

Abstract
Microtubule organization and dynamics are essential during axon and dendrite formation and maintenance in neurons. However, little is known about the regulation of microtubule dynamics during synaptic development and function in mammalian neurons. Here, we present evidence that the microtubule plus-end tracking protein CLASP2 (cytoplasmic linker associated protein 2) is a key regulator of axon and dendrite outgrowth that leads to functional alterations in synaptic activity and formation. We found that CLASP2 protein levels steadily increase throughout neuronal development in the mouse brain and are specifically enriched at the growth cones of extending neurites. The short-hairpin RNA-mediated knockdown of CLASP2 in primary mouse neurons decreased axon and dendritic length, whereas overexpression of human CLASP2 caused the formation of multiple axons, enhanced dendritic branching, and Golgi condensation, implicating CLASP2 in neuronal morphogenesis. In addition, the CLASP2-induced morphological changes led to significant functional alterations in synaptic transmission. CLASP2 overexpression produced a large increase in spontaneous miniature event frequency that was specific to excitatory neurotransmitter release. The changes in presynaptic activity produced by CLASP2 overexpression were accompanied by increases in presynaptic terminal circumference, total synapse number, and a selective increase in presynaptic proteins that are involved in neurotransmitter release. Also, we found a smaller increase in miniature event amplitude that was accompanied by an increase in postsynaptic surface expression of GluA1 receptor localization. Together, these results provide evidence for involvement of the microtubule plus-end tracking protein CLASP2 in cytoskeleton-related mechanisms underlying neuronal polarity and interplay between microtubule stabilization and synapse formation and activity.
Introduction
Neurons are highly polarized cells that are composed of two structurally and functionally distinct cell processes, an axon and dendrites (Craig and Banker, 1994). During development, axon specification depends on the outgrowth or elongation of one of the immature neuritic processes, and the ability to perform axonal polarization is crucial for synaptic transmission (Bradke and Dotti, 2000). Intracellular mechanisms that regulate axon outgrowth depend primarily on the reorganization of the cytoskeleton, including actin filaments and microtubules that are involved in transport of cellular cargo, cell polarization, and regulation of cell motility. The heterogeneous family of plus-end tracking proteins (+TIPs) specifically accumulate at the plus-ends of microtubules, strategically placing them to control microtubule dynamics, growth directionality, and crosstalk between microtubules and the actin cytoskeleton, all of which are essential for neurite growth (Akhmanova and Hoogenraad, 2005; Basu and Chang, 2007; Akhmanova and Steinmetz, 2008). Among the +TIPs, cytoplasmic linker associated proteins (CLASPs) are widely conserved in fungi, plants, and animals (Galjart, 2005; Bratman and Chang, 2008). Mammalian CLASPs are represented by two closely related homologs, a ubiquitously expressed CLASP1 and the brain-specific CLASP2 (Akhmanova et al., 2001). CLASPs accumulate asymmetrically near the growing plus-ends of microtubules toward the leading edges of migrating fibroblasts, suggesting that CLASPs are required for persistent motility (Akhmanova et al., 2005; Wittmann and Waterman-Storer, 2005). Also, CLASP2 has been shown to mediate asymmetric microtubule nucleation at the Golgi apparatus and is crucial for establishing the continuity and morphology of the Golgi apparatus (Efimov et al., 2007; Miller et al., 2009).
Relatively little is known about the role of CLASP in the mammalian nervous system. Genetic evidence placed Orbit/MAST, the Drosophila ortholog of CLASP, as acting downstream from Slit to induce growth cone repulsion and inhibit axon growth (Lee et al., 2004). Recently, CLASP2 was shown to both promote and restrict axon growth cones, suggesting that the opposing roles of CLASP are rooted in its microtubule-binding activities (Hur et al., 2011). Here, we characterize the role of CLASP2 in neuronal development, specifically during axon and dendrite formation and maintenance, synapse formation, and functional plasticity. We found that CLASP2 protein levels increased steadily during neuronal development and were enriched in growth cones of developing neurites of primary mouse hippocampal neurons. CLASP2 short-hairpin RNA (shRNA) expressing neurons displayed a significant decrease in axon elongation and dendritic branching and length. Conversely, overexpression of CLASP2 in primary neurons caused formation of multiple axons, enhanced dendritogenesis, and Golgi condensation, implicating CLASP2 in neuronal morphogenesis. In addition, we found that overexpression of CLASP2 increased both spontaneous release of neurotransmitters and surface levels of glutamate receptor 1 (GluA1) at excitatory synapses, suggesting that CLASP2 regulates synaptic function. Quantification of synaptic proteins showed that overexpression of CLASP2 caused a selective increase in presynaptic proteins involved in the synaptic vesicle fusion machinery, suggesting that the increase contributes to the CLASP2-mediated presynaptic transmission phenotype.
Materials and Methods
Plasmids.
Full-length human CLASP2 (National Center for Biotechnology Information reference sequence NM_015097.2) was PCR amplified from a full-length cDNA clone (ImaGenes) and subcloned into pEGFP–C1 (Clontech) to create an N-terminal enhanced green fluorescence protein (EGFP) tag. EGFP–CLASP2 and EGFP control were then subcloned into the lentiviral vector pFUW and sequence verified. The shRNA constructs for mouse CLASP2 include GCATCAGTCCTTTCAACAAGT and GAACTTGAAGAGACGTTAAAT, and control shRNA CCGCAGGTATGCACGCGT are all cloned into the pLKO.1 vector (Addgene plasmid 10879).
Primary neuronal cultures and lentiviral infection.
Primary hippocampal neuronal cultures were prepared from newborn mice of either sex. Neurons were dissociated with trypsin for 10 min at 37°C, triturated, and plated onto coverslips coated with Matrigel (Ho et al., 2006, 2008). Neurons were maintained in culture at 37°C in a humidified incubator with 5% CO2. Recombinant lentiviruses were produced by transfecting HEK293T cells with plasmids for EGFP–CLASP2, shRNA constructs, or control protein in combination with viral enzymes and envelope proteins (pRSV/REV, pMDLg/RRE, and pVSVG) using FuGENE6 reagent (Roche) as described previously (Ho et al., 2006, 2008). Conditioned medium containing lentivirus was harvested after 48 h, centrifuged at 1000 × g for 10 min, and stored at −80°C. Neurons were infected with lentivirus at 1 d in vitro (DIV). All animal experiments were performed according to Boston University Institutional Animal Care and Use Committee guidelines.
Immunocytochemistry and electron microscopy.
Primary hippocampal neurons on coverslips were fixed in cold absolute methanol at −20°C for 10 min, permeabilized, and blocked in 10% goat serum and 0.1% saponin in PBS. Primary incubation was performed in 5% goat serum in PBS overnight at 4°C. After washing with PBS, cells were incubated with goat anti-rabbit or goat anti-mouse secondary antibodies conjugated to Alexa Fluor-546 or Alexa Fluor-633 (Invitrogen). Labeled cells were washed in PBS, mounted in ProLong Gold with DAPI (Invitrogen), and imaged with a Carl Zeiss MZ6 fluorescence microscope or a FluoView FV10i scanning confocal microscope. For electron microscopy, coverslips were fixed in 2.5% glutaraldehyde and 0.1 m sodium cacodylate buffer, pH 7.2, and examined by standard procedures (Rosahl et al., 1995).
Image analysis and quantification.
