

Abstract
Clinical graft-versus-host disease (GVHD) symptoms are the result of a complex set of interactions between cellular and soluble factors. One of the key soluble factors is the proinflammatory cytokine, TNF-a, which participates in the initiating events that culminate in GVHD as well as amplifies the disease process once established. The importance of TNF-a in this process has been supported by a series of clinical experiments demonstrating strong correlation between TNF receptor-1 levels and GVHD. TNF-a has both indirect effects, through activating and proliferation pathways of T cells, the main cellular effector of GVHD, and direct effects leading to apoptosis, on GVHD target tissues. Accordingly, TNF-a has been used as a therapeutic target in experimental GVHD prevention and treatment strategies with promising clinical results. TNF-a can be pharmacologically inhibited using soluble TNF receptors or monoclonal antibodies. The optimal dosing and duration of TNF inhibition to prevent or treat GVHD remains under investigation.
Keywords: Tumor necrosis factor, Graft-versus-host disease, Allogeneic, Bone marrow transplantation, Treatment, Prevention
Introduction
The introduction in the early 1990s of a new paradigm for graft-versus-host disease (GVHD) pathophysiology1 that incorporated both cellular factors (such as donor T cells) and soluble factors (such as TNF-α) led to major changes in the overall approach to acute GVHD diagnosis, prevention, and management. Over the past two decades, this model of GVHD pathophysiology has been continuously updated as new experimental data became incorporated into the overall understanding of GVHD2. The key to understanding this model of GVHD pathophysiology rests on two key underlying principles. First, clinical GVHD symptoms reflect the culmination of exaggerated but normal inflammatory mechanisms mediated by donor lymphocytes transplanted into the recipient where they function appropriately, given the foreign environment they encounter. Second, recipient tissue damage as a consequence of underlying disease, prior infections, and/or the transplant conditioning regimen facilitates the process of donor lymphocyte activation against host tissue.2 The production of proinflammatory cytokines and chemokines by damaged recipient tissues that that then increase the expression of receptors on antigen presenting cells (APCs) involved in the cross-presentation of polypeptide proteins, such as minor histocompatibility antigens, to the donor lymphocytes that mediate GVHD is a key step in this process. GVHD pathophysiology can best be conceptualized in three sequential steps or phases: (1) activation of the APCs; (2) donor T cell activation, proliferation, differentiation and migration; and (3) target tissue destruction (Figure 1).
Figure 1.
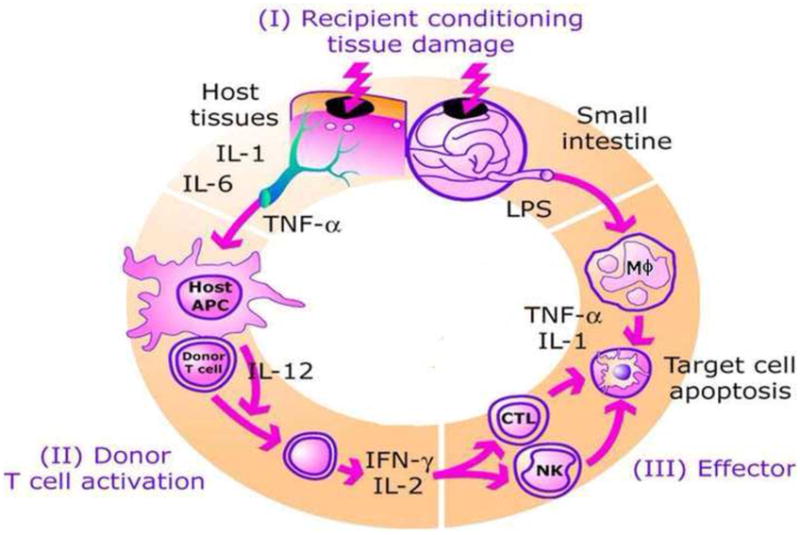
Model of graft versus host disease. GVHD develops in three phases. (I) Release of inflammatory cytokines activate antigen presenting cells. (II) Donor T cells are activated by host antigen presenting cells. (III) Cellular and plasma effectors cause target organ apoptosis.
