

Abstract
The synapse is a localized neurohumoral contact between a neuron and an effector cell and may be considered the quantum of fast intercellular communication. Analogously, the postsynaptic neurotransmitter receptor may be considered the quantum of fast chemical to electrical transduction. Our understanding of postsynaptic receptors began to develop about a hundred years ago with the demonstration that electrical stimulation of the vagus nerve released acetylcholine and slowed the heart beat. During the past 50 years, advances in understanding postsynaptic receptors increased at a rapid pace, owing largely to studies of the acetylcholine receptor (AChR) at the motor endplate. The endplate AChR belongs to a large superfamily of neurotransmitter receptors, called Cys-loop receptors, and has served as an exemplar receptor for probing fundamental structures and mechanisms that underlie fast synaptic transmission in the central and peripheral nervous systems. Recent studies provide an increasingly detailed picture of the structure of the AChR and the symphony of molecular motions that underpin its remarkably fast and efficient chemoelectrical transduction.
I. INTRODUCTION
When an action potential (AP) reaches a motor nerve terminal (FIGURE 1A), the accompanying depolarization releases the contents of about 100 ACh-containing synaptic vesicles into the synaptic cleft (147). Within a fraction of a millisecond, the ACh diffuses across the 100-nm width of the synaptic cleft where it binds to and rapidly opens several thousand postsynaptic AChR channels (117). Because the AChR ion channel selects cations over anions, the cell depolarizes, and if the membrane potential reaches threshold, a muscle AP is generated and contraction follows. ACh is removed from the synaptic cleft in less than a millisecond (139, 221), through both enzymatic hydrolysis and diffusion, but the postsynaptic depolarization, or excitatory postsynaptic potential (EPSP), declines to baseline over several additional milliseconds (FIGURE 1B); the rapid rise and the slower exponential decline of the EPSP are determined by the rates at which the AChR switches among resting, intermediate, and open states (17, 164). Evolution has apparently fine-tuned the rates of these interstate transitions so the EPSP concludes before the end of the AP, resulting in only one AP for each EPSP.
FIGURE 1.

A: electron micrograph of rat neuromuscular junction treated with peroxidase-conjugated α-bungarotoxin to label postjunctional AChRs. (From Engel et al. Neurology 27: 307–325, 1977, with permission.) B: time course of endplate potential and current. Horizontal bar, 2 ms; vertical bar, 40 nA (EPC) or 1 mV (EPP). (From Barrett EF and Magleby KL. Biology of Cholinergic Function, edited by A. M. Goldberg and I. Hanin. New York: Raven, 1976, p. 29–100, with permission.)
Two main gene superfamilies encode fast acting postsynaptic receptors: pentameric Cys-loop receptors (75) and tetrameric glutamate receptors (168). The two superfamilies are not related, but receptors in each family assemble from homologous subunits, and thus exhibit either five- or fourfold rotational symmetry. Subunits from the Cys-loop superfamily contain a signature sequence of 13 residues flanked by cysteine residues that form a disulfide bond, creating the eponymous Cys-loop. Cys-loop receptors can be either excitatory or inhibitory; are activated by the small molecule neurotransmitters ACh, GABA, glycine, or serotonin; and assemble from either one kind of subunit to form homopentamers or from several kinds of subunits to form heteropentamers. Although Cys-loop receptors are found only in eukaryotes, their ultimate ancestor is prokaryotic (262). The prokaryotic ancestors are also pentameric and exhibit a similar protein scaffold (25, 26, 118, 119), despite <10% sequence identity, but they lack the two cysteine residues that form a Cys-loop. Instead, the prokaryotic ancestors harbor an analogous structure similar to the Cys-loop of eukaryotes.
The endplate AChR from skeletal muscle has become an exemplar member of the Cys-loop receptor superfamily owing to both its inherent biological properties and its technical advantages. Endplate AChRs localize opposite motor nerve terminals and aggregate at a density of some 10,000/μm2 (117, 149), enabling detection by electron microscopy and microelectrode recording. Snake venom α-neurotoxins bind pseudo-irreversibly to AChRs and serve as labels to quantify AChR number and track cellular and subcellular locations (48). The electric organ of the Torpedo ray is the richest natural source of AChRs and yields milligram amounts of protein ideal for structural and biochemical studies (171). The embryologic origin of the electric organ is the same as skeletal muscle, but the cells lack contractile filaments and form thin, flattened disks that stack in tall columns aligned side by side. Muscle AChRs are heteropentamers composed of α-, β-, and δ-subunits, plus either the fetal γ-subunit or the adult ε-subunit (257), although some animals, such as Torpedo, lack the ε-subunit (263). The subunits assemble in a sequence of oligomerization steps with intervening conformational changes (101) that minimize multiple or incorrect subunit stoichiometries, rendering assembly highly specific. Elementary currents through endplate AChRs exhibit large and highly uniform amplitudes, with the temporal sequences of current pulses well described by Markov models containing a small number of stable states (235), enabling quantification of transition rate constants for elementary reaction steps within the activation process.
II. MECHANISM OF AChR ACTIVATION
Since the realization that nerve-released ACh creates a brief short circuit across the postsynaptic membrane (86), an enduring quest has been to define the underlying physical mechanism. Over several decades, a series of powerful technical breakthroughs helped attain our current mechanistic understanding. The first was introduction of the microelectrode (188), which enabled transmembrane voltage to be measured in sufficiently large cells. In a method called iontophoresis, an electrical microjet filled with an ionizable agonist allowed rapid and spatially precise delivery of agonist by passing a suitable outward current (187). Because changes in voltage across a cell membrane contain both ionic and capacitative contributions, the two-electrode voltage clamp was developed to eliminate the capacitative current and register only the ionic current (121).