To determine axon length, hippocampal neurons were infected at 1 DIV, fixed 24 h later, and processed for the pan-axonal neurofilament marker SMI-312 immunofluorescence. Quantitative immunofluorescence to determine the length of each individual axon was performed using NIH ImageJ software (http://imagej.nih.gov/ij/). For axon polarization, the number of axons per neuron was determined using SMI-312 immunostaining at 4 and 9 DIV. For phosphatidylinositol 3-kinase (PI3K) inhibitor treatment, primary neurons were treated with LY 294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one] at 1 DIV in a final concentration of 20 μm (Cell Signaling Technology). To measure neuronal branching, neurons were infected at 1 DIV, fixed at 5 and 15 DIV, and processed for microtubule-associated protein 2 (MAP2) immunostaining. The number and length of all neuronal processes was measured using NIH ImageJ software with the Sholl plug-in (http://imagej.nih.gov/ij/). To assess Golgi morphology during neuronal development, we infected primary neurons at 1 DIV and performed immunocytochemistry for the cis-Golgi marker GM130 in combination with MAP2 at 5 DIV. Golgi morphology was defined as “stacked” when the Golgi appeared condensed and nearly circular in shape and “ribbon” when extended so that the length exceeded the width by more than fourfold. Golgi with intermediate phenotypes (representing <5%) were excluded from the analysis. To assess synapse area and density, primary neurons infected at 1 DIV were processed at 14 DIV for immunocytochemistry for the presynaptic protein synapsin and the postsynaptic protein postsynaptic density 95 (PSD95). An equal number of comparable processes in each condition were assessed using the colocalization plug-in for NIH ImageJ (http://imagej.nih.gov/ij/). Synaptic parameters on digital electron micrographs were quantified based on the following criteria: total number of synaptic vesicles, number of docked vesicles, number of docked vesicles within 150 nm of the active zone, presynaptic terminal circumference and area, and length of the PSD.
Quantitative protein analyses.
For protein quantifications, brain tissue from mice at various postnatal stages was isolated and homogenized in PBS containing 10 mm EDTA and protease inhibitors. Brain proteins (25–40 μg) were separated by SDS-PAGE, and immunoblotting was performed as described previously (Ho et al., 2006). For protein quantifications in neuronal cultures, high-density neocortical cultures at various DIV were harvested by washing neurons with cold PBS, followed by the addition of reducing sample buffer. SDS-PAGE and immunoblotting was performed using standard methods (Ho et al., 2006). For GluA1 surface expression, neuronal cultures were incubated with 1 mg/ml sulfo-NHS-LC-Biotin (Thermo Fisher Scientific) in PBS for 20 min on ice, quenched with Tris-buffered saline (150 mm NaCl and 50 mm Tris, pH 7.5), and lysed with RIPA buffer (65 mm Tris, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 0.5% Na-deoxycholate, 1 mm EDTA, pH 8.0, 50 mm NaH2PO4, 50 mm NaF, 10 mm Na4P2O7, 1 mm Na3VO4, and protease inhibitor cocktail). Lysates were clarified by centrifugation at 14,000 × g for 20 min at 4°C. Supernatants containing 100 μg of protein were incubated with RIPA-equilibrated NeutrAvidin beads (50% slurry; Thermo Fisher Scientific) overnight at 4°C with gentle rocking to precipitate biotinylated proteins. Beads were washed several times with RIPA buffer and spun at 800 × g for 1 min. Proteins were eluted from the NeutrAvidin beads by boiling for 10 min in reducing sample buffer, resolved by SDS-PAGE, and immunoblotted for GluA1. All data are means ± SEMs.
Electrophysiological analyses.
Synaptic transmission was recorded from neurons cultured at 14–17 DIV using whole-cell voltage-clamp techniques. Recordings were obtained with an Axopatch 200A amplifier and Clampex 10.0 software (Molecular Devices). Synaptic recordings were filtered at 1 kHz and sampled at 10 kHz. Electrode solution for the pipette contained the following (in mm): 105 Cs-MeSO3, 10 CsCl, 5 NaCl, 10 HEPES, 0.2 EGTA, 4 Mg-ATP, and 0.3 Na2GTP, pH 7.4 (300 mOsm). Spontaneous event recordings were performed in the presence of 1 μm tetrodotoxin and 50 μm d-2-amino-5-phosphonovaleric acid. For recording miniature EPSCs (mEPSCs), 20 μm bicuculline was added to the bath. For mIPSCs, 20 μm 6-cyano-7-nitroquinoxaline-2,3-dione was added.
Antibodies.
Rabbit CLASP2 (Novagen), mouse GM130 (BD Biosciences), rabbit MAP2 (Millipore Bioscience Research Reagents), mouse SMI-312 (Covance), rabbit β-tubulin (Covance), mouse α-tubulin (Sigma), mouse PSD95 (clone K28/43; University of California, Davis/National Institutes of Health NeuroMab Facility), mouse GluA1 (kind gift from Dr. H. Man, Boston University, Boston, MA), and rabbit synapsin (P610), rabbit synaptobrevin (P939), rabbit syntaxin (438B), and rabbit synaptosomal-associated protein, 25 kDa (SNAP25) (P913; kind gifts from Dr. T. C. Südhof, Stanford University, Stanford, CA).
Statistical analyses.
We used a paired Student's t test, ANOVA, or Fisher's exact test as appropriate.
Results
CLASP2 expression during neuronal development
Previous experiments have shown that CLASP1 is expressed in all tissues, whereas CLASP2 is specifically enriched in the brain (Akhmanova et al., 2001). To examine the localization of CLASP2 during neuronal development, we characterized the endogenous distribution of CLASP2 in hippocampal neuronal cultures by immunofluorescence microscopy. Staining with CLASP2, SMI-312 (a pan-axonal neurofilament marker), and α-tubulin detected CLASP2 expression along the length of all processes, including intense labeling of growth cones at the leading tip of developing neurites at 4–5 DIV (Fig. 1A,B). This result supports previous evidence of CLASP2 as a +TIP and is consistent with a role for CLASP2 in growth cone dynamics of neurons (Akhmanova and Steinmetz, 2008; Hur et al., 2011). Additional staining for α-tubulin demonstrates that CLASP2 is also highly expressed in astrocytes, suggesting that CLASP2 plays a role in the microtubule dynamics of glia (Fig. 1C).
Figure 1.

Localization and expression of CLASP2 in the developing brain. A, Representative images of endogenous CLASP2 enriched in the growing tip of a growth cone of mouse hippocampal neurons at 4–5 DIV double labeled with anti-CLASP2 (green) and anti-SMI-312 (red) antibodies. Representative images of hippocampal neuron (B) and a glial cell at 4–5 DIV double labeled with anti-CLASP2 (green) and anti-α-tubulin (red) antibody (C). Scale bars: A, 2.5 μm; B, 16 μm; C, 15 μm. D, Protein expression levels of CLASP2, MAP2, SMI-312, and β-tubulin during postnatal development examined by immunoblotting of total mouse brain lysate at the indicated ages (Adult = 3 months). E, Expression levels of CLASP2 was examined by immunoblotting of total lysate from 7–14 DIV cortical neurons infected with control, human CLASP2, or CLASP2 shRNA lentivirus.
To examine the role of CLASP2 in vivo, we probed mouse brain homogenates at different postnatal days for CLASP2, dendritic (MAP2), and axonal (SMI-312) markers (Fig. 1D). CLASP2 expression was detected at postnatal day 1 (P1) to P2 and strikingly increased by 4.5-fold at P5 and stabilizing in the adult brain. The present results also show that MAP2 was synthesized at the earliest time point examined at P1 and that MAP2 protein levels remained similar throughout development. Conversely, protein expression of the axonal protein marker SMI-312 markedly increased into adulthood.