Phase I: Activation of APCs
The first step involves the activation of APCs by the underlying disease and the transplant conditioning regimen. As mentioned above, host tissues respond to damage by producing “danger” signals, including proinflammatory cytokines (such as TNF-α), chemokines, and increased expression of adhesion molecules, MHC antigens and costimulatory molecules on host APCs.3–6
Phase II: Donor T Cell Activation
In the second phase of GVHD, donor T cells proliferate and differentiate in response to host APCs. This step is augmented by enhanced interaction at the immune synapse between APCs like dendritic cells and T cells when exposed to pro-inflammatory cytokines like TNF-α7. Once T cells are activated, a rapid intracellular biochemical cascade ensues resulting in increased production of large amounts of Th1 cytokines such as IFN-γ, IL-2 and TNF-α. The traditional focus of pharmacologic GVHD prevention strategies has been interference with IL-2 production leading to the widespread clinical use of calcineurin inhibitors such as cyclosporine and tacrolimus8,9. Daclizumab, a humanized monoclonal antibody against the interleukin 2 (IL-2) receptor, has been tested as a treatment for acute GVHD. In a randomized trial of 102 patients with new onset acute GVHD, patients treated with high dose corticosteroids plus either daclizumab or placebo, found no difference in GVHD response rates10. The trial was halted early due to inferior survival in the daclizumab treated patients who experienced high rates of both GVHD-related and relapse-related deaths. This finding was difficult to explain but one theory was that antibody targeting of the IL-2-receptor may have eliminated critical populations of conventional T cells (required for graft-versus-malignancy effect) and regulatory T cells (required for GVHD control). In fact, newer data indicate that IL-2 plays an important role in the generation and maintenance of regulatory T cells, suggesting that prolonged interference with IL-2 may actually prevent the development of long term tolerance after allogeneic HCT.11
Phase III: Cellular and Inflammatory Effector Phase
In the third phase of GVHD, cellular mediators such as cytotoxic T lymphocytes (CTLs) and NK cells and inflammatory mediators such as TNF-α, IFN-γ, IL-1 synergize to amplify local tissue injury and further promote inflammation and target tissue destruction12,13. Important among these mediators is the microbial product, lipopolysaccharide (LPS), which can leak through intestinal mucosa or skin damaged by the pre-HCT conditioning regimen. LPS can stimulate the secretion of inflammatory cytokines through toll-like receptors (TLRs)5,14,15. Among the acute GVHD target organs, the gastrointestinal tract appears especially susceptible to damage from TNF-α, creating an amplification loop that propagates the “cytokine storm” characteristic of acute GVHD5. Inhibition of TNF-α in experimental allogeneic HCT models ameliorated the apoptosis characteristic of GVHD related damage to the GI tract16,17.
In summary, TNF-α plays a role in all phases of GVHD pathophysiology from the early phase of host APC activation through to both direct tissue damage (as its name implies) and indirect tissue damage mediated by cytotoxic lymphocytes.
HUMAN STUDIES OF TNF-α IN GVHD
TNF-α in GVHD Prediction and Prevention
The experimental data supporting a key role for TNF-α in the pathophysiology of GVHD has been supported by human studies of allogeneic HCT recipients. Although human studies do not allow for the careful dissection of each GVHD phase described by the model described above, observations from human studies have been consistent with the expected results predicted by the GVHD model. Chief among these are clinical studies that have developed strong evidence supporting the correlation between TNF-α levels following hematopoietic cell transplantation (HCT) and GVHD18–20. The largest of these studies measured TNF-receptor-1 (TNFR1) levels pre-transplant and on day 7 post-transplant in 438 recipients of myeloablative related and unrelated hematopoietic cell transplants (HCT). TNFR1 levels have become accepted as a surrogate for direct measurement of TNF-α. TNF-α binds to its receptors, TNFR1 and TNFR2 on the surface of multiple cell types21. After binding, the receptor-ligand complex is shed into the plasma where it can be measured by enzyme-linked immunosorbent assay (ELISA). In this clinical study, the magnitude of change in the early post-transplant TNFR1 levels compared to pre-transplant baseline levels was performed by creating ratios of the day 7 level divided by the baseline level. These day 7 TNFR1 ratios were then used to compare groups of patients. This strategy simplified the handling of the wide variability in pre-transplant baseline levels, which otherwise would have made statistical comparisons more difficult. Several important findings, consistent with predictions from