

Abstract
A number of web tools are available for the prediction and identification of target microRNAs (miRNAs). The choice, availability, validity and selection of an optimal yet appropriate tool are a challenge for the design of high throughput assays with promising miRNA targets. The current trends and challenges for target microRNAs (miRNAs) prediction, identification and selection is described in this review.
Keywords: pre-miRNA, miRNA, biogenesis, algorithms, in-silico biology
Background
MicroRNAs, the tiny molecules that fine-tune gene expression, were first discovered in 1993; Victor Ambros made a startling discovery [1]. With implications for the treatment of cancer, diabetes and brain disorders miRNAs play a vital role in the human genome. MicroRNAs (miRNAs) are endogenous ~22 nt RNAs that do play important regulatory roles in animals and plants by targeting mRNAs for cleavage or translational repression. The microRNA biogenesis has been well characterized as illustrated in Figure 1 [2].
Figure 1.
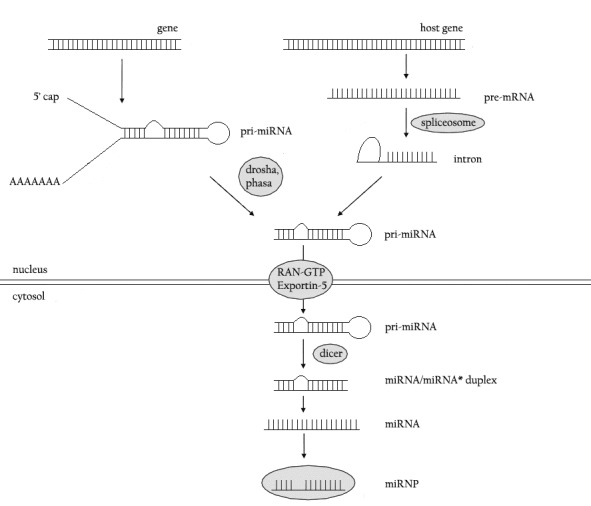
MicroRNA biogenesis.
miRNA – the name
A system of nomenclature has been adopted and names are designated to specific miRNAs before publication of their discovery as hundreds of human miRNAs and thousands across other species are available as of now [3, 4]. Experimentally confirmed microRNAs are given a number that is attached to the prefix mir followed by a dash e.g. mir-123. The uncapitalised mir- refers to the pre-miRNA and the capitalised miR- refers to the mature form. miRNAs with similar structures bar at 1 or 2 nucleotides are annotated to show their similar structure with added lower case letter e.g. miR-1a and miR-1b.
It is possible for miRNAs at different loci to produce the same miRNA and these are show with additional number eg miR-1-1 and miR-1-2. Names are even preceded by the annotation for the species they are observed in e.g. Homo sapiens = hsa-miRxxx. Others include viral v-mirNA and Drosphila d-miRNA. microRNAs originating for the 3' end or 5' end are denoted with a -3p or 5p suffix e.g. miR-142-5p, miR-142-3p.
Biogenesis
Most microRNA genes are found in the intergenic regions or in anti-sense oritentation to certain genes containing their own miRNA gene promoter and regulatory units. [5–8] However, as much as 40% are said to lie in the introns and non-protein coding DNA or even rarely in exons. These are usually, though not exclusively, found in the sense orientation and thus usually show a concurrent transcription and regulation expression profile originating from a common promoter with their host genes [9–14].
Other microRNA genes showing a common promoter include 42-48% of all miRNAs originating from polycistronic units contaning 2-7 discrete loops from which mature miRNAs are processed, though this does not necessarily mean the mature miRNAs of a family will be homologous in structure and function [6, 15]. The promoters mentioned have been shown to have some similarities in their motifs to promoters of other class II (meaning transcribed by POL II) genes such as protein coding genes [6, 16]. The DNA template is not the final word on mature miRNA production: 6% of human miRNAs show RNA editing, the site specific modification of RNA sequences to yield products different to those encoded by their DNA. This increases the diversity and scope of miRNA action beyond that implicated from the genome.