Lentiviral infection of primary cortical neurons was used to determine the effects of CLASP2 overexpression or knockdown on neurite development and maintenance. In primary cortical neurons treated with control GFP, CLASP2 protein levels are detected as early as 3 DIV and steadily increase from 7 to 14 DIV (Fig. 1E). Lentiviral-induced human CLASP2 overexpression increased CLASP2 levels 2- to 3.5-fold higher at 7–11 DIV compared with endogenous levels. By 14 DIV, the levels of overexpressed CLASP2 were comparable with control levels (at 108%). Conversely, we found that CLASP2 shRNA efficiently downregulated CLASP2 protein expression as early as 3 DIV and resulted in a significant 90% knockdown by 7 DIV. We also investigated the distribution of overexpressed CLASP2 in primary hippocampal neurons. When CLASP2 is overexpressed in neurons, CLASP2 was found to accumulate along the whole length of microtubules, colocalizing with both axonal and dendritic markers (data not shown). These results indicate that overexpression did not produce aberrant localization of CLASP2 compared with endogenous CLASP2 expression. We conclude that CLASP2 protein levels gradually increase during neuronal development and that CLASP2 is specifically enriched at microtubule growing tips during neurite polarization and outgrowth.
CLASP2 regulates neurite growth
To investigate the role of CLASP2 on axon polarization and dendritic branching, we compared primary hippocampal neurons infected at 1 DIV with control shRNA, human CLASP2, or CLASP2 shRNA expressing lentivirus by immunofluorescence. At 2 DIV, quantitative analysis showed that overexpression of human CLASP2 resulted in a significant 20% increase in axon length (Fig. 2A,B). The shRNA-mediated knockdown of CLASP2 decreased axon length by 20% compared with control-infected neurons. To determine whether the decrease in axon length resulted specifically from CLASP2 knockdown, we coexpressed CLASP2 shRNA with human CLASP2 cDNA, which rescued the CLASP2 shRNA phenotype (Fig. 2A,B).
Figure 2.

CLASP2 increases axon length and induces multiple axon formation. A, Hippocampal neurons infected at 1 DIV with control, human CLASP2, CLASP2 shRNA, or CLASP2 shRNA coexpressing human CLASP2 were fixed at 2 DIV and stained with SMI-312 (axonal marker). Scale bar, 20 μm. B, Quantitative analysis showed that human CLASP2 significantly increased axon length at 2 DIV (n = 49 neurons for each group; *p < 0.05, **p < 0.001). C, Hippocampal neurons infected with human CLASP2 were fixed at 4 DIV and stained with SMI-132 showed multiple axons compared with control and CLASP2 shRNA (n = 62 neurons for each group; ***p < 0.0001). Neuronal polarity phenotypes were categorized into two groups: single axon (white bar) and multiple axons (black bar).
In addition, we found that 38% of neurons overexpressing human CLASP2 had multiple SMI-312-positive axons when compared with control neurons that predominantly extend a single primary axon at 2 DIV (Fig. 2C). This increase was further pronounced at 9 DIV when 55% of CLASP2-overexpressing neurons exhibited more than one SMI-positive axon (data not shown). Conversely, CLASP2 shRNA knockdown did not affect the number of polarized axon-bearing neurons. Coexpression of CLASP2 shRNA with human CLASP2 cDNA was similar to control-infected neurons.
Next, we examined the time course of dendrite development from neurons infected with control shRNA, human CLASP2, or CLASP2 shRNA. We found that overexpression of human CLASP2 significantly increased the total number of MAP2-positive processes at 5 DIV, with extensive dendrite outgrowth in both primary and secondary dendritic branching (Fig. 3A). To examine the complexity of the dendritic arbor and the overall pattern of arborization, we used Sholl analysis to examine global changes in neuronal morphology (Fig. 3B). Sholl analysis revealed that overexpression of human CLASP2 shifted the distribution upward and to the right compared with control neurons, indicating both an increase in number and length of the neurites (Fig. 3C,D). Conversely, the shRNA knockdown of CLASP2 greatly reduced dendritic growth. To confirm the specificity of the observed effects, CLASP2 knockdown phenotype was rescued by overexpressing human CLASP2. Overexpression of human CLASP2 fully reversed the negative effects of CLASP2 shRNA on total neurite length but not the number of MAP2-positive processes (Fig. 3C,D). Overall, these results suggest that CLASP2 has an important role in axon polarization, neurite growth, and dendritic branching.
Figure 3.
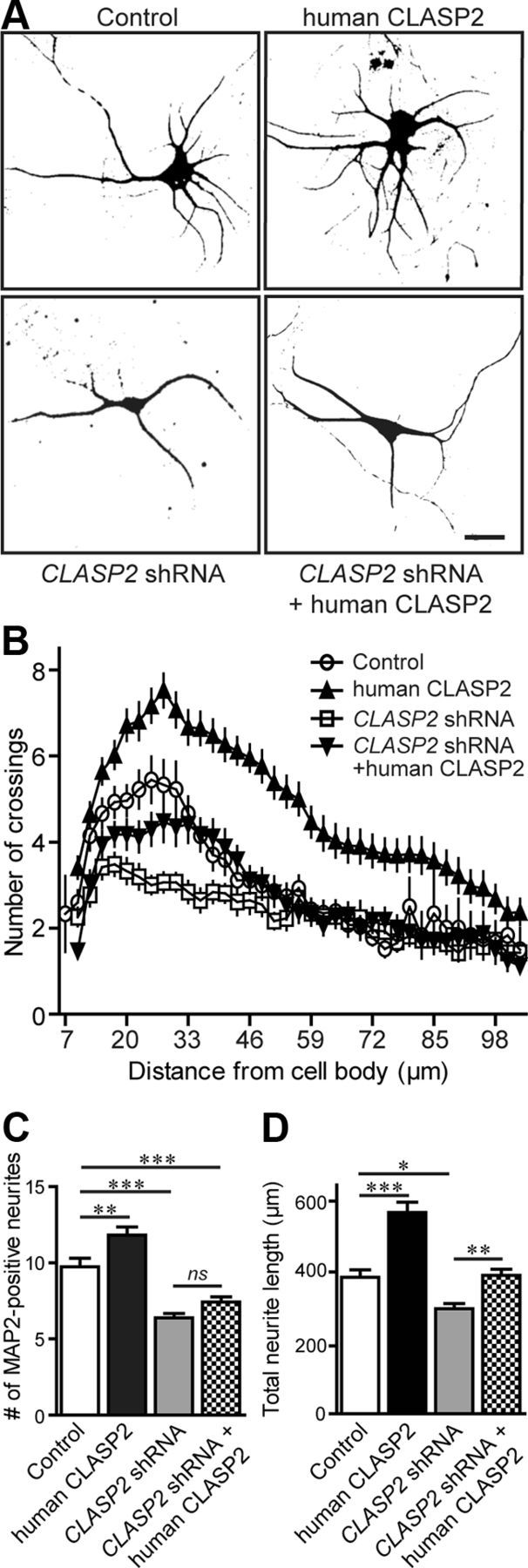
CLASP2 regulates dendritic growth and branching. A, Hippocampal neurons infected at 1 DIV with control, human CLASP2, CLASP2 shRNA, or CLASP2 shRNA coexpressing human CLASP2 were fixed at 5 DIV and stained with MAP2 (dendritic marker). Scale bar, 20 μm. B, Sholl analysis based on the number of dendritic crossings distributed over the distance from the cell body (n = 17 neurons for each group). CLASP2 significantly increased the number of MAP2-positive neurites (C) (n = 32 for each group; **p < 0.01, ***p < 0.001; ns, not significant) and the total length of all neurites (D) (n = 19 for each group; *p < 0.05, **p < 0.01, ***p < 0.001).