the GVHD model derived from animal studies, were made. First, patients who never developed clinically significant GVHD (grade 0-I) nearly doubled their TNFR1 levels by the end of the first week post-transplant (day 7 TNFR1 ratio 1.91, n=269). The increase in TNFR1 levels as a result of tissue damage related to intense chemotherapy/radiation therapy as part of the pre-HCT conditioning regimen occurred as predicted in prior animal and human studies4,18. In patients who do not develop serious complications, the levels return to near baseline. Second, patients who went on to develop clinically significant GVHD had higher day 7 TNFR1 ratios (grade II, day7 TNFR1 ratio 2.32, n=83 and grade III/IV day 7 TNFR1 ratio 2.92, n=86). The correlation of day 7 TNFR1 ratios with GVHD severity was highly statistically significant (p<0.001). In this study, the median onset of GVHD was day 30 in related donor recipients and day 20 in unrelated donor recipients. Therefore, the magnitude of increase in TNFR1 levels was able to be measured 2–3 weeks in advance of the median onset of clinical symptoms, thereby suggesting that the events that culminate in significant GVHD are underway well in advance of their detection by such factors as skin rash, gastrointestinal symptoms, or jaundice. The third important finding was the strong correlation between day 7 TNFR1 levels, time to onset of GVHD, transplant-related mortality (TRM), and overall survival. Patients in the highest quartile of day 7 TNFR1 ratios (day 7 TNFR1 ratio >2.5) developed GVHD earlier and more often, had higher rates of TRM, and were less likely to be alive one-year after HCT than the other patients. The correlation between the day 7 TNFR1 ratio and these clinical outcomes was highly statistically significant (p<0.001) for each of the outcomes. Because donor type (related vs unrelated) is a well-known predictor for these same clinical outcomes, it is noteworthy that the day 7 TNFR1 ratio correlated with these important outcomes in both related (n=267) and unrelated donor (n=171) HCT recipients when these groups were analyzed separately, although the difference in survival at one-year did not meet statistical significance in the related donor recipients. In multivariate analysis, which included donor type, degree of HLA-match, recipient age, gender, disease and disease status at transplant, and conditioning regimen, the day 7 TNFR1 ratio was an independent predictor of GVHD, TRM, and overall survival when treated as either a continuous variable or using a threshold day 7 TNFR1 ratio. The hazard ratios using a day 7 TNFR1 ratio threshold are shown in Table 1. Day 7 TNFR1 ratios continued to correlate with clinical outcomes when children were analyzed separately20.
Table 1.
Multivariate model with hazard ratios for time to GVHD, time to TRM, and overall survival using day TNFR1 ratio threshold (>2.5) [from reference 19]
| GVHD | TRM | Overall survival | |||||
|---|---|---|---|---|---|---|---|
| Variable | Category | HR | P | HR | P | HR | p |
| Day 7 TNFR1 ratio | ≤2.5 | Ref | - | Ref | - | Ref | - |
| >2.5 | 2.44 | <0.001 | 2.40 | <0.001 | 1.39 | 0.03 | |
| Donor type | Related | Ref | - | Ref | - | Ref | - |
| Unrelated | 1.87 | <0.001 | 3.25 | <0.001 | 1.49 | 0.006 | |
| HLA-matched | Yes | Ref | - | Ref | - | Ref | - |
| No | 1.93 | 0.01 | 1.09 | 0.79 | 1.25 | 0.28 | |
| Recipient age* | 1.05 | 0.35 | 1.28 | 0.002 | 1.10 | 0.03 | |
| Donor/recipient gender | Male/male | Ref | - | Ref | - | Ref | - |
| Female/male | 1.29 | 0.19 | 1.26 | 0.39 | 1.02 | 0.90 | |
| Male/female | 0.63 | 0.04 | 0.69 | 0.19 | 0.88 | 0.46 | |
| Female/female | 0.80 | 0.54 | 0.75 | 0.57 | 1.01 | 0.98 | |
| Disease** | Nonmalignant | Ref | - | Ref | - | Ref | - |
| Standard risk | 1.65 | 0.36 | 0.96 | 0.96 | 4.26 | 0.05 | |
| Advanced risk | 1.74 | 0.31 | 1.31 | 0.70 | 7.67 | 0.005 | |
| Conditioning regimen | Non-TBI | Ref | - | Ref | - | Ref | - |
| TBI | 1.14 | 0.63 | 0.61 | 0.20 | 1.18 | 0.45 | |
TBI = total body irradiation
For recipient age, HR reflects the change in hazard for every ten years
Standard risk: lymphoma with chemosensitive disease, leukemia in complete remission, or untreated myelodysplastic syndrome. Advanced risk: leukemia not in remission, lymphoma with stable or progressive disease at time of transplantation