Detection of elementary channel-like currents through the AChR was first achieved by combining the two-electrode voltage clamp with measurements of current through an independent extracellular micropipette (189). However, the method was technically difficult, and the signal-to-noise ratio was low, limiting the detected dwell times to tens of milliseconds and longer. Continued refinement of the method, however, led to the discovery of the giga-ohm seal that forms when a heat-polished micropipette is applied to a bare or cleaned cell membrane followed by gentle suction (107). The ability to form giga-ohm seals, combined with increasingly sophisticated electronic circuit designs (236), gave rise to the present-day patch-clamp method for recording currents through single ion channels. When a giga-ohm seal is established, the vast majority of the current flows through the membrane patch and into the patch-clamp circuitry, rather than being lost through the seal. The patch-clamp electronics also allowed direct application of a transmembrane voltage, thus eliminating the need for an independent voltage clamp. The patch clamp increased temporal resolution of single-channel currents to the present day minimum duration of ~10 μs, and also permitted formation of cell-free membrane patches at the tip of the recording pipette (107). Development of techniques for rapid microperfusion, combined with cell-free membrane patches, allowed agonist application and response detection on time scales approaching that of synaptic transmission (35, 165).
Patch-clamp recording yielded a wealth of experimental data, but a practical limitation was the time-consuming detection of thousands of unitary current pulses required to decipher the underlying states and transition rate constants. Over several years, commercial computer software was developed to measure the amplitudes and durations of the current pulses, and the stochastic nature of single-channel dwell times was established (144, 169). A general theoretical framework was established to relate single-channel dwell times to rate constants in kinetic schemes containing small numbers of stable states (62), and free software was developed to fit the schemes to the experimental dwell times (211) (http://www.qub.buffalo.edu/; http://www.ucl.ac.uk/pharmacology/dc.html).
More than 50 years ago, the ionic basis of the EPSP was established in the wake of classical studies of the nerve impulse, which arises from a transient voltage-dependent permeability increase selective for sodium ions followed by a separate voltage-dependent permeability increase selective for potassium ions (120). Analogous studies, combining voltage clamping with ion substitution, revealed that the EPSP arose from simultaneous permeability increases to sodium and potassium ions, unlike the nerve impulse (160, 259). In further contrast to the nerve impulse, the EPSC showed a linear current-voltage relationship, indicating the permeability change did not depend on membrane voltage. The quantized nature of the permeability increase came to light by close inspection of responses elicited by low concentrations of ACh, which exhibited fluctuations about the mean response that substantially exceeded the background noise (9, 140). Fourier analyses of the fluctuations disclosed the amplitude and average duration of the elementary ACh-induced responses, but could not discern the shape of the responses. Introduction of the patch clamp, however, immediately revealed rectangular elementary current pulses, implying a channel mechanism, in accord with the rosette-like structures in postsynaptic membranes detected by electron microscopy (117).
A. The del Castillo and Katz Mechanism
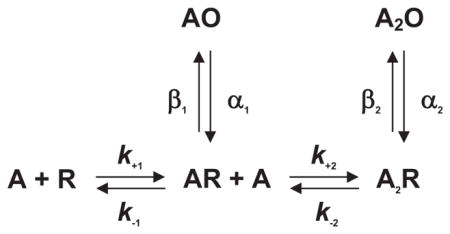
The simplest mechanism of receptor activation is a one-step reaction in which binding of ACh directly produces the active agonist-receptor complex. However, this mechanism could not account for the observations that full and partial agonists elicited different maximal responses and that a weak agonist could act as a competitive inhibitor of a strong agonist (72). By analogy to Michaelis-Menten enzyme theory, del Castillo and Katz proposed that when an agonist binds to the AChR, the initial complex is inactive, but it then isomerizes to the active agonist-receptor complex
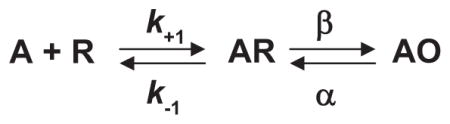
where A is the agonist, R is the resting receptor with the channel closed, AR is the inactive complex with the channel closed, AO is the active complex with the channel open, k−1 is the agonist association rate constant, k−1 is the agonist dissociation rate constant, β is the channel opening rate constant, and α is the channel closing rate constant. According to this mechanism, full and partial agonists differ in their ability to promote the isomerization step, with slow forward or rapid reverse rate constants giving a partial agonist, and rapid forward or slow reverse rate constants giving a full agonist; for a pure competitive antagonist, the isomerization step does not occur. A natural consequence of distinct binding and isomerization steps was that the EC50, or the agonist concentration that produces a half-maximal response, depends jointly on the dissociation constant for agonist binding (K = k−1/k−1) and the efficacy of the isomerization step (θ = β/α) (61).