CLASP2 controls neuronal Golgi morphology
Previous experiments have shown that CLASP2 localizes to the Golgi in both fibroblast and epithelial cells and is crucial for establishing continuity and proper morphology of the Golgi complex (Akhmanova et al., 2001; Efimov et al., 2007); however, little is known about the role of CLASP2 on the Golgi complex in neurons. To examine CLASP2 distribution in the Golgi of hippocampal neurons, we double labeled for CLASP2 and the Golgi marker GM130 and found that endogenous CLASP2 localization overlapped with GM130 (Fig. 4A).
Figure 4.

CLASP2 regulates Golgi morphology in neurons. A, Representative images of anti-CLASP2 (green) and anti-GM130 (red) antibodies showing colocalization of endogenous CLASP2 with Golgi complex in a hippocampal neuron at 5 DIV. B, Neurons treated at 1 DIV with control, human CLASP2, or CLASP2 shRNA lentivirus were stained for GM130 (red), MAP2 (cyan), and DAPI (blue) to assess ribbon, stacked, and fragmented Golgi morphology in hippocampal neurons at 5 DIV. Scale bars: A, 1.5 μm; B, 10 μm. C, CLASP2 led to a significant increase in the percentage of neurons with a stacked Golgi phenotype compared with those with a ribbon-shaped Golgi (n = 100 for each group; ***p < 0.0001). D, The Golgi is highly fragmented in CLASP2 knockdown neurons (n = 100 for each group; ***p < 0.0001).
In mammalian cells, CLASP2 is required for the regulation of microtubules originating from the Golgi apparatus in a centrosome-independent manner (Efimov et al., 2007). In addition, Golgi-derived CLASP-dependent microtubules control proper morphology of the Golgi complex, which is essential for cell polarization and motility in retinal pigment epithelial cells (Miller et al., 2009). To determine whether CLASP2 regulates morphology of the Golgi complex in neurons, we examined cultured hippocampal neurons at 4 DIV that were infected with control shRNA, human CLASP2, or CLASP2 shRNA expressing lentivirus. In the control condition, the Golgi in ∼65% of all neurons appear condensed and stacked near the nucleus, whereas the other 35% appear elongated with a continuous ribbon structure extending into the dendritic process (Fig. 4B,C). We found that overexpression of human CLASP2 led to 80% of the Golgi assuming a stacked conformation rather than Golgi deployment into dendrites (Fig. 4C). CLASP2 shRNA knockdown did not affect the percentage of ribbon and stacked Golgi phenotype compared with control. Coexpression of CLASP2 shRNA with human CLASP2 cDNA was similar to control-infected neurons.
At 4 DIV, we also found that the Golgi is highly fragmented in CLASP2 knockdown neurons (Fig. 4B,D), indicative of impaired membrane fusion. CLASP2 knockdown neurons contained a significant 1.9-fold increase in the number of fragments compared with control neurons. Overexpression of human CLASP2 fully reversed the fragmented effects of CLASP2 shRNA on the Golgi. Together, these data suggest that CLASP2 controls the morphology and organization of the Golgi complex in hippocampal neurons.
CLASP2-mediated Golgi morphology correlates with dendritogenesis
Because Golgi morphology has been implicated in different aspects of neuronal polarity and dendritic development (Hanus and Ehlers, 2008), we examined whether changes in CLASP2-mediated Golgi morphology coincides with dendritic outgrowth and branching. We examined cultured hippocampal neurons that were infected with control GFP or CLASP2 expressing lentivirus and double labeled with MAP2 and GM130 antibodies. We found that the increase in branching attributable to CLASP2 overexpression was restricted to neurons with a stacked Golgi phenotype (Fig. 5). These data suggest that the effects of CLASP2 on Golgi morphology have an important role in maintaining dendritic development and branching.
Figure 5.
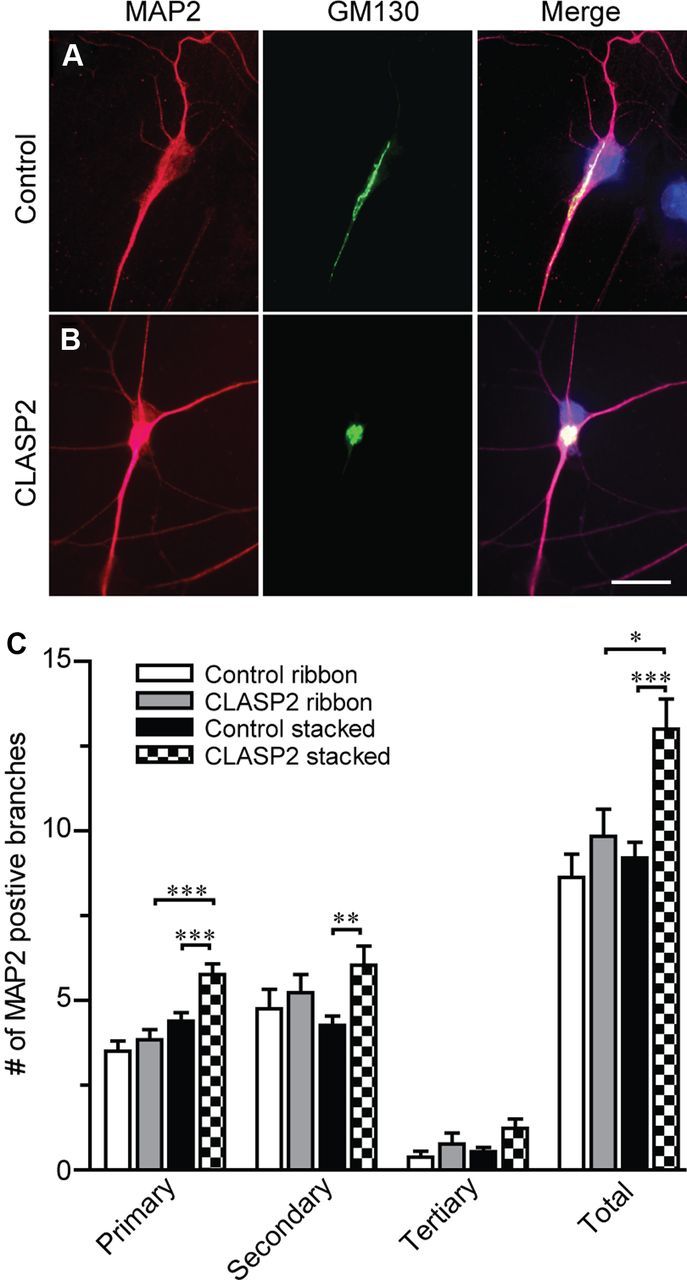
CLASP2 increases neuritic branching in neurons with stacked but not ribbon Golgi phenotypes. A, B, Primary hippocampal neurons infected at 1 DIV with control or CLASP2 were fixed at 4 DIV and immunostained for MAP2 (red) and GM130 (green). Scale bar, 23 μm. C, All primary, secondary, tertiary, and total number of MAP2-positive branches were counted and separated by treatment and Golgi phenotype (n > 13 for each group; *p = 0.029, **p = 0.004, ***p < 0.0009).
CLASP2 regulates spontaneous synaptic transmission
To determine whether the morphological changes in neuronal growth and branching lead to functional alterations in synaptic transmission, we performed whole-cell recordings to monitor the frequency and amplitude of spontaneous miniature events from hippocampal neurons at 14–17 DIV that were infected with control shRNA, human CLASP2, or CLASP2 shRNA expressing lentivirus. Neurons overexpressing human CLASP2 showed a 4.6-fold increase in miniature event frequency in excitatory synapses compared with control neurons, suggesting that CLASP2 alters excitatory presynaptic neurotransmitter release machinery (Fig. 6A,B). In addition, we found a small but significant 1.3-fold increase in miniature event amplitude between CLASP2 and control neurons, suggesting that CLASP2 overexpression also has a smaller effect on postsynaptic receptors (Fig. 6A,C). Interestingly, we found no changes in frequency and amplitude in mIPSCs, suggesting that the effects of CLASP2 are specific and restricted to the development and function of excitatory synapses (Fig. 6D–F).