In the above studies, the analyses were restricted to recipients of myeloablative conditioning, a setting in which tissue damage and subsequent TNF-α release is most likely to occur. Accordingly, one would expect a reduction in GVHD following reduction of conditioning intensity, a finding that has been observed across several studies although in some work the reduction in GVHD has been primarily observed as a delay in GVHD onset22–24. Not surprisingly, correlations between day 7 TNFR1 levels and GVHD have also been observed in studies of non-myeloablative conditioning25. In a study of 106 patients who received non-myeloablative conditioning regimens of 2 Gy TBI alone (n=15), 2 Gy TBI plus fludarabine 90 mg/m2 (n=73), or 4 Gy TBI plus fludarabine 90 mg/m2 (n=18), TNFR1 levels increased significantly from baseline to day 7 (p<0.0001) with the largest increases in the day 7 TNFR1 ratio seens in those conditioned with the highest dose of radiation. Multivariate analysis confirmed the independent correlation of the day 7 TNFR1 ratio with GVHD incidence and severity, but a statistically significant relationship between day 7 TNFR1 ratios and survival was not observed. These findings are supported by a study from the United States National Cancer Institute where percentage of monocytes producing TNF-α were serially measured in 17 patients undergoing reduced intensity allogeneic HCT26. Patients who developed GI GVHD had rises in the percentage of TNF-α producing monocytes, measured by flow cytometry, preceding the clinical development of GVHD and peaking at the time of GVHD onset. In further support, the level of TNF-α messenger RNA levels in T cells climbed in the period preceding the onset of GVHD, peaking at the onset of GVHD in 8 of 9 patients who developed grade II-IV GVHD but not in 8 allogeneic patients who did not develop clinically significant GVHD, 1 syngeneic HCT recipient, or 4 autologous controls27. Taken together, plasma protein levels, RNA transcription levels, and flow cytometry results all convincingly support the premise that rises in TNF-α precede the onset of the GVHD and peak at the time of its development. The subsequent declines in TNF-α may be attributed to GVHD treatment
These findings suggest that it may be possible to predict GVHD occurrence based on early measurement of GVHD biomarkers such as TNFR1. Although measurements of TNFR1 alone appear to be inadequate for this purpose, recent unpublished work from the Paczesny laboratory at the University of Michigan suggests that using multiple GVHD biomarkers may allow for GVHD prediction. In this study, plasma samples were prospectively collected at three time points (day 0, 7, and 14) from 513 unrelated donor HCT patients transplanted over a 10 year time period. The patients were randomly divided into training and validation sets and the plasma samples were then assayed for levels of three GVHD biomarkers that had been previously validated in other studies – IL-2-receptor-α (IL2Rα, TNFR1, and elafin)28,29. A logistic regression model using the levels of the three biomarkers at days 0 and 7 was able to risk stratify patients into high and low risk groups for future development of GVHD. Patients who were categorized as low risk and had not yet developed GVHD were re-analyzed at day 14 for the purpose re-assessing their risk for GVHD development. The model was able to correctly predict the occurrence or absence of grade II-IV GVHD in 66% of patients. A clinical trial is being designed to pre-emptively treat GVHD (similar to the practice currently used to preemptively treat cytomegalovirus infections in allogeneic HCT recipients) using these results. In other unpublished work, etanercept has been used to augment GVHD prophylaxis and thereby improve survival after myeloablative unrelated donor HCT in a phase II clinical trial of 71 patients conducted at the University of Michigan and Loyola University Medical Center. All patients received chemotherapy-alone conditioning regimens. Etanercept (0.4 mg/kg, max 25 mg was given twice weekly from the start of conditioning to day 56 in addition to standard GVHD prophylaxis using tacrolimus and methotrexate (5 mg/m2 on days 1, 3, 6, 11). When compared to 29 case-matched controls who received standard GVHD prophylaxis alone the etanercept treated patients had only a 21% increase in TNFR1 levels at day 7 which was significantly lower than the control patients (p=0.002) although baseline TNFR1 levels were similar in both groups. The incidence of grade II–IV GVHD was 47%, similar to expected, but response to treatment with high dose corticosteroids was high (73%), non-relapse mortality at six months was low (14%) and two-year overall survival was high (55%). These results await validation in a larger multicenter randomized study, which is currently planned through the Blood and Marrow Transplant Clinical Trials Network. In a small study of 19, mostly related donor, myeloablative HCT recipients, infliximab was given prior to conditioning and then on days 0, +7, +14, +28, and +42 together with standard cyclosporine and methotrexate prophylaxis30. The cumulative incidence of GVHD grades II-IV (37%) was no different than that observed in 30 case-matched historical controls. There was no improvement in survival and there were more bacterial and fungal infections in the infliximab treated patients compared to case control patients. The small size of the study makes interpretation difficult, but this study does not increase enthusiasm for incorporation of infliximab in GVHD prevention strategies.