Transcription
In the nucleus, polymerase II (POL II) is used to transcribe microRNA encoding parts of the genome often through binding to a promoter found near the sequence destined to be the hairpin loop of the pre-miRNA [6, 16]. This produces a transcript that is capped at the 5' end, polyadenylated to give a (poly)A tail and spliced to form pri-miRNA several hundred to thousand bp in size. Curiously, some pri-miRNAs have been shown to be able to coordinately express both miRNAs and mRNAs, when the stem loop precursor is found in the 3' UTR of an mRNA [16]. Uncommonly, polymerase III (POL III) is speculated to be used instead of POL II when transcribing microRNA that have upstream -Alu, -tRNA, Mammalian Wide Interspersed Repeat (MWIR) promoter units [17].
Nuclear processing and export
Pri-miRNA's are processed by the microprocessor complex consisting of drosha and its cofactor DGCR8 into pre-miRNAs. Pri-miRNA contains at least 1 (up to 6 when transcribed from polycistronic units) ~70 nucleotide hairpin loop structures, there is a potential for a single pri-miRNA to house many miRNAs. The hairpin loops have >40 nucleotide flanking RNA sequences necessary for efficient processing. These are recognised by the DiGeorge Syndrome Critical Region 8 (DGCR8), the cofactor to drosha, DGCR8 is a dsRNA binding nuclear protein that recognizes the hairpin loop of the primiRNA and orientates the catalytic RNAse III domain of drosha for cleavage. This cleavage occurs around 11 nucleotides from their base (2 helical RNA turns into the stem) by Drosha, a RNAse III type dsRNA specific endonuclease, to form premiRNA. Together, drosha and DGCR8 (the invertebrate equivalent is Pasha) form the microprocessor complex [18]. The microprocessor complex introduces staggered cuts to the ends of the hairpin loop arms resulting in a 2 nucleotide overhand on the 3' end and phosphate on the 5' end to produce a premiRNA of ~ 70 nt in length. Mostly, one arm of the hairpin loop is destined to become the mature miRNA, though rarely a mature miRNA may be produced from either arm eg Mir-458- 3p/mir-458-5p and mir-202/mir-202* with the asterisk applying to less predominantly expressed transcript.
There is evidence that pre-miRNAs can be produced without having to undergo the microprocessor machinery if they are directly spliced from the introns in which they reside These miRNAs are called mirtrons and have traditionally been thought to only exist in drosphila and c elegans. Recently however, mammalian mirtons that even show conservation between species have even been discovered [19].
Pri-miRNA can also be subject to RNA editing wherein the miRNA processing or specificity is altered through adenosine deaminase acting on RNA (ADAR) enzymes catalysing adenosine to inosine transitions, the most common form of RNA editing in metazoans [20]. RNA editing has been shown to occur in 6% of miRNAs, even altering the specificity of miRNAs when it was observed in the seed region of miR-376, though this is only present in the CNS [21].
RNA editing of microRNA can also prevent their processing, as seen in the pri-miR-142 editing leading to degradation by the tudor SN protein (a RISC component) and thus avoiding of the drosha pathway [22]. Overall, this offers many implications in expanding the already complicated role in genetic expression that are covered in more detail than this paper has to opportunity to do in an excellent review [23]. The nuclear membrane protein exportin 5 recognises the 2 nucleotide overhang on the 3 end of the pre-miRNA [24] and then transports it into the cytoplasm using ran-guanine triphosphatase (Ran-GTP) [25].
Cytoplasmic processing
In the cytoplasm the pre-miRNA is cleaved by another RNAse III type double stranded endonuclease called Dicer. Dicer cleavage of pre-miRNA results in an imperfect miRNA:miRNA duplex around 20-25 nucleotides in size containing the mature miRNA strand and its opposite complementary miRNA strand [26]. Dicer is associated with the cofactors immunodeficiency virus (HIV) transactivating response RNA binding protein (TRBP) and protein activator of the interferon induced protein kinase PACT that physically bring the TRBP-PACT-dicer complex into contact with Ago2 to form the RNA Induced Silencing (RISC) loading apparatus. Dicer processing of the premiRNA is thought to be coupled to the unwinding of the duplex to produce a mature miRNA which Ago2 binds to, forming the active miRISC complex. The mature miRNA then guides the RISC to target sites in order to induce silencing [26, 27]. The precise sequence of events is difficult to elucidate and still under debate.
miRNAs in plants
Plants contain significant quantities of miRNAs derived from various miRNA biogenesis pathways. Many of these miRNAs play regulatory roles in plants. Analysis revealed that numerous miRNAs in corn, rice and soybean seeds have high sequence similarity to animal genes. However, exogenous RNA is considered to be unstable within the gastrointestinal tract of many animals, thus limiting potential for any adverse effects from consumption of dietary RNA.