The del Castillo and Katz mechanism depicted one agonist binding site per receptor, but evidence quickly emerged for more than one site. By applying small iontophoretic doses of ACh to frog endplates, Katz and Thesleff (141) found that the depolarizing response as a function of ACh dose had a sigmoid rather than a linear start, indicating a positively cooperative response. Subsequent studies rapidly applied known concentrations of agonist and used voltage clamping to directly monitor changes in membrane conductance. The results revealed Hill plots with limiting slopes of two, confirming the cooperative nature of the response (3, 74, 78). The findings could not be explained by a mechanism with only one agonist binding step, but instead required at least two
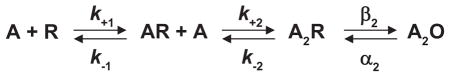
In the special case of identical binding sites the association and dissociation rate constants are related by statistical factors as follows: k+1 = 2k+2 and k−2 = 2k−1; in the following sections, statistical factors are omitted to avoid assumptions about binding site identity. The extended del Castillo and Katz mechanism held for many decades, and often serves as the starting point for contemporary analyses.
Once single-channel currents could be registered with high temporal resolution, channel opening events were found to be interrupted by brief transitions to the closed, baseline current (FIGURE 2). Both the original and the extended del Castillo and Katz mechanisms predicted that channel openings from fully occupied receptors would exhibit such brief closings due to sojourns in the A2R state, and that at low concentrations of agonist, they would be too brief to arise from dissociation of agonist to form AR followed by rebinding of agonist to form A2R. Thus the kinetic properties of the brief interruptions were related in a simple way to elementary rate constants within the mechanism. The mean duration of the interruptions was predicted to equal the sum of the channel opening and agonist dissociation rate constants (β2 + k−2), and the number of interruptions per burst of openings was predicted to follow a geometric distribution with a mean equal to the ratio of channel opening to agonist dissociation rate constants (β2/k−2) (62, 64). The reciprocal of the mean open time gave the rate constant for channel closing (α2). The maximum probability of channel opening then followed from the ratio of the channel opening rate constant to the sum of the channel opening plus closing rate constants [β2/(β2 + α2)].
FIGURE 2.

Patch-clamp recording of a single acetylcholine receptor (AChR) channel activation episode elicited by a low concentration of ACh applied to adult human endplate AChRs expressed in 293 HEK cells (S. M. Sine and N. Mukhtasimova, unpublished data). Channel openings are upward deflections. Membrane potential, −120 mV; Gaussian filter, 20 kHz.
Experimental measurements were soon quantitatively analyzed according to the extended del Castillo and Katz mechanism. For AChRs at frog endplates, channel openings exhibited a mean duration of 1.4 ms, were interrupted by brief closings with a mean duration of 20 μs, and occurred at a frequency of 1.9 interruptions per burst of openings; these measured parameters translated to a channel opening rate constant of 31,000 s−1, an agonist dissociation rate constant of 8,200 s−1, a channel closing rate constant of 700 s–1, and maximum probability of channel opening of 0.97 (63, 64). For fetal mouse AChRs expressed in a clonal cell line, the major class of brief closings exhibited a mean duration of 45 μs and a frequency of 2.7 interruptions/burst, giving a channel opening rate constant of 16,000 s−1, an agonist dissociation rate constant of 6,000 s−1, a channel closing rate constant of 73 s−1, and a maximum channel open probability of 0.99 (248). Quantitative differences between the two sets of rate constants could have arisen because the frog endplate receptors contained the adult ε-subunit, whereas the mouse receptors contained the fetal γ-subunit. A third study examined receptors from cultured Xenopus myocytes, in which ACh elicited channel opening of low-conductance fetal and high-conductance adult receptors (10). The high-conductance channels exhibited rate constants closer to those observed from the frog endplate, while the low-conductance channels exhibited rate constants closer to those from fetal mouse muscle. The overall findings suggested the channel opening and closing steps were faster for adult than for fetal receptors, but more importantly, the extended del Castillo and Katz mechanism could describe activation of both receptor types.
When agonists with different efficacy were compared, however, observations of burst fine structure diverged. Brief interruptions recorded from frog endplate AChRs differed for different agonists, in agreement with expectations of the extended del Castillo and Katz mechanism (63). However, brief interruptions recorded from fetal mouse AChRs were similar for different agonists (248). Furthermore, for fetal mouse AChRs, infrequent channel openings elicited by a competitive antagonist exhibited burst fine structure similar to that observed for strong agonists (247). Differences between adult frog and fetal mouse receptors might have explained the diverging results, but some 20 years later, a deeper mechanistic explanation emerged (150, 185).
Subsequently, the extended del Castillo and Katz mechanism was fitted to temporal sequences of single-channel dwell times recorded over a wide range of agonist concentrations, yielding complete sets of the elementary rate constants. The theoretical foundation for these analyses was laid by Colquhoun and Hawkes (62), who provided a general quantitative framework to predict single-channel open and closed dwell times from a specified mechanism. The experimental design was further aided by desensitization, a process that inactivated the majority of receptors in the patch of membrane, resulting in recordings in which episodes of many successive channel openings, all from one receptor, were flanked by prolonged quiescent periods (223). After removal of the long quiescent periods, the extended del Castillo and Katz mechanism could be fitted to the sequences of closely spaced open and closed dwell times. Use of a range of agonist concentrations allowed sampling of all states in the mechanism, from un-liganded to doubly-liganded, enabling estimates of rate constants for agonist association and dissociation for each binding site and for opening and closing of the channel. Also, high concentrations of agonist were found to block the open channel (197, 249), and the fitting yielded rate constants for agonist blocking and unblocking.