TNF-α in GVHD Treatment
The GVHD pathophysiology model above predicts that TNF-α, and by corollary TNFR1 levels, will be elevated at the onset of GVHD. Multiple studies over the past 15 years have confirmed the accuracy of this prediction26,28,31,32. Based on these results, several clinical studies have targeted TNF-α as part of a treatment strategy for acute GVHD. Although there are several ways to inhibit TNF-α, most of the clinical trials have used either etanercept, a soluble dimeric TNF-α receptor 2, that competes for TNF-α binding with cellular receptors, or infliximab, a monoclonal antibody that binds TNF-α. A major difference between etanercept and infliximab is that infliximab can induce systemic elimination of monocytes and macrophages that express membrane-bound TNF, whereas etanercept does not33. This elimination of monocytes and macrophages may explain, at least in part, the high rates of invasive Aspergillus infections seen in some studies using infliximab34,35, but not in clinical trials of etanercept31,36. Additionally, changes in fungal prophylaxis to include agents with Aspergillus coverage may also explain some of the differences observed in terms of undesirable outcomes with these two different TNF-inhibitors. These differences in mechanism of action may also explain some of the differences in clinical outcomes seen when using these drugs.
TNF-inhibition has shown some promise as a treatment for new onset acute GVHD. In one study, 61 patients with new onset acute GVHD grades II–IV were prospectively treated with daily high dose corticosteroids (methylprednisolone 2 mg/kg/d) and etanercept (0.4 mg/kg/dose, maximum dose 25 mg) twice weekly for eight weeks31. A high rate of complete resolution of GVHD symptoms (69%) was observed by day 28 after initiation of treatment, which compares favorably to the expected 35% rate previously reported in the literature when using high dose corticosteroids alone37. When compared to 99 contemporaneous case-matched patients with GVHD grades II–IV treated initially with high dose corticosteroids alone, the etanercept treated subjects had a statistically superior rate of resolution of GVHD symptoms (69% vs 33%; p<0.001) and superior survival at six months from GVHD onset (69% vs 55%; p= 0.08), although this latter finding did not meet statistical significance. Interestingly, the apparent benefit of etanercept was most clearly seen in recipients of unrelated donor HCT, a group of patients for whom the higher rate of early resolution of GVHD appeared to translate into a survival advantage at six months post treatment initiation.(73% vs 52%, p=0.05). In contrast, although related donor HCT recipients treated with etanercept were more likely to quickly resolve their GVHD recipients than similar patients treated with high dose steroids alone, ultimately large proportions of both groups achieved a complete response to treatment. Thus, it was not surprising that there was no survival advantage observed for related donor recipients whose new onset acute GVHD was treated with the combination of etanercept and high dose steroids. In all patients, TNFR1 levels were elevated at the onset of GVHD and significantly declined in those whose GVHD responded to treatment. Similar results, were seen when TNF-inhibition with etanercept was incorporated into the treatment of new onset acute GVHD. In a multicenter prospective study, 180 patients with new onset acute GVHD were randomized to eceive methylprednisolone 2 mg/kg per day plus either etanercept, mycophenolate mofetil (MMF), denileukin diftitox (denileukin), or pentostatin36. The study was intended to select one agent for a prospective, randomized, placebo controlled trial of GVHD treatment and was therefore not powered, nor did it detect, any statistically significant differences between the four drugs tested. Patients who randomized to the etanercept arm had lower rates of early complete resolution of GVHD symptoms compared to the single center study or the other three drugs tested, but ultimately similar response rates were achieved with all agents. Survival at nine months from initiation of treatment was best for MMF (64%), while etanercept, denileukin, and pentostatin treated patients all experienced essentially identical rates of survival (47%, 49%, and 47%, respectively). Importantly, in these studies there has been no indication of an increase in the rate of infectious complications or relapse, and no serious complications were attributed to the use of etanercept. In a prospective, randomized study of 63 patients treated with high dose corticosteroids±infliximab (10 mg/kg/dose weekly for four doses), patients on the infliximab arm experienced high rates of resolution (55%) or improvement (7%) in GVHD symptoms by day 28 from initiation of treatment, but the steroid alone arm performed equally well, and no statistically significant differences between the treatment groups was found for either response to treatment or survival38.
