also enable this enzyme to mediate sugar transfer to chemically distinct acceptor atoms for industrial application. Further, characterization and structural study of these glycosyltransferases would help to understand these difficult chemical reactions and facilitate biotechnological development for utilizing these novel UGTs.
Figure 4.
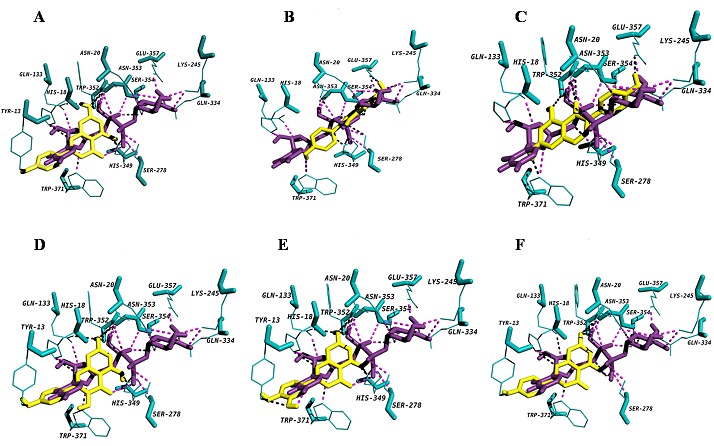
Diagram showing (A) apigenin (B) diadzein (C) genistein (D) kaempferol (E) luteolin and (F) naringenin docked into the proposed binding pockets. UDP-glucose is shown in deep violet purple colour and acceptors are shown in yellow colour. Hydrogen bonding interactions are indicated by dashed lines.
Conclusion
Flavonoids are natural source of antioxidants with various medicinal properties and synthesised by glycosylation which is catalysed by GTs. In this study, 3D structure of WsFGT was developed using available plant UGTs as templates which revealed GT-B fold. Docking analysis with different substrates identified important amino acids for catalytic function. However, this model is predictive and structure needs to be confirmed experimentally. The proposed model may be useful to understand glycosylation mechanism of flavonoid glucosides. Elucidation of the 3D structure of WsFGT will lead to inform design of novel biocatalysts for the synthesis and modification of plant natural products.
Supplementary material
Acknowledgments
SKRJ is thankful to Council of Scientific and Industrial Research (CSIR) for senior research fellowship.
Footnotes
Citation:Jadhav et al, Bioinformation 8(19): 943-949 (2012)
References
- 1.D Bowles, et al. Curr Opin Plant Biol. 2005;8:254. doi: 10.1016/j.pbi.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 2.P Chaturvedi, et al. Appl Biochem Biotechnol. 2011;165:47. doi: 10.1007/s12010-011-9232-0. [DOI] [PubMed] [Google Scholar]
- 3.BS Hong, et al. J Biochem Mol Biol. 2007;40:870. doi: 10.5483/bmbrep.2007.40.6.870. [DOI] [PubMed] [Google Scholar]
- 4.JH Kim, et al. Biosci Biotechnol Biochem. 2006;70:1471. doi: 10.1271/bbb.60006. [DOI] [PubMed] [Google Scholar]
- 5.CM Gachon, et al. Trends Plant Sci. 2005;10:542. doi: 10.1016/j.tplants.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 6.A Chang, et al. Proc Natl Aca Sci. 2011;108:17649. [Google Scholar]
- 7.R Udayakumar, et al. Int J Mol Sci. 2009;10:2367. doi: 10.3390/ijms10052367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.UM Unligil, JM Rini. Curr Opin Struct Biol. 2000;10:510. doi: 10.1016/s0959-440x(00)00124-x. [DOI] [PubMed] [Google Scholar]
- 9.SA Osmani, et al. Plant Physiol. 2008;148:1295. doi: 10.1104/pp.108.128256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.HM Berman, et al. Nucleic Acids Res. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MA Larkin, et al. Bioinformatics. 2007;23:2947. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 12.LJ McGuffin, et al. Bioinformatics. 2000;16:404. [Google Scholar]
- 13.A Sali, TL Blundell. J Mol Biol. 1993;234:779. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 14.EF Pettersen, et al. J Comput Chem. 2004;25:1605. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 15.RA Laskowski, et al. J Biomol NMR. 1996;8:477. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 16.C Colovos, TO Yeates. Protein Sci. 1993;2:1511. [Google Scholar]
- 17.M Wiederstein, MJ Sippl. Nucleic Acids Res. 2007;35:407. [Google Scholar]
- 18.MF Sanner. J Mol Graph Model. 1999;17:57. [PubMed] [Google Scholar]
- 19.O Trott, AJ Olson. J Comput Chem. 2010;3:455. [Google Scholar]
- 20.K Kherraz, et al. Bioinformation. 2011;6:115. [Google Scholar]
- 21.AL Morris, et al. Proteins. 1992;12:345. [Google Scholar]
- 22.A Noguchi, et al. Plant Cell. 2009;21:1556. doi: 10.1105/tpc.108.063826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.X Wang, et al. FEBS Lett. 2009;583:3303. doi: 10.1016/j.febslet.2009.09.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.