Figure 6.

CLASP2 increases spontaneous synaptic activity at excitatory but not inhibitory synapses. A, Sample traces showing mEPSC of control, human CLASP2, CLASP2 shRNA, or CLASP2 shRNA coexpressing human CLASP2 at 14–17 DIV. B, Bar graph of mEPSC frequency revealed a significant 4.6-fold increase in miniature frequency in neurons infected with human CLASP2 compared with control (n ≥ 14 for each group; *p < 0.05, ***p < 0.0001). C, Bar graph of mEPSC amplitude demonstrating a significant increase in mEPSC amplitude in neurons infected with human CLASP2 (n ≥ 14 for each group; *p < 0.05). D, Sample traces showing mIPSCs of control and overexpressed human CLASP2. E, No changes in mIPSC frequency were detected in neurons infected with human CLASP2 compared with control (n ≥ 10 for each group). F, Bar graph of mIPSC amplitude showed no changes in miniature amplitude in neurons infected with human CLASP2 (n ≥ 10 for each group).
We further examined the effects of CLASP2 shRNA knockdown on mEPSCs and found a 1.5-fold decrease in miniature event frequency in excitatory synapses compared with control neurons, suggesting a change in the presynaptic release apparatus (Fig. 6A–C). No changes in the amplitude of mEPSCs were observed in CLASP2-mediated knockdown neurons. We next tested whether overexpression of human CLASP2 rescued the effects of CLASP2 shRNA on mEPSCs. Overexpression of human CLASP2 fully restored the miniature event frequency in excitatory synapses.
CLASP2 effects on synaptic morphology
The electrophysiological results raise the possibility that the number of vesicles docked at the active zone is increased in CLASP2 overexpressing hippocampal neurons. To test this, we examined the ultrastructure of cultured neurons infected with control GFP or human CLASP2 expressing lentivirus by electron microscopy at 14 DIV (Fig. 7A; Table 1). Analysis of control synapses revealed presynaptic nerve terminals with clustered synaptic vesicles at the active zone and a filamentous PSD. Synapses of CLASP2-overexpressing neurons exhibited no significant change in the number of synaptic vesicles that are either docked at the active zone or in close proximity, indicative of the readily releasable pool of synaptic vesicles. However, we observed a significant 28% increase in presynaptic terminal circumference in neurons overexpressing human CLASP2, which may be a consequence of increased vesicle fusion with the plasma membrane and could be a cause for the functional effect in spontaneous synaptic transmission.
Figure 7.

CLASP2 regulates synaptic structure and synaptic protein levels. A, Ultrastructural analysis by electron microscopy of synaptic structure in hippocampal neuronal cultures infected with control and overexpressed human CLASP2 at 14 DIV. Representative images of asymmetric excitatory synapses of control and CLASP2 infected neurons. Scale bar, 500 nm. B, Hippocampal neuronal processes infected with control, human CLASP2, CLASP2 shRNA, or CLASP2 shRNA coexpressing human CLASP2 at 14 DIV stained with synapsin (Syn; green) and PSD95 (red). Scale bar, 4 μm. C, D, CLASP2 increases overall synapse number and area of colocalization of synapsin and PSD95 in neuronal processes at 14 DIV (*p < 0.05, ***p < 0.0001). E, Western blotting of CLASP2, various synaptic markers, and β-tubulin in response to control, human CLASP2, CLASP2 shRNA, or CLASP2 shRNA coexpressing human CLASP2 (rescue) in cortical neurons. F, We detected a significant increase of surface (S) compared with total (T) endogenous GluA1 receptor subunit in cultured cortical neurons that overexpressed CLASP2 at 14 DIV using a biotinylation assay (left, immunoblot; right, quantification). Total GluA1 levels were determined from input lysates, whereas surface GluA1 was determined after elution from NeutrAvidin precipitation (*p = 0.04).
Table 1.
Quantitative ultrastructural analysis of asymmetric neuronal synapses
| Parameter | Control | CLASP2 | p value |
|---|---|---|---|
| Presynaptic terminal area (μm2) | 2.13 ± 0.23 | 2.82 ± 0.29 | p = 0.074 |
| Presynaptic terminal circumference (μm) | 2.61 ± 0.15 | 3.34 ± 0.23 | p = 0.012* |
| Length of PSD (μm) | 0.33 ± 0.04 | 0.42 ± 0.06 | p = 0.187 |
| Docked vesicles per active zone | 1.86 ± 0.36 | 1.94 ± 0.37 | p = 0.882 |
| Vesicles 150 nm of the active zone | 10.07 ± 0.9 | 8.64 ± 0.80 | p = 0.241 |
| Vesicles per terminal | 57.97 ± 5.5 | 48.18 ± 4.7 | p = 0.180 |
| Vesicle density | 4.71 ± 0.89 | 8.12 ± 1.60 | p = 0.073 |
All data are presented as means ± SEM. Control, n = 29; CLASP2, n = 33.
*Indicates significance.
We next examined whether overexpression of CLASP2 alters the number and size of synapses. We performed immunofluorescence labeling using antibodies against the presynaptic protein synapsin and the postsynaptic marker PSD95 on hippocampal neurons infected with control shRNA, human CLASP2, or CLASP2 shRNA at 14 DIV (Fig. 7B). We measured the number and the size of synapses defined by the colocalization of synapsin and PSD95 and found that CLASP2 overexpression led to a 2.5-fold increase in the number of synapses and a 25% increase in synapse area (Fig. 7C,D). Conversely, the shRNA-mediated knockdown of CLASP2 decreased the number of synapses by 25% compared with control-infected neurons, whereas the synapse area remained the same. Overexpression of human CLASP2 cDNA rescued the CLASP2 shRNA phenotype. Together, these data suggest that CLASP2 regulates synaptic morphology, leading to functional alterations in synaptic transmission.
CLASP2 regulates synaptic protein levels
To examine whether changes in synaptic protein composition contribute to the CLASP2-mediated synaptic phenotype, we measured synaptic protein levels in cultured neurons overexpressing CLASP2 from 5 to 13 DIV. Quantitative immunoblotting uncovered pronounced changes in a subset of presynaptic proteins during the formation of synapses and clustering of synaptic vesicles at 9 DIV (Fig. 7E) (Basarsky et al., 1994; Kavalali et al., 1999; Mozhayeva et al., 2002). This included a selective 15% increase in the levels of the presynaptic vesicular protein synapsin in CLASP2-overexpressed neurons. In addition, we found an increase in synaptobrevin (33%) and SNAP25 (27%) but not syntaxin, presynaptic proteins essential for the synaptic vesicle fusion machinery. Overexpression of CLASP2 did not affect postsynaptic proteins, such as PSD95 and excitatory glutamate receptor GluA1 expression (Fig. 7E,F). The early increase in the subset of presynaptic proteins suggests that overexpression of CLASP2 causes early synaptic remodeling and/or synaptogenesis. CLASP2 knockdown selectively decreased synapsin, syntaxin, and PSD95 levels, which was rescued by overexpressed human CLASP2. We found no changes in β-tubulin levels in any of the groups examined. These results suggest that CLASP2 specifically regulates synaptic proteins, possibly through stabilization of the cytoskeleton to directly affect synaptic activity.