Other studies, usually small prospective trials testing infliximab, have shown some promise for TNF-inhibition as a treatment for steroid-refractory GVHD34,39–42. When taken together, this published literature contains over 100 patients with steroid-refractory GVHD treated with TNF-inhibition. Reported response rates range from 52–82% with a median reported response rate of 63% in this difficult to treat population. Complete responses were seen in the majority of responders and long-term survival was around 40%. Interestingly, and particularly with the infliximab treated patients, steroid refractory gastrointestinal GVHD appeared to respond best to the addition of anti-TNF therapy to the treatment regimen. Infection rates were high as expected in this patient population.
SUMMARY.
TNF-α is implicated in all stages of GVHD pathophysiology, making anti-TNF therapy an attractive modality to explore in GVHD prevention and treatment. Clinical trials in this area continue to produce some promising results, but overall none of the outcomes have been definitively supportive of the utility of TNF-inhibition in the management of GVHD. The most encouraging results to date have been with the use of infliximab to treat steroid-refractory gastrointestinal GVHD, where the response rates have been good and consistent across multiple small studies. As a prevention or early treatment strategy, TNF-inhibition remains an area of exploration. Fortunately, larger, more definitive clinical trials are planned with accrual expected to begin this year. Perhaps the greatest implication of TNF-α in the pathogenesis of GVHD may be its role as a predictive GVHD biomarker together with other biomarkers, which could then allow for pre-emptive treatment strategies.
References
- 1.Antin JH, Ferrara JL. Cytokine dysregulation and acute graft-versus-host disease. Blood. 1992;80:2964–2968. [PubMed] [Google Scholar]
- 2.Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373:1550–1561. doi: 10.1016/S0140-6736(09)60237-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matzinger P. The danger model: A renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 4.Xun CQ, Thompson JS, Jennings CD, Brown SA, Widmer MB. Effect of total body irradiation, busulfan-cyclophosphamide, or cyclophosphamide conditioning on inflammatory cytokine release and development of acute and chronic graft-versus-host disease in H-2-incompatible transplanted SCID mice. Blood. 1994;83:2360–2367. [PubMed] [Google Scholar]
- 5.Hill GR, Ferrara JL. The primacy of the gastrointestinal tract as a target organ of acute graft-versus-host disease: Rationale for the use of cytokine shields in allogeneic bone marrow transplantation. Blood. 2000;95:2754–2759. [PubMed] [Google Scholar]
- 6.Hill GR, Crawford JM, Cooke KR, Brinson YS, Pan L, Ferrara JL. Total body irradiation and acute graft-versus-host disease: The role of gastrointestinal damage and inflammatory cytokines. Blood. 1997;90:3204–3213. [PubMed] [Google Scholar]
- 7.Bollyky PL, Evanko SP, Wu RP, et al. Th1 cytokines promote T-cell binding to antigen-presenting cells via enhanced hyaluronan production and accumulation at the immune synapse. Cell Mol Immunol. 2010;7:211–220. doi: 10.1038/cmi.2010.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ratanatharathorn V, Nash RA, Przepiorka D, et al. Phase III study comparing methotrexate and tacrolimus (prograf, FK506) with methotrexate and cyclosporine for graft-versus-host disease prophylaxis after HLA-identical sibling bone marrow transplantation. Blood. 1998;92:2303–2314. [PubMed] [Google Scholar]
- 9.Nash RA, Antin JH, Karanes C, et al. Phase III study comparing methotrexate and tacrolimus with methotrexate and cyclosporine for prophylaxis of acute graft-versus-host disease after marrow transplantation from unrelated donors. Blood. 2000;96:2062–2068. [PubMed] [Google Scholar]
- 10.Lee SJ, Zahrieh D, Agura E, et al. Effect of up-front daclizumab when combined with steroids for the treatment of acute graft-versus-host disease: results of a randomized trial. Blood. 2004;104:1559–1564. doi: 10.1182/blood-2004-03-0854. [DOI] [PubMed] [Google Scholar]