This August, Monsanto people searched for plant miRNAs sequences in public sRNA datasets from various tissues of mammals, chicken and insects. They revealed that plant miRNAs were present in the animal sRNA datasets, and significantly miR168 was extremely over-represented. Furthermore, all or nearly all (>96%) miR168 sequences were monocot derived for most datasets, including datasets for two insects reared on dicot plants in their respective experiments. To investigate if plantderived miRNAs, including miR168, could accumulate and move systemically in insects, they conducted insect feeding studies for three insects including corn rootworm, which has been shown to be responsive to plant-produced long double-stranded RNAs [28].
miRNA biogenesis in plants differs from metazoan biogenesis mainly in the steps of nuclear processing and export. Instead of being cleaved by two different enzymes, once inside and once outside the nucleus, both cleavages of the plant miRNA is performed by a Dicer homolog, called Dicer-like1 (DL1). DL1 is only expressed in the nucleus of plant cells, which indicates that both reactions take place inside the nucleus. Before plant miRNA:miRNA* duplexes are transported out of the nucleus its 3' overhangs are methylated by a RNA methyltransferaseprotein called Hua-Enhancer1 (HEN1). The duplex is then transported out of the nucleus to the cytoplasm by a protein called Hasty (HST), an Exportin 5 homolog, where they disassemble and the mature miRNA is incorporated into the RISC [29].
miRNA and viruses
A microRNA is about 17 to 24 nucleotides, and its function is to dampen or shut down the production of proteins in the body. There are hundreds of different types of microRNAs in animals. It's been known for many years that when a virus such as influenza infects respiratory cells there is an immediate antiviral response at the cellular level -- the first barrier for protecting the body from the virus. Most of the changes that occur are a result of antiviral gene expression. The expression of transcription activators by human herpesvirus-6 DNA is believed to be regulated by viral miRNA [30].
Target Finder tools – the machine guns
The current edition of miRBase [31] (version 19) lists more than 21000 microRNAs. Clearly, a challenge ahead will be to identify the targets of all these small RNAs. Since, each microRNA can target several mRNA sequences, this will be a daunting task and will require the aid of in-silico target finder tools. However, most typical Bioinformatics tools require longer sequences than the ~20 nucleotides offered by microRNAs. In addition, microRNAs are only partially complementary to their mRNA target sequences, which make identifying them even more difficult. Nevertheless, a number of algorithms have been developed which can be used for microRNA target prediction.
miRecords [32] is resource for animal miRNA-target interactions developed at the University of Minnesota. miRecords consists of two components. The Validated Targets component is a large, high-quality database of experimentally validated miRNA targets resulting from meticulous literature curation. The Predicted Targets component of miRecords is an integration of predicted miRNA targets produced by 11 established miRNA target prediction programs.
PicTar [33] is an algorithm for the identification of microRNA targets. This searchable website provides details (3' UTR alignments with predicted sites, links to various public databases etc) regarding microRNA target predictions in vertebrates, several Drosophila species, and C. elegans.
miRanda [34] is an algorithm for finding genomic targets for microRNAs. This algorithm has been written in C and is available as an open-source method under the GPL. MiRanda was developed at the Computational Biology Center of Memorial Sloan-Kettering Cancer Center. This software will be further developed under the open source model, coordinated by Anton Enright and Chris Sander.
TargetScan [35] predicts microRNA targets. They are essentially the 3'UTR targets, with a few changes arising from updated gene boundary definitions from the April 2005 UCSC genome browser mapping of RefSeq mRNAs to the hg17 human genome assembly. To avoid difficulties in browser display, the few predictions spanning splice junctions are excluded.
RNAhybrid [36] tool finds the minimum free energy hybridization of a long and a short RNA. The hybridization is performed in a kind of domain mode, ie. the short sequence is hybridized to the best fitting part of the long one. The tool is primarily meant as a means for microRNA target prediction.
DIANA MicroT Analyzer [37] this is a tool for prediction of MicroRNA targets. The program reports a signal to noise ratio and a precision score which help in the evaluation of the significance of the predicted results.
RegRNA [38] is an integrated web server for identifying the homologs of Regulatory RNA motifs and elements against an input mRNA sequence. Both sequence homologs and structural homologs of regulatory RNA motifs can be identified.