The increased mEPSC amplitude induced by CLASP2 overexpression (Fig. 6C) strongly suggests alterations in postsynaptic receptors and a role for CLASP2 in the trafficking of receptors to synapses. We therefore examined GluA1 receptor subunit expression at the cell surface as assessed by biotinylation (Fig. 7F). All surface proteins in neurons were biotinylated, washed, lysed, precipitated with immobilized NeutrAvidin–agarose beads, and probed with a GluA1 receptor antibody by Western blotting. We found a 50% increase in the levels of surface GluA1 receptor subunits in CLASP2 overexpressing neurons compared with controls, indicating that the regulated expression and surface insertion of receptors could account for the increase in mEPSC amplitude induced by CLASP2 overexpression (Fig. 7F).
CLASP2 is required for PI3K-induced axon outgrowth
Previous findings indicate that PI3K signaling through glycogen synthase kinase 3β (GSK3β) is an important mediator for CLASP2 function (Akhmanova et al., 2001; Wittmann and Waterman-Storer, 2005; Kumar et al., 2009; Watanabe et al., 2009; Al-Bassam et al., 2010). Also, PI3K–GSK3β signaling underlies axon elongation and guidance (Kim et al., 2011). To determine whether CLASP2 is important for PI3K-induced axon elongation, we treated neurons with LY 294002, a specific inhibitor for PI3K. As expected, overexpression of human CLASP2 resulted in a significant 49% increase in axon length (Fig. 8A,B). The PI3K inhibitor decreased axon length by 17% compared with control-infected neurons. Overexpression of human CLASP2 in the presence of PI3K inhibitor completely rescued the axon growth defect. These data suggest that CLASP2 is required for PI3K-induced axon polarization.
Figure 8.

CLASP2 is required for PI3K-induced axon elongation. A, Hippocampal neurons infected at 1 DIV with control, human CLASP2, or treated with PI3K inhibitor LY 294002 or PI3K inhibitor LY 294002 coexpressing human CLASP2 were fixed at 2 DIV and stained with SMI-312 (axonal marker). Scale bar, 20 μm. B, Quantitative analysis showed that overexpressed human CLASP2 significantly increased axon length at 2 DIV and rescued the axonal growth defect in neurons treated with LY 294002 (n = 47 neurons for each group; **p < 0.01, ***p < 0.001).
Discussion
In the present study, we show that the microtubule plus-end binding protein CLASP2 is an important regulator of neuronal polarity and synaptic function. We found that endogenous CLASP2 protein expression steadily increased during neuronal development and was present in both axonal and somato-dendritic compartments (Fig. 1). CLASP2 downregulation in primary neurons impairs axon elongation and dendritic branching, which was rescued by human CLASP2. Conversely, overexpression of CLASP2 induced the growth of multiple axons, enhanced dendritic branching, and ultimately to functional alterations in synaptic structure and function (Figs. 2–7).
CLASP2 was initially identified as a binding partner of the cytoplasmic linker protein of 170 kDa (CLIP-170) in motile fibroblasts and as an important regulator in cell polarity by stabilizing microtubules at their plus ends (Akhmanova et al., 2001; Galjart, 2005). The asymmetric distribution of CLASP in regulating cellular polarity was shown to be mediated by PI3K signaling through GSK3β. The spatially activated PI3K signaling is conveyed downstream through localized inhibition of GSK3β activity that enhances CLASP2 binding to microtubules (Akhmanova et al., 2001; Wittmann and Waterman-Storer, 2005; Kumar et al., 2009; Watanabe et al., 2009; Al-Bassam et al., 2010). Interestingly, these two spatially coupled kinases have been shown to control axon growth via regulation of the +TIP adenomatous polyposis coli (APC) (Zhou et al., 2004). Recent studies also show that CLASP2 is enriched in growth cones and supports axon regeneration by stabilizing the growing ends of axonal microtubules downstream of GSK3β (Hur et al., 2011). In line with other +TIPs such, as APC (Zhou et al., 2004) and CLIPs (Neukirchen and Bradke, 2011), our data show that overexpression of CLASP2 in primary hippocampal neurons promotes axon elongation and induces growth of multiple axons per neuron (Fig. 2). Also, we show that PI3K-driven axon elongation depends on microtubule dynamics of CLASP2 (Fig. 8).
In addition to increasing axon outgrowth during neuronal polarization, CLASP2 overexpression induced dendritic development. Overexpression of CLASP2 significantly increased dendritic length and the total number of MAP2-positive processes at 5 DIV with extensive dendrite outgrowth in both primary and secondary dendritic branching (Fig. 3). These results strongly suggest that CLASP2 plays an active role in regulating the initial stabilization of both axons and dendrites; however, the cellular and molecular mechanisms underlying their specific targeting to the growing plus-ends of neurites remain elusive. The differential regulation of +TIP interactions with microtubule ends and the composition of the microtubule cytoskeleton can influence whether a process becomes an axon or a dendrite as a result of either specific motor-based loading or regional control of posttranslational modifications (Akhmanova and Steinmetz, 2008). For instance, axon-specific microtubules are uniformly oriented with plus-ends pointing outward that efficiently recruit kinesin-1 motor-driven transport, whereas the microtubule orientations of mixed polarity in dendrites facilitate dynein motor entry (Kapitein and Hoogenraad, 2011). Axonal and dendritic microtubule arrays also differ in their patterns of posttranslational modification. For example, acetylated and detyrosinated microtubules are enriched in axons and selectively recruit kinesin-1 motor proteins (Witte and Bradke, 2008; Konishi and Setou, 2009). Because CLASP2 is regulated by the PI3K–GSK3β signaling pathway, it is possible that phosphorylation is a molecular switch capable of inducing rapid changes in the microtubule dynamic between axons and dendrites.
In non-neuronal cells, there is a substantial Golgi-associated pool of CLASPs, and previous results indicate that CLASP2 promotes microtubule nucleation from the trans-Golgi network, leading to asymmetry of microtubule arrays in polarized cells (Efimov et al., 2007). In addition, Golgi-derived CLASP-dependent microtubules are crucial for establishing continuity and proper morphology of the Golgi complex, which is essential for cell polarization and motility (Miller et al., 2009). Microtubules nucleated at the Golgi are preferentially oriented toward the leading edge and direct polarized trafficking in motile cells similar to the microtubule-dependent trafficking pathway during axon specification (Hoogenraad et al., 2001; Witte et al., 2008; Miller et al., 2009). In neurons, the Golgi apparatus has also been implicated in neuronal polarity in which the position of the Golgi and the adjoined centrosome correlates with newly emerging axons (Zmuda and Rivas, 1998; de Anda et al., 2005, 2010). Specialized Golgi outposts that populate exclusively along dendrites have been shown to regulate extension and retraction of dendritic branching (Horton et al., 2005; Ye et al., 2007; Hanus and Ehlers, 2008). Here, we found that CLASP2 colocalizes with the Golgi marker GM130 in primary neurons and that increasing CLASP2 expression leads to increased Golgi condensation (Fig. 4). We also found that CLASP2 overexpression led to increased neuritic branching only in neurons with stacked as opposed to ribbon Golgi morphology (Fig. 5), suggesting that CLASP2 mediates specific changes in the Golgi that stabilize the cytoskeleton to promote branching.