The first complete set of rate constants for AChR activation was obtained for cloned Torpedo AChRs expressed in fibroblasts in the presence and absence of calcium. In the presence of calcium, the rate constant for channel opening was rapid, 45,000 s−1, while the rate constant for channel closing was slower, 8,000 s−1, predicting a maximum open probability of 0.85 (241). The association rate constants were ~10-fold slower than the limit imposed by diffusion, but at the high concentration of ACh achieved during synaptic transmission, the agonist binding and channel opening steps were predicted to be similarly rapid, with neither process rate limiting. Rate constants for ACh dissociation from the two binding sites differed by ~100-fold, indicating the sites bound agonist with different affinities. Two contemporaneous studies, using fetal mouse receptors and a narrower range of agonist concentrations, reached similar conclusions (127, 297); the rate constant for channel opening was at least 28,000 s−1, the agonist association rate constants were ~10-fold slower than the diffusion limit, agonist affinity for the two binding sites differed by 30- to 500-fold, and the maximum channel open probability approached unity.
B. Development of a Cyclic Mechanism
Three additional insights led to a general mechanistic description of AChR activation. Two of these insights originated from Katz and Thesleff’s study of desensitization at the motor endplate (141), a phenomenon in which the response to agonist diminishes if the agonist concentration is maintained for a sufficiently long time. The first insight was based on the large extent of desensitization that develops following steady-state exposure to agonist, suggesting the desensitized state bound agonist more tightly than the resting state. Subsequent measurements showed that agonist affinity increased in a time-dependent manner (212, 281, 284, 285) and that the onset of the affinity increase paralleled the onset of functional desensitization (114, 251). The second insight arose from the observation that the rate of recovery from desensitization exceeded the rate of onset of desensitization elicited by a low concentration of agonist; this observation could not be explained by a sequential mechanism, but instead was explained by the following cyclic mechanism
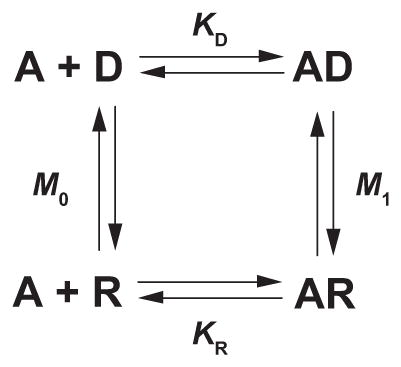
where A is the agonist, R is the resting state, D is the desensitized state, KR and KD are the agonist dissociation constants for the two states, and M0 and M1 are the interstate equilibrium constants. The cyclic mechanism was a seminal breakthrough because, in the words of Katz and Thesleff, “a proportion of receptors is present in a refractory form, and on account of its very high affinity, will preferentially absorb small quantities of applied ACh.” Although Katz and Thesleff came to this realization in the context of desensitization, it would become a core tenet in a general mechanism of receptor activation.
A third mechanistic insight is credited to Monod, Wyman, and Changeux (MWC) who proposed a general mechanism to account for positive cooperativity in oligomeric enzymes (182). The MWC mechanism united three key principles. The first was that an allosteric protein adopts two distinct states, active and inactive, that interconvert in the absence of activator. The second was that the active state binds the activator with higher affinity than the inactive state. These two principles were analogous to those in Katz and Thesleff’s cyclic mechanism for desensitization. The third principle was unique and held that in switching from the inactive to the active state, protomers within the oligomer, acting one upon another, preserve molecular symmetry, meaning that all protomers maintain the same energetic state and thus change from one state to another in a concerted manner. The resulting MWC mechanism was general in that it applied to oligomers containing any number of protomers, and the extent of cooperativity, measured by the slope of the Hill plot, depended only on the number of protomers, the equilibrium between active and inactive states in the absence of activator, and the ratio of activator affinities for the two states. Applied to an AChR with two agonist binding sites, the MWC mechanism yielded
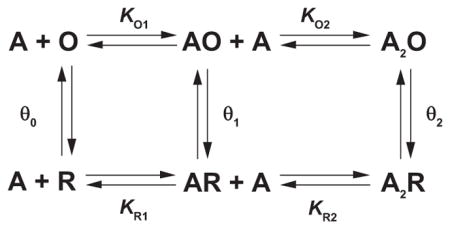
where R is the resting state, O is the open channel state, the θn are interstate equilibrium constants, and the other terms are as defined before. Notice that the extended del Castillo and Katz mechanism is imbedded within the two-site MWC mechanism. According to the MWC mechanism, an agonist’s fundamental action is to increase the inherently small equilibrium constant between closed and open states, defined by θ0, with occupancy of each binding site increasing the corresponding θn. Furthermore, the degree of positive cooperativity depends on both θ0 and the ratio of dissociation constants KRn/KOn. Because transition between the closed and open states is concerted, a key expectation is the absence of intermediates with one binding site in the low-affinity state and the other site in the high-affinity state.
The two-site MWC mechanism has been evaluated, both qualitatively and quantitatively, in light of experimental measurements of agonist-activated single-channel currents. Qualitative support included the findings that the AChR channel opened infrequently and for brief durations in the absence of agonist (130), subsaturating concentrations of agonist elicited kinetically distinct classes of brief, singly-liganded and long, doubly-liganded openings, and the ratio of brief to long openings diminished as the agonist concentration was increased (127, 172). Quantitative tests paralleled those in which the extended del Castillo and Katz mechanism was fitted to single-channel dwell times that gave estimates of elementary transition rate constants. However, transitions of a single AChR between unliganded closed and open states could not be quantified, and direct transitions between successive open states could not be distinguished. Thus most studies fitted the following subset of the two-site MWC mechanism to single-channel dwell times.