- 11.Zeiser R, Nguyen VH, Beilhack A, et al. Inhibition of CD4+CD25+ regulatory T-cell function by calcineurin-dependent interleukin-2 production. Blood. 2006;108:390–399. doi: 10.1182/blood-2006-01-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welniak LA, Blazar BR, Murphy WJ. Immunobiology of allogeneic hematopoietic stem cell transplantation. Annu Rev Immunol. 2007;25:139–170. doi: 10.1146/annurev.immunol.25.022106.141606. [DOI] [PubMed] [Google Scholar]
- 13.Ferrara JL, Deeg HJ. Graft-versus-host disease. N Engl J Med. 1991;324:667–674. doi: 10.1056/NEJM199103073241005. [DOI] [PubMed] [Google Scholar]
- 14.Cooke KR, Hill GR, Crawford JM, et al. Tumor necrosis factor- alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J Clin Invest. 1998;102:1882–1891. doi: 10.1172/JCI4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 16.Brown GR, Lindberg G, Meddings J, Silva M, Beutler B, Thiele D. Tumor necrosis factor inhibitor ameliorates murine intestinal graft-versus-host disease. Gastroent. 1999;116:593–601. doi: 10.1016/s0016-5085(99)70181-2. [DOI] [PubMed] [Google Scholar]
- 17.Stuber E, Buschenfeld A, von Freier A, Arendt T, Folsch UR. Intestinal crypt cell apoptosis in murine acute graft versus host disease is mediated by tumour necrosis factor alpha and not by the FasL-Fas interaction: effect of pentoxifylline on the development of mucosal atrophy. Gut. 1999;45:229–235. doi: 10.1136/gut.45.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holler E, Kolb HJ, Moller A, et al. Increased serum levels of tumor necrosis factor alpha precede major complications of bone marrow transplantation. Blood. 1990;75:1011–1016. [PubMed] [Google Scholar]
- 19.Choi SW, Kitko CL, Braun T, et al. Change in plasma tumor necrosis factor receptor 1 levels in the first week after myeloablative allogeneic transplantation correlates with severity and incidence of GVHD and survival. Blood. 2008;112:1539–1542. doi: 10.1182/blood-2008-02-138867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitko CL, Paczesny S, Yanik G, et al. Plasma elevations of tumor necrosis factor-receptor-1 at day 7 postallogeneic transplant correlate with graft-versus-host disease severity and overall survival in pediatric patients. Biol Blood Marrow Transpl. 2008;14:759–765. doi: 10.1016/j.bbmt.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vandenabeele P, Declercq W, Beyaert R, Fiers W. Two tumour necrosis factor receptors: structure and function. Trends Cell Biol. 1995;5:392–399. doi: 10.1016/s0962-8924(00)89088-1. [DOI] [PubMed] [Google Scholar]
- 22.Levine JE, Uberti JP, Ayash L, et al. Lowered-intensity preparative regimen for allogeneic stem cell transplantation delays acute graft-versus-host disease but does not improve outcome for advanced hematologic malignancy. Biol Blood Marrow Transpl. 2003;9:189–197. doi: 10.1053/bbmt.2003.50012. [DOI] [PubMed] [Google Scholar]
- 23.Mielcarek M, Martin PJ, Leisenring W, et al. Graft-versus-host disease after nonmyeloablative versus conventional hematopoietic stem cell transplantation. Blood. 2003;102:756–762. doi: 10.1182/blood-2002-08-2628. [DOI] [PubMed] [Google Scholar]
- 24.Couriel DR, Saliba RM, Giralt S, et al. Acute and chronic graft-versus-host disease after ablative and nonmyeloablative conditioning for allogeneic hematopoietic transplantation. Biol Blood Marrow Transplant. 2004;10:178–185. doi: 10.1016/j.bbmt.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Willems E, Humblet-Baron S, Dengis O, Seidel L, Beguin Y, Baron F. Elevations of tumor necrosis factor receptor 1 at day 7 and acute graft-versus-host disease after allogeneic hematopoietic cell transplantation with nonmyeloablative conditioning. Bone Marrow Transplant. 45:1442–1448. doi: 10.1038/bmt.2009.360. [DOI] [PubMed] [Google Scholar]
- 26.Fowler DH, Foley J, Whit-Shan Hou J, et al. Clinical “cytokine storm” as revealed by monocyte intracellular flow cytometry: correlation of tumor necrosis factor alpha with severe gut graft-versus-host disease. Clin Gastroenterol Hepatol. 2004;2:237–245. doi: 10.1016/s1542-3565(04)00011-4. [DOI] [PubMed] [Google Scholar]