Microtubules act as the main cytoskeletal tracks for transport of materials to and from the synapse (Conde and Caceres, 2009; Hoogenraad and Bradke, 2009; Goellner and Aberle, 2012). However, little is known about the role of the cytoskeletal machinery, specifically +TIPs, on synapse formation and function in mammals. Our results indicate that CLASP2 has a functional role in spontaneous neurotransmission (Fig. 6). The shRNA knockdown of CLASP2 reduced miniature event frequency in excitatory synapses, whereas overexpression of CLASP2 caused an increase in both miniature event frequency and amplitude specifically in excitatory synapses, suggesting that CLASP2 regulates both presynaptic neurotransmitter release machinery and postsynaptic receptor trafficking. The current changes in spontaneous neurotransmission could be attributable to CLASP2 effects on axon and dendritic outgrowth, exclusively targeting excitatory neurons that have been shown to vary greatly in their somatic, dendritic, and axonal morphologies compared with inhibitory neurons (Markram et al., 2004). Interestingly, we found that CLASP2 induced ultrastructural changes in asymmetric excitatory synapses that support the presynaptic changes we observed electrophysiologically (Fig. 7). Quantitative electron microscopy revealed a selective increase in presynaptic terminal circumference that may be a consequence of increased vesicle fusion with the plasma membrane. In line with the ultrastructural data, the size and number of synapses as assessed by synapsin–PSD95 colocalization was significantly enhanced in neurons overexpressing CLASP2.
We surveyed several synaptic proteins to determine whether CLASP2 has selective effects on the composition of the synapse. We found that CLASP2 overexpression caused pronounced increases in presynaptic (synapsin, synaptobrevin, and SNAP25) but not postsynaptic (PSD95, GluR1) synaptic protein expression (Fig. 7). Recent studies demonstrate that, although the presynaptic complex that gives rise to either action potential-evoked or spontaneous neurotransmitter release uses the same molecular machinery, they rely on distinct molecular interactions of the same components for normal function (Ramirez and Kavalali, 2011). Structure–function analyses of neuronal SNARE proteins (syntaxin, synaptobrevin, and SNAP25) revealed key differences between molecular interactions that give rise to spontaneous versus evoked fusion (Ramirez and Kavalali, 2011). For example, loss of SNAP25 or synaptobrevin essentially abolishes calcium-dependent evoked release but leaves spontaneous fusion release intact, suggesting a role for alternate SNAREs in mediating spontaneous release (Schoch et al., 2001; Washbourne et al., 2002; Bronk et al., 2007). The CLASP2-mediated increase in synapsin, synaptobrevin, and SNAP25 may provide possible mechanistic detail of the functional increase in evoked mEPSCs because alterations in expression levels of these proteins have been shown to cause calcium-dependent evoked neurotransmitter release (Lu et al., 1992; Rosahl et al., 1995; Schoch et al., 2001; Washbourne et al., 2002; Bronk et al., 2007).
At the postsynaptic membrane, we found that CLASP2 plays an important role in glutamate receptor trafficking, including an increase in surface levels of GluA1 in CLASP2 overexpressing neurons. This raises the possibility that CLASP2 functions as a factor controlling the delivery of synaptic components to synaptic terminals (Fig. 7). Previously, +TIPs have been shown to play important roles in positioning neurotransmitter receptors and ion channels. APC and end-binding protein 1 (EB1) have been shown to be involved in assembly and stabilization of α3 nicotinic acetylcholine receptor at the postsynaptic side of cholinergic synapses by anchoring microtubules (Temburni et al., 2004). EB1 was also shown to target voltage-gated potassium (Kv1) channels to axons (Gu et al., 2006). In principal, localized remodeling of the cytoskeleton through +TIPs could achieve the spatial remodeling required for synapse-specific and activity-dependent synaptic plasticity. It is possible that binding of CLASP2 to microtubules or other end-binding proteins is important for the phenotypes we observed, but additional studies will be necessary to investigate this possibility.
In summary, our results suggest that the control of microtubule organization by CLASP2 may represent a mechanism for regulated growth cone motility and synaptic growth and plasticity. We find that increased CLASP2 expression leads to accelerated and enhanced neuron and synaptic development. Although the cellular and molecular mechanism remains to be determined, CLASP2 has a number of specific binding domains that allow for protein interactions at the Golgi, cytoskeleton, and the growing tips of neurites. The enhanced neuronal development may therefore arise as a result of altered Golgi function, increased trafficking along microtubules, or increased vesicle fusion attributable to stabilization of the cytoskeleton. The specific CLASP2 domains responsible for these functional changes will be important to address in the future.
Footnotes
This work was supported by National Institutes of Health Grant K01 AG027311 (A.H.) and Alzheimer's Association Grant IIRG-09-130591 (A.H.). We thank Drs. T. C. Südhof from Stanford University and H. Man from Boston University for antibodies, Dr. X. Liu for the electron microscopy services, and advice from the Center for Cellular and Molecular Imaging at Yale University. We are grateful to Dr. Maria Matos and Sarah Sullivan for comments and advice.
The authors declare no competing financial interests.
References
- Akhmanova A, Hoogenraad CC. Microtubule plus-end-tracking proteins: mechanisms and functions. Curr Opin Cell Biol. 2005;17:47–54. doi: 10.1016/j.ceb.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Steinmetz MO. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 2008;9:309–322. doi: 10.1038/nrm2369. [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Hoogenraad CC, Drabek K, Stepanova T, Dortland B, Verkerk T, Vermeulen W, Burgering BM, De Zeeuw CI, Grosveld F, Galjart N. Clasps are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell. 2001;104:923–935. doi: 10.1016/s0092-8674(01)00288-4. [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Mausset-Bonnefont AL, van Cappellen W, Keijzer N, Hoogenraad CC, Stepanova T, Drabek K, van der Wees J, Mommaas M, Onderwater J, van der Meulen H, Tanenbaum ME, Medema RH, Hoogerbrugge J, Vreeburg J, Uringa EJ, Grootegoed JA, Grosveld F, Galjart N. The microtubule plus-end-tracking protein CLIP-170 associates with the spermatid manchette and is essential for spermatogenesis. Genes Dev. 2005;19:2501–2515. doi: 10.1101/gad.344505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Bassam J, Kim H, Brouhard G, van Oijen A, Harrison SC, Chang F. CLASP promotes microtubule rescue by recruiting tubulin dimers to the microtubule. Dev Cell. 2010;19:245–258. doi: 10.1016/j.devcel.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basarsky TA, Parpura V, Haydon PG. Hippocampal synaptogenesis in cell culture: developmental time course of synapse formation, calcium influx, and synaptic protein distribution. J Neurosci. 1994;14:6402–6411. doi: 10.1523/JNEUROSCI.14-11-06402.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu R, Chang F. Shaping the actin cytoskeleton using microtubule tips. Curr Opin Cell Biol. 2007;19:88–94. doi: 10.1016/j.ceb.2006.12.012. [DOI] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. Establishment of neuronal polarity: lessons from cultured hippocampal neurons. Curr Opin Neurobiol. 2000;10:574–581. doi: 10.1016/s0959-4388(00)00124-0. [DOI] [PubMed] [Google Scholar]
- Bratman SV, Chang F. Mechanisms for maintaining microtubule bundles. Trends Cell Biol. 2008;18:580–586. doi: 10.1016/j.tcb.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Bronk P, Deák F, Wilson MC, Liu X, Südhof TC, Kavalali ET. Differential effects of SNAP-25 deletion on Ca2+-dependent and Ca2+-independent neurotransmission. J Neurophysiol. 2007;98:794–806. doi: 10.1152/jn.00226.2007. [DOI] [PubMed] [Google Scholar]
- Conde C, Cáceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 2009;10:319–332. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- Craig AM, Banker G. Neuronal polarity. Annu Rev Neurosci. 1994;17:267–310. doi: 10.1146/annurev.ne.17.030194.001411. [DOI] [PubMed] [Google Scholar]
- de Anda FC, Pollarolo G, Da Silva JS, Camoletto PG, Feiguin F, Dotti CG. Centrosome localization determines neuronal polarity. Nature. 2005;436:704–708. doi: 10.1038/nature03811. [DOI] [PubMed] [Google Scholar]
- de Anda FC, Meletis K, Ge X, Rei D, Tsai LH. Centrosome motility is essential for initial axon formation in the neocortex. J Neurosci. 2010;30:10391–10406. doi: 10.1523/JNEUROSCI.0381-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimov A, Kharitonov A, Efimova N, Loncarek J, Miller PM, Andreyeva N, Gleeson P, Galjart N, Maia AR, McLeod IX, Yates JR, 3rd, Maiato H, Khodjakov A, Akhmanova A, Kaverina I. Asymmetric CLASP-dependent nucleation of noncentrosomal microtubules at the trans-Golgi network. Dev Cell. 2007;12:917–930. doi: 10.1016/j.devcel.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galjart N. CLIPs and CLASPs and cellular dynamics. Nat Rev Mol Cell Biol. 2005;6:487–498. doi: 10.1038/nrm1664. [DOI] [PubMed] [Google Scholar]
- Goellner B, Aberle H. The synaptic cytoskeleton in development and disease. Dev Neurobiol. 2012;72:111–125. doi: 10.1002/dneu.20892. [DOI] [PubMed] [Google Scholar]
- Gu C, Zhou W, Puthenveedu MA, Xu M, Jan YN, Jan LY. The microtubule plus-end tracking protein EB1 is required for Kv1 voltage-gated K+ channel axonal targeting. Neuron. 2006;52:803–816. doi: 10.1016/j.neuron.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Hanus C, Ehlers MD. Secretory outposts for the local processing of membrane cargo in neuronal dendrites. Traffic. 2008;9:1437–1445. doi: 10.1111/j.1600-0854.2008.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho A, Morishita W, Atasoy D, Liu X, Tabuchi K, Hammer RE, Malenka RC, Südhof TC. Genetic analysis of Mint/X11 proteins: essential presynaptic functions of a neuronal adaptor protein family. J Neurosci. 2006;26:13089–13101. doi: 10.1523/JNEUROSCI.2855-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho A, Liu X, Südhof TC. Deletion of Mint proteins decreases amyloid production in transgenic mouse models of Alzheimer's disease. J Neurosci. 2008;28:14392–14400. doi: 10.1523/JNEUROSCI.2481-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad CC, Bradke F. Control of neuronal polarity and plasticity–a renaissance for microtubules? Trends Cell Biol. 2009;19:669–676. doi: 10.1016/j.tcb.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Hoogenraad CC, Akhmanova A, Howell SA, Dortland BR, De Zeeuw CI, Willemsen R, Visser P, Grosveld F, Galjart N. Mammalian Golgi-associated Bicaudal-D2 functions in the dynein-dynactin pathway by interacting with these complexes. EMBO J. 2001;20:4041–4054. doi: 10.1093/emboj/20.15.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton AC, Rácz B, Monson EE, Lin AL, Weinberg RJ, Ehlers MD. Polarized secretory trafficking directs cargo for asymmetric dendrite growth and morphogenesis. Neuron. 2005;48:757–771. doi: 10.1016/j.neuron.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Hur EM, Saijilafu, Lee BD, Kim SJ, Xu WL, Zhou FQ. GSK3 controls axon growth via CLASP-mediated regulation of growth cone microtubules. Genes Dev. 2011;25:1968–1981. doi: 10.1101/gad.17015911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein LC, Hoogenraad CC. Which way to go? Cytoskeletal organization and polarized transport in neurons. Mol Cell Neurosci. 2011;46:9–20. doi: 10.1016/j.mcn.2010.08.015. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Klingauf J, Tsien RW. Activity-dependent regulation of synaptic clustering in a hippocampal culture system. Proc Natl Acad Sci U S A. 1999;96:12893–12900. doi: 10.1073/pnas.96.22.12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YT, Hur EM, Snider WD, Zhou FQ. Role of GSK3 signaling in neuronal morphogenesis. Front Mol Neurosci. 2011;4:48. doi: 10.3389/fnmol.2011.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Setou M. Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat Neurosci. 2009;12:559–567. doi: 10.1038/nn.2314. [DOI] [PubMed] [Google Scholar]
- Kumar P, Lyle KS, Gierke S, Matov A, Danuser G, Wittmann T. GSK3beta phosphorylation modulates CLASP-microtubule association and lamella microtubule attachment. J Cell Biol. 2009;184:895–908. doi: 10.1083/jcb.200901042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Engel U, Rusch J, Scherrer S, Sheard K, Van Vactor D. The microtubule plus end tracking protein Orbit/MAST/CLASP acts downstream of the tyrosine kinase Abl in mediating axon guidance. Neuron. 2004;42:913–926. doi: 10.1016/j.neuron.2004.05.020. [DOI] [PubMed] [Google Scholar]
- Lu B, Greengard P, Poo MM. Exogenous synapsin I promotes functional maturation of developing neuromuscular synapses. Neuron. 1992;8:521–529. doi: 10.1016/0896-6273(92)90280-q. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- Miller PM, Folkmann AW, Maia AR, Efimova N, Efimov A, Kaverina I. Golgi-derived CLASP-dependent microtubules control Golgi organization and polarized trafficking in motile cells. Nat Cell Biol. 2009;11:1069–1080. doi: 10.1038/ncb1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozhayeva MG, Sara Y, Liu X, Kavalali ET. Development of vesicle pools during maturation of hippocampal synapses. J Neurosci. 2002;22:654–665. doi: 10.1523/JNEUROSCI.22-03-00654.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neukirchen D, Bradke F. Cytoplasmic linker proteins regulate neuronal polarization through microtubule and growth cone dynamics. J Neurosci. 2011;31:1528–1538. doi: 10.1523/JNEUROSCI.3983-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Kavalali ET. Differential regulation of spontaneous and evoked neurotransmitter release at central synapses. Curr Opin Neurobiol. 2011;21:275–282. doi: 10.1016/j.conb.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- Temburni MK, Rosenberg MM, Pathak N, McConnell R, Jacob MH. Neuronal nicotinic synapse assembly requires the adenomatous polyposis coli tumor suppressor protein. J Neurosci. 2004;24:6776–6784. doi: 10.1523/JNEUROSCI.1826-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Benditó G, Molnár Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. 2002;5:19–26. doi: 10.1038/nn783. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Noritake J, Kakeno M, Matsui T, Harada T, Wang S, Itoh N, Sato K, Matsuzawa K, Iwamatsu A, Galjart N, Kaibuchi K. Phosphorylation of CLASP2 by GSK-3beta regulates its interaction with IQGAP1, EB1 and microtubules. J Cell Sci. 2009;122:2969–2979. doi: 10.1242/jcs.046649. [DOI] [PubMed] [Google Scholar]
- Witte H, Bradke F. The role of the cytoskeleton during neuronal polarization. Curr Opin Neurobiol. 2008;18:479–487. doi: 10.1016/j.conb.2008.09.019. [DOI] [PubMed] [Google Scholar]
- Witte H, Neukirchen D, Bradke F. Microtubule stabilization specifies initial neuronal polarization. J Cell Biol. 2008;180:619–632. doi: 10.1083/jcb.200707042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann T, Waterman-Storer CM. Spatial regulation of CLASP affinity for microtubules by Rac1 and GSK3beta in migrating epithelial cells. J Cell Biol. 2005;169:929–939. doi: 10.1083/jcb.200412114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye B, Zhang Y, Song W, Younger SH, Jan LY, Jan YN. Growing dendrites and axons differ in their reliance on the secretory pathway. Cell. 2007;130:717–729. doi: 10.1016/j.cell.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Zmuda JF, Rivas RJ. The Golgi apparatus and the centrosome are localized to the sites of newly emerging axons in cerebellar granule neurons in vitro. Cell Motil Cytoskeleton. 1998;41:18–38. doi: 10.1002/(SICI)1097-0169(1998)41:1<18::AID-CM2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]