The strategy was to record single-channel currents from end-plate AChRs, human or mouse, activated by a wide range of ACh concentrations, and to fit this mechanism to all the data simultaneously (113, 201, 275; FIGURE 3). Rate constants for ACh association were found to be rapid, ranging from 1 to 5 × 108 M−1·s−1 at a temperature of 21°C, and differed by no more than threefold between the two binding sites from the same receptor or between binding sites from the two species of receptors. For human receptors, agonist dissociation rate constants differed by four- to fivefold between the two binding sites, whereas for mouse receptors no difference was detected. For both species of receptors, the agonist dissociated rapidly from at least one binding site, with rate constants from 13,000 to 25,000 s−1. For mono-liganded receptors, the channel opening rate constant β1 was slow, 60–250 s−1, while the channel closing rate constant α1 was faster, 3,000–10,000 s−1, giving a singly-liganded gating equilibrium constant, θ1, from 0.01 to 0.1; precision in these parameters was limited because brief, singly-liganded channel openings were relatively infrequent over the agonist concentration range that allowed clear identification of clusters of single-channel openings. For doubly-liganded receptors, the channel opening rate constant β2 was rapid, 43,000–53,000 s−1, while the channel closing rate constant α2 was slower, 1,700–2,600 s−1, giving a gating equilibrium constant, θ2, from 20 to 30; these parameters were obtained with good precision because long, doubly-liganded channel openings were plentiful at all agonist concentrations. Applying the principle of detailed balancing to the two-site MWC mechanism, using the relationship θ2 = θ1(KR2/KO2), yielded an ACh dissociation constant for the doubly-liganded open state, KO2, from 10 to 115 nM, some 200–3,000 smaller than KR2.
FIGURE 3.

Single-channel currents through adult human AChRs elicited by the indicated concentrations of ACh. The extended del Castillo and Katz mechanism was fitted simultaneously to the global set of closed and open dwell times for the indicated concentrations of ACh. [From Mukhtasimova et al. (184).]
Estimating all the parameters in the two-site MWC mechanism would require measurements of channel opening of a single AChR in the absence of agonist. However, although channel openings in the absence of agonist are detectable, the number of AChRs in a given patch of membrane is unknown. The number of AChRs in a patch of membrane was estimated from the density of radiolabeled α-bungarotoxin binding sites together with estimates of membrane area based on the measured pipette resistance. Combining these estimates with measurements of spontaneous channel opening yielded a gating equilibrium constant, θ0, from 3 × 10−7 to 5×10−6 (127,130). Two additional sources of uncertainty were expected to affect these estimates. The first was that spontaneous channel openings exhibited multiple exponential components (128, 172), indicating multiple open states, suggesting θ0 was overestimated. The second arises from the tendency of proteins to adhere to glass, and thus only a portion of receptors in the membrane patch may be electrically accessible (255), suggesting θ0 was further overestimated. Disregarding these uncertainties and taking θ0 = 1 × 10−6, θ1 = 0.01–0.1 and KR1 = 10 μM, the relationship θ1 = θ0(KR1/KO1) yields KO1 for ACh from 1 to 10 nM.
To summarize, when viewed according to the two-site MWC mechanism, AChR activation produces a strong depolarization with a rapid onset and a rapid offset, two key functional requirements of the endplate EPSP. A strong depolarization is accomplished by the large fraction of AChRs that activate in response to ACh; the AChR is essentially inactive in the absence of agonist, with θ0 ~1 × 10−6, and is almost fully active in its presence, with θ2 ~25. This 25 million-fold shift in the closed-open equilibrium requires the agonist to bind to the open state with very high affinity, but the presence of two rather than one binding site provides enough binding energy to achieve such a shift (129). If the AChR contained only one binding site, and the dissociation constant of ACh for the active state KO1 was 10 nM, the relationship θ1 = θ0(KR1/KO1) predicts a low-affinity dissociation constant KR1 of 0.1 M, which would require 10-fold greater synaptic concentrations of ACh (147). However, with two binding sites the relationship is θ2 = θ0(KR1/KO1)(KR2/KO2), and a 25 million-fold increase in θ0 is achievable with KR1 and KR2 values of tens of micromolar. The requirement of rapid EPSP onset is met by the high rate of ACh association, only 10-fold slower than the diffusion limit, combined with the rapid rate of channel opening. The requirement of rapid EPSP offset is met by a high rate of ACh dissociation from one of the two binding sites, which also favors a large KR2 and increases the ratio KR2/KO2, further promoting a large extent of channel opening.
C. Mechanisms With Intermediates Between Closed and Open States
The two-site MWC mechanism views channel gating as a pure two-state reaction between a closed and an open state, but recent studies revealed a transient intermediate between these states. The first evidence emerged from fitting kinetic mechanisms to sequences of single-channel dwell times obtained from the glycine receptor (38), another member of the Cys-loop receptor superfamily. The closed and open dwell times, obtained over a wide range of glycine concentrations, were not well described by a mechanism in which the resting state made a direct transition to the open state, but instead were described by including an intervening closed state. The receptor was thus envisioned to flip from the resting to an intermediate closed state before the final closed to open transition could occur. Subsequent studies showed that whereas full and partial agonists differed in their ability to form the closed state intermediate, the two classes of agonists had similar abilities to promote transition from the intermediate closed to the open state (150). In other words, the ultimate closed to open transition did not depend on agonist efficacy. For a receptor with two agonist binding sites, the flip mechanism is given by
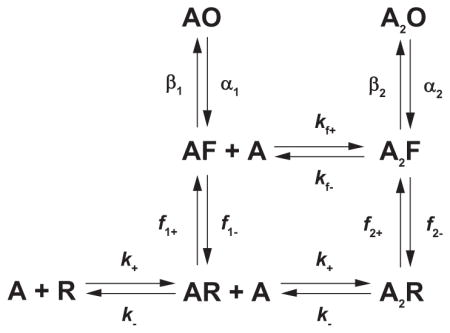
where F is the intermediate flip state, f−n are the forward flip rate constants, f−n are the backward flip rate constants, and the other terms are as defined before.