RNA22 [39] is a pattern-based method developed by IBM group, for the identification of microRNA-target sites and their corresponding RNA/RNA complexes.
TarBase [40] is a database of experimentally supported targets. TarBase reveals significantly more experimentally supported targets than even recent reviews claim, thereby providing a comprehensive data set from which to assess features of miRNA targeting that will be useful for the next generation of target prediction programs.
psRNATarget [41] is a Plant Small RNA Target Analysis Server. A plant small RNA (including microRNAs) target analysis server, which features two important analysis functions:
reverse complementary matching between miRNA and target transcript using a proven scoring schema, and
target site accessibility evaluation by calculating unpaired energy (UPE) required to “open” secondary structure around miRNA's target site on mRNA.
PsRNATarget incorporates recent discoveries in plant miRNA target recognition, e.g. it distinguishes translational and post-transcriptional inhibition, and it reports the number of miRNA/target site pairs that may affect miRNA binding activity to target transcript. psRNA Target is replacing miRU (Plant microRNA Potential Target Finder) by the same group.
miRDB [42] is an online database for miRNA target prediction and functional annotations. All the targets were predicted by a bioinformatics tool MirTarget2, which was developed by analyzing thousands of genes impacted by miRNAs with an SVM learning machine. Common features associated with miRNA target binding have been identified and used to predict miRNA targets. miRDB hosts predicted miRNA targets in five species: human, mouse, rat, dog and chicken.
IPA's microRNA Target Filter [43] a new microRNA target prioritization tool that is available within IPA - The latest that adds to the list. It contains content from TarBase, TargetScan, miRecords, and micro-RNA related findings from the published literature. It also includes filtering tools that helps quickly sort through thousands of microRNA targets to find the ones most biologically relevant dataset or experiment.
HMMER [44] searches sequence databases for homologs of protein sequences, and for making protein sequence alignments. It implements methods using probabilistic models called profile Hidden Markov Models (profile HMMs).
Conclusion
Targeting miRNAs have received new impetus in recent years. The computational miRNA target finders, that exist may vary in the algorithm used; and one can state opinions about the strengths or weaknesses of each particular algorithm, the fact of the matter is that all methods fall substantially short of capturing the full detail of physical, temporal, and spatial requirements of biologically significant miRNA::mRNA interactions. The performance of current tools is largely dependent on the overall number of predicted targets (hits). Some tools may be very efficient in predicting true targets sites (High sensitivity) but at the same time display an extremely large number of overall hits (Low specificity). In contrast other tools display an overall high specificity but a very low sensitivity. It is evident that there is great need for a more sophisticated target prediction tool that achieves a balance between sensitivity and specificity. Performing this large scale scan of all miRNAs in the online registry and all the genes of the human genome, will allow us to compare our tool with other available tools on a large scale basis. The results will be stored on an online database that will allow users to query the database using miRNA name or gene name. This will shed some light into new gene targets for miRNAs and the molecular processes that they regulate. Finally experimental verification of computational predictions will be the ultimate step in revealing the molecular pathways of miRNA regulation and characterizing their involvement in disease.
Finding true miRNAs in-silico is still a big task [45]. A better insight for future growths on miRNA gene search can likely be attained by considering what is not a miRNA. A given location in the genome does not contain a miRNA if:
A stem–loop structure liable for effective processing by all partakers in the miRNA biogenesis pathway is not found;
it cannot regulate a target gene in a physiologically relevant manner
it is not effectively transcribed.
Widely used modes focused on the first case and the other two are still awaiting attention.
Another question that becomes more important when many authors argue that the identification of well conserved and phylogenetically extensive miRNAs is reaching its saturation is whether non-conserved, presumably more exotic, miRNA precursors would be processed as such in different organisms that may have small yet important differences in their processing pathways. The elucidation of this question is crucial to methods which try to generalize from pre-miRNAs taken from several different species.
Acknowledgments
This review is made possible through the help and support of academic advisors, teachers, publishers, family and friends. The author thanks Prof. Dr. Mrutyunjay Suar, Director and Head, KIIT School of Biotechnology (KSBT), KIIT University. Thanks are also to Dr. Rajanikanta Mahapatra, Faculty Associate, KIIT School of Biotechnology (KSBT), KIIT University.
Footnotes
Citation:Das, Bioinformation 8(17): 841-845 (2012)
References
- 1.RC Lee, et al. Cell. 1993;75:843. [Google Scholar]
- 2.ND Mendes, et al. Nucleic Acids Res. 2009;37:2419. doi: 10.1093/nar/gkp145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.V Ambros, et al. RNA. 2003;9:277. [Google Scholar]
- 4.S Griffiths-Jones, et al. Nucleic Acids Res. 2006;34:D140. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.NC Lau, et al. Science. 2001;294:858. [Google Scholar]
- 6.Y Lee, et al. EMBO J. 2004;23:4051. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.M Lagos-Quintana, et al. Science. 2001;294:853. [Google Scholar]
- 8.RC Lee, V Ambros. Science. 2001;294:862. [Google Scholar]
- 9.M Mraz, et al. Blood. 2012;119:2110. [Google Scholar]
- 10.A Rodriguez, et al. Genome Res. 2004;14:1902. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.X Cai, et al. RNA. 2004;10:1957. [Google Scholar]
- 12.MJ Weber. FEBS J. 2005;272:59. doi: 10.1111/j.1432-1033.2004.04389.x. [DOI] [PubMed] [Google Scholar]
- 13.YK Kim, VN Kim. EMBO J. 2007;26:775. doi: 10.1038/sj.emboj.7601512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.S Baskerville, DP Bartel. RNA. 2005;11:241. [Google Scholar]
- 15.Y Altuvia, et al. Nucleic Acids Res. 2005;33:2697. doi: 10.1093/nar/gki567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.X Zhou, et al. PLoS Comput Biol. 2007;3:e37. doi: 10.1371/journal.pcbi.0030037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.M Faller, F Guo. Biochim Biophys Acta. 1779;663 doi: 10.1016/j.bbagrm.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.RI Gregory, et al. Methods Mol Biol. 2006;342:33. doi: 10.1385/1-59745-123-1:33. [DOI] [PubMed] [Google Scholar]
- 19.E Berezikov, et al. Mol Cell. 2007;28:328. [Google Scholar]
- 20.L Valente, K Nishikura. Prog Nucleic Acid Res Mol Biol. 2005;79:299. doi: 10.1016/S0079-6603(04)79006-6. [DOI] [PubMed] [Google Scholar]
- 21.Y Kawahara, et al. Science. 2007;315:1137. [Google Scholar]
- 22.W Yang, et al. Nat Struct Mol Biol. 2006;13:13. [Google Scholar]
- 23.M Ohman. Biochimie. 2007;89:1171. doi: 10.1016/j.biochi.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 24.Y Zeng, BR Cullen. Nucleic Acids Res. 2004;32:4776. doi: 10.1093/nar/gkh824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MT Bohnsack, et al. RNA. 2004;10:185. [Google Scholar]
- 26.E Lund, JE Dahlberg. Cold Spring Harb Symp Quant Biol. 2006;71:59. doi: 10.1101/sqb.2006.71.050. [DOI] [PubMed] [Google Scholar]
- 27.X Ji, et al. Curr Top Microbiol Immunol. 2008;320:99. doi: 10.1007/978-3-540-75157-1_5. [DOI] [PubMed] [Google Scholar]
- 28.Y Zhang, et al. BMC Genomics. 2012;13:381. doi: 10.1186/1471-2164-13-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.C Lelandais-Brière, et al. Curr Genomics. 2010;11:14. doi: 10.2174/138920210790217918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.L Tuddenham, et al. J Virol. 2012;86:1638. doi: 10.1128/JVI.05911-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. www.mirbase.org.
- 32. www.mirecords.biolead.org.
- 33. www.pictar.mdc-berlin.de.
- 34. www.microrna.org.
- 35. www.targetscan.org.
- 36. www.bibiserv.techfak.uni-bielefeld.de/rnahybrid.
- 37. www.diana.cslab.ece.ntua.gr.
- 38. www.regrna.mbc.nctu.edu.tw.
- 39. www.cbcsrv.watson.ibm.com/rna22.html.
- 40. www.diana.cslab.ece.ntua.gr/tarbase.
- 41. www.plantgrn.noble.org/psRNATarget.
- 42. www.mirdb.org/miRDB.
- 43. www.ingenuity.com/products/IPA/microRNA.html.
- 44. www.hmmer.janelia.org.
- 45.N Das, et al. BIOMIRROR. 2012;3:1/bm-2229260212. [Google Scholar]