- 27.Ritchie D, Seconi J, Wood C, Walton J, Watt V. Prospective monitoring of tumor necrosis factor alpha and interferon gamma to predict the onset of acute and chronic graft-versus-host disease after allogeneic stem cell transplantation. Biology of Blood and Marrow Transplantation. 2005;11:706–712. doi: 10.1016/j.bbmt.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 28.Paczesny S, Krijanovski OI, Braun TM, et al. A biomarker panel for acute graft-versus-host disease. Blood. 2009;113:273–278. doi: 10.1182/blood-2008-07-167098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paczesny S, Braun T, Levine JE, et al. Elafin is a biomarker of graft-versus-host disease of the skin. Science Translational Medicine. 2010;2:50–57. doi: 10.1126/scitranslmed.3000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamadani M, Hofmeister CC, Jansak B, et al. Addition of infliximab to standard acute graft-versus-host disease prophylaxis following allogeneic peripheral blood cell transplantation. Biol Blood Marrow Transplant. 2008;14:783–789. doi: 10.1016/j.bbmt.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levine JE, Paczesny S, Mineishi S, et al. Etanercept plus methylprednisolone as initial therapy for acute graft-versus-host disease. Blood. 2008;111:2470–2475. doi: 10.1182/blood-2007-09-112987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imamura M, Hashino S, Kobayashi H, Kubayashi S, Hirano S. Serum Cytokine Levels in Bone Marrow Transplantation: Synergistic Interaction of Interleukin-6, Interferon-γ, and Tumor Necrosis Factor-α in Graft-Versus-Host Disease. Bone Marrow Transpl. 1994;13:745–751. [PubMed] [Google Scholar]
- 33.Ehlers S. Tumor necrosis factor and its blockade in granulomatous infections: differential modes of action of infliximab and etanercept? Clin Infect Dis. 2005;41 (Suppl 3):S199–203. doi: 10.1086/429998. [DOI] [PubMed] [Google Scholar]
- 34.Couriel D, Saliba R, Hicks K, et al. Tumor necrosis factor-alpha blockade for the treatment of acute GVHD. Blood. 2004;104:649–654. doi: 10.1182/blood-2003-12-4241. [DOI] [PubMed] [Google Scholar]
- 35.Marty FM, Lee SJ, Fahey MM, et al. Infliximab use in patients with severe graft-versus-host disease and other emerging risk factors of non-Candida invasive fungal infections in allogeneic hematopoietic stem cell transplant recipients: a cohort study. Blood. 2003;102:2768–2776. doi: 10.1182/blood-2003-01-0267. [DOI] [PubMed] [Google Scholar]
- 36.Alousi AM, Weisdorf DJ, Logan BR, et al. Etanercept, mycophenolate, denileukin or pentostatin plus corticosteroids for acute graft vs. host disease: a randomized phase II trial from the BMT CTN. Blood. 2009;114:511–517. doi: 10.1182/blood-2009-03-212290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacMillan ML, Weisdorf DJ, Wagner JE, et al. Response of 443 patients to steroids as primary therapy for acute graft-versus-host disease: comparison of grading systems. Biol Blood Marrow Transpl. 2002;8:387–394. doi: 10.1053/bbmt.2002.v8.pm12171485. [DOI] [PubMed] [Google Scholar]
- 38.Couriel DR, Saliba R, de Lima M, et al. A phase III study of infliximab and corticosteroids for the initial treatment of acute graft-versus-host disease. Biol Blood Marrow Transplant. 2009;15:1555–1562. doi: 10.1016/j.bbmt.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Busca A, Locatelli F, Marmont F, Ceretto C, Falda M. Recombinant human soluble tumor necrosis factor receptor fusion protein as treatment for steroid refractory graft-versus-host disease following allogeneic hematopoietic stem cell transplantation. Am J Hematol. 2007;82:45–52. doi: 10.1002/ajh.20752. [DOI] [PubMed] [Google Scholar]
- 40.Sleight BS, Chan KW, Braun TM, Serrano A, Gilman AL. Infliximab for GVHD therapy in children. Bone Marrow Transplant. 2007;40:473–480. doi: 10.1038/sj.bmt.1705761. [DOI] [PubMed] [Google Scholar]
- 41.Patriarca F, Sperotto A, Damiani D, et al. Infliximab treatment for steroid-refractory acute graft-versus-host disease. Haematologica. 2004;89:1352–1359. [PubMed] [Google Scholar]
- 42.Jacobsohn DA, Hallick J, Anders V, McMillan S, Morris L, Vogelsang GB. Infliximab for steroid-refractory acute GVHD: A case series. Am J Hematol. 2003;74:119–124. doi: 10.1002/ajh.10392. [DOI] [PubMed] [Google Scholar]