The Flip mechanism challenged the prevailing mechanistic theory of agonist efficacy. For both the del Castillo and Katz and two-site MWC mechanisms, maximal channel open probability, a measure of agonist efficacy, is given by Popen = θ2apparent/(1 + θ2apparent). For the Flip mechanism, the same relationship holds but with θ2apparent = θ2F2/(1 + F2), F2 the equilibrium constant for the second flip reaction and θ2 the true channel gating equilibrium constant. Thus, for the del Castillo and Katz and two-site MWC mechanisms, agonist efficacy is determined solely by the apparent channel gating equilibrium constant θ2apparent, but for the Flip mechanism it is determined jointly by the flip equilibrium constant F2 and the true channel gating equilibrium constant θ2.
The Flip mechanism was further tested for its ability to describe single-channel dwell times from human adult end-plate AChRs activated by the full agonist ACh and the partial agonist tetramethylammonium (TMA; Ref. 150). The two agonists differed in their ability to promote the flip reaction, by analogy to the studies of glycine receptors, whereas they were similar in their ability to promote the closed to open reaction; for ACh, F2 was 3.8, but for TMA was 0.14; for ACh, θ2 was 34 and for TMA was 28. A recent study of choline, a product of ACh hydrolysis at the motor endplate and a very weak agonist, revealed F2 = 0.006 and θ2 = 16 (151). Thus from the weakest to the strongest agonist, F2 changed by a factor of 633, whereas θ2 changed by a factor of 2.1. Comparison of ACh and choline showed that the forward flip step f2+ increased 115-fold, whereas the reverse flip step f2− decreased 4-fold. Thus agonist efficacy depends primarily on the agonist’s ability to destabilize the resting closed state, rather than to stabilize the flip state.
Although the Flip mechanism has a stable intermediate between resting and open states, it maintains two of the three tenets of the MWC mechanism. First, affinity of agonist for the flip state is higher than that for the resting state, although it remains lower than that for the open state. Second, transition from the resting to the flip state is concerted; intermediates with one binding site flipped and the other not flipped are not included. The third tenet, not considered in the Flip mechanism, is whether the flip state forms in the absence of agonist.
If the Flip mechanism was not introduced to account for the kinetic complexities of glycine receptor activation, a long time might have elapsed before it was considered for the endplate AChR. The reason is that the extended del Castillo and Katz and MWC mechanisms appeared to adequately describe single-channel dwell time distributions for the AChR over a wide range of ACh concentrations (FIGURE 3). However, several qualitative observations could not be reconciled by the del Castillo and Katz and MWC mechanisms. Neither mechanism could explain why agonists with widely different efficacies exhibited similar brief interruptions of long-lived channel openings (247, 248), nor could they explain multiple exponential components of unliganded channel openings (102, 103, 128, 172). A recent study, however, provided a mechanistic framework that accounted for these observations (185). When hydrophilic substitutions were installed into the AChR pore, spontaneous channel opening increased to measureable levels, and the single-channel events exhibited multiple exponential components of both closed and open dwell times. To account for these observations, unliganded, closed receptors were proposed to isomerize to a primed closed state before the channel could open.
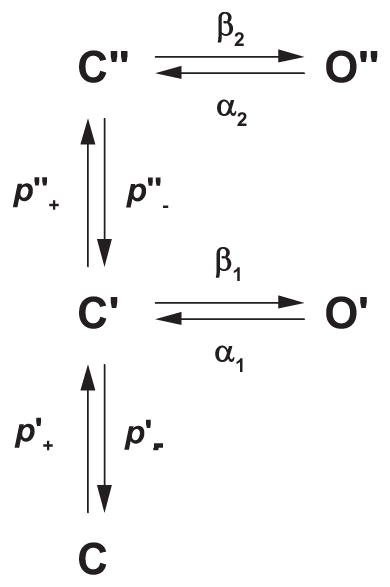
where C is the resting closed state, C′ is the singly primed state, C″ is the doubly primed state, p+ is the priming rate constant, and p− is the unpriming rate constants. The singly-primed state C′ gives rise to brief openings O′, whereas the doubly-primed state C″ gives rise to long openings O″. These unliganded long openings were interrupted by brief closings to state C″, and these interruptions exhibited the same mean duration and frequency of occurrence as those within long openings elicited by the agonist. Remarkably, these observations imply that transitions from the brief closed to the long open state are agonist independent. Incorporating two agonist occupancy steps into the preceding mechanism yields
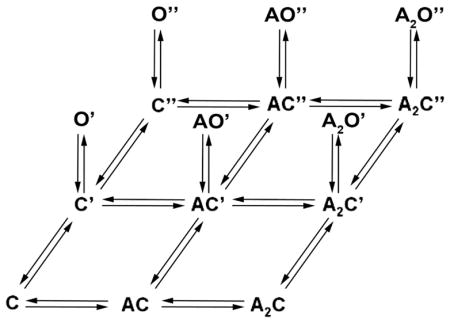
where the closed states are arrayed in three columns, one for each degree of agonist occupancy, and in three rows, one for each degree of priming. Each priming step corresponds to a change in one agonist binding site, with singly-primed receptors eliciting brief openings, and doubly-primed receptors eliciting long openings.
The Primed mechanism depicts sequential rather than concerted priming of the agonist binding sites, unlike the MWC and Flip mechanisms. However, two other tenets of the MWC mechanism are maintained: both primed and open states form in the absence of agonist, and these states bind agonist with higher affinity compared with the resting state. Because the Primed mechanism was developed to describe activation of a mutant receptor, a tacit assumption was that the same core mechanism of activation operated in both the wild-type and mutant receptors, but with different values of the transition rate constants. According to this view, kinetic studies of mutant AChRs hold the potential to unmask state transitions that occur with low or negligible probability in the wild-type receptor.
III. AChR STRUCTURE
Owing to the AChR’s large size, oligomeric quaternary structure and location within the cell membrane, structure determination by x-ray crystallography has remained elusive. Thus multidisciplinary approaches, both direct and indirect, have been brought to bear on AChR structure determination. Electron microscopy of negatively stained or rapidly frozen synaptic membranes revealed the overall shape and size of the AChR (177, 265), and combined with subunit-specific antibodies or α-neurotoxins, addressed the question of the location of subunits within the oligomer (136, 146, 299). Affinity purification of the AChR, using α-neurotoxin or a quaternary ammonium ligand coupled to a solid support (162, 170), enabled purification followed by characterization by standard biochemical approaches, including SDS-PAGE (163), density gradient sedimentation (220), and NH2-terminal sequencing (217). Once the AChR subunits were cloned, models of subunit folding in the cell membrane emerged (57, 195). Cloning also paved the way for development of expression systems and application of site-directed mutagenesis to decipher structure-function relationships. Locations of the subunits within the oligomer were deduced using expression systems that allowed deliberate omission of the subunits, combined with ligand binding measurements that revealed subunit-subunit juxtaposition (23, 240). Analogously, mutagenesis combined with ligand binding measurements showed that the ligand binding sites contained multiple protein loops, and further, that each subunit within the oligomer was built upon a common protein scaffold (238, 243). Covalent labeling by site-specific ligands, followed by proteolytic digestion and peptide sequencing, provided direct evidence for residue localization within regions such as the ligand binding site and pore (70, 94, 206, 219). Cryoelectron microscopy (cryo-EM) applied to two-dimensional arrays of AChRs from Torpedo yielded images with an initial resolution of 18 Å (177, 265). However, further refinements of the method culminated in the current state of the art resolution of 4 Å, sufficient to localize α-carbon atoms and large side chains (271). In the past 10 years, x-ray crystallography yielded high-resolution structures of modules of the AChR, including acetylcholine binding protein (AChBP), homologous to the AChR extracellular domain, and the extracellular domain of the α1-subunit (32, 73). More recently, complete x-ray crystal structures were obtained for two prokaryotic relatives of the AChR (25, 118, 119).
A. Early Advances in AChR Structure
Initial investigations used direct methods to visualize AChR structure. After postsynaptic membranes were negatively stained or rapidly frozen, deep etched, and rotary shadowed, electron microscopy revealed densely packed rosettes, each with a diameter of 80 Å when viewed from the synaptic cleft (117). In a serendipitous technical breakthrough, conditions were found in which isolated Torpedo synaptic membranes formed long tubes with the AChRs arrayed in a two-dimensional crystalline lattice (36). Electron microscopy of these tubes, embedded in amorphous ice, revealed that the unit AChR was an elongated cylinder with an extracellular projection of 65 Å, a transmembrane span of 30 Å, and an intracellular projection of 30 Å (177, 265). Each cylinder was joined to another identical structure to form a dimer through disulfide bonded cysteine residues at the COOH termini of the δ-subunits, a structural motif unique to electric rays (263). Evidence that the cylindrical structures were genuine AChRs included their selective location in postsynaptic membranes (117), their coincidence with α-neurotoxin binding (89), a similar size and shape to the purified AChR protein imaged by electron microscopy (27, 80, 143, 292), and functional reconstitution of purified AChRs in lipid bilayers (161).
In parallel with the ultrastructural studies, protein composition of the AChR was determined by sedimentation fractionation of Torpedo synaptic membranes followed by detergent solubilization and affinity purification, all in the presence of protease inhibitors. Molecular weight of the intact AChR oligomer was determined by analytical ultracentrifugation in which the contribution of bound detergent was neutralized by density matching (220), and also by gel electrophoresis after the subunits were cross-linked using a bifunctional reagent (21); molecular masses of the AChR monomer and dimer were found to be 250 and 500 kDa, respectively. Subunits within the monomer remained tightly associated even in the presence of concentrations of non-ionic detergents sufficient to dissolve the cell membrane, but they dissociated in the presence of the ionic detergent sodium dodecyl sulfate; gel electrophoresis under denaturing conditions revealed four polypeptide chains with molecular masses 40 (α), 50 (β), 60 (γ), and 65 (δ) kDa. Given the molecular masses of the subunits and of the monomeric AChR, subunit stoichiometry was proposed to be α2βγδ. This stoichiometry agreed with results from preparative electrophoresis of each subunit followed by quantification of protein content (163), and was further verified by quantitative NH2-terminal sequencing of the subunits (217).
Over a span of several years, improvements in cryo-EM applied to tubular arrays of Torpedo AChRs increased the resolution from the initial 18 Å to 9 Å (270). This improvement was achieved chiefly by averaging more electron microscopic images. At 9 Å resolution, individual subunits of the AChR pentamer were visible as long rods positioned approximately normal to the cell membrane. The extracellular projection formed a wide vestibule freely accessible to solvent, whereas the contiguous transmembrane region formed a narrower lumen with a constriction presumed to form the barrier to ion flow (FIGURE 4A). Still further improvement was achieved by employing a high-voltage field emission electron microscope with a helium-cooled stage, and by computational correction of image distortions inherent to the tubular crystalline lattice (178). The resulting resolution of 4.6 Å disclosed a series of twisted β-strands within the extracellular wall of each subunit, visible as crescents of densities when viewed from the synaptic cleft, as well as cytoplasmic projections of each subunit that contained apertures through which permeating ions could pass after exiting the channel.
FIGURE 4.

Progression of AChR atomic structure determination. A: Torpedo AChR at 9 Å resolution obtained by cryo-EM (270), with permission from N. Unwin. B: crystal structure of AChBP at 2.7 Å resolution (32; PDB code 1I9B). C: Torpedo AChR at 4 Å resolution (271; PDB code 2BG9). D: GLIC at 3.3 Å resolution (PDB code 3EHZ).
Cloning of the AChR subunits provided the most detailed information at the time regarding the chemical composition of the AChR. Cloning was possible in part due to NH2-terminal sequencing of the four AChR subunits, which allowed synthesis of oligonucleotides to probe a Torpedo cDNA library for open reading frames encoding the subunits (195). The sequence of the α-subunit emerged first (193), followed by that of the γ-subunit (57), and then the remaining subunit sequences (195). Alignment of the primary sequences revealed a high degree of sequence homology among the subunits, indicating they evolved from a common ancestor.
Plots of residue hydrophobicity against residue number were used to build models of subunit folding in the cell membrane (57, 194). The first half of each subunit was hydrophilic, whereas the following span included four sections of hydrophobic residues, each long enough to traverse the membrane as an α-helix. An extended stretch of hydrophilic residues, predicted to form an amphipathic α-helix, was located between the third and fourth hydrophobic regions. All the models portrayed the large NH2-terminal segment as the main constituent of the extracellular ligand binding domain, but a debate developed about which segment formed the lining of the ion channel. A prevailing view was that to account for rapid flux of ions through the AChR channel, the ion translocation pathway should be hydrophilic. Apart from the large NH2-terminal region, the only other hydrophilic region was the amphipathic helix, which emerged as a candidate for the channel lining (294). However, the amphipathic helix was the least conserved segment of all the subunits, contrary to the expectation that a conserved structure should form the ion translocation pathway.
The identity of residues that formed the ligand binding site also remained enigmatic. Early studies using sulfhydryl-reactive reagents showed that a readily reducible disulfide bond was present at the ligand binding site (137), and the α-subunit was the only one that contained vicinal cysteine residues within the NH2-terminal domain. However, the eponymous Cys-loop was also suggested to form the binding site (58), even though it was present in all the subunits and 6 of the 13 residues within the Cys-loop were the same in each subunit. The models of subunit folding were subsequently tested by mutagenesis combined with measurements of receptor function, site-directed photolabeling followed by microsequencing and substituted cysteine accessibility measurements.
Positioning of subunits within the AChR pentamer was initially addressed by electron microscopy of two-dimensional arrays of Torpedo AChRs bound with α-neurotoxin. Two α-neurotoxin molecules were known to bind to each pentamer, presumably because it contained two α-subunits, and α-neurotoxin bound to peptides derived from the α-subunit but not the other subunits (291). The resulting electron micrographs showed that the two α-subunits were not adjacent, but were separated by a non-α-subunit (299). Furthermore, the intervening subunit was not the δ-subunit, which was identifiable in electron micrographs because it joined pairs of AChR monomers (108), indicating either the β- or the γ-subunit was the intervening subunit. From this point, the evidence diverged; chemical cross-linking of AChR monomers via the β-subunit indicated this subunit was not the intervening subunit (136), but electron microscopy of the AChR bound with antibodies against β- and γ-subunits suggested the β-subunit was the intervening subunit (146).
The advent of expression systems resolved the subunit positioning question. Binding sites for small ligands could be formed from combinations of α- and either γ- or δ-subunits, but not α- and β-subunits (23). Evidence that the complexes formed true ligand binding sites was that the α-γ complexes bound the competitive antagonist d-tubocurarine with high affinity, the α-δ complexes bound with low affinity, and the two affinities were similar to the high- and low-affinity components observed in the native AChR. These studies had the limitations that coexpression of pairs of subunits yielded incomplete oligomers that were retained within the cytoplasm, and the β-subunit was not present. However, the picture clarified when combinations of three subunits were coexpressed (240). When α-, β-, and γ-subunits were coexpressed, pentameric assemblies formed with stoichiometry α2βγ2; the pentamers were transported to the cell surface and they bound dimethyl-d-tubocurarine with a high-affinity dissociation constant that coincided with the high-affinity component in the native AChR. Conversely, when α-, β-, and δ-subunits were coexpressed, cell-surface, pentameric assemblies again formed, with stoichiometry α2βδ2, and they bound dimethyl-d-tubocurarine with a low-affinity dissociation constant that coincided with the low-affinity component in the native AChR. The β-subunit, common to both types of pentamers, emerged as a structural subunit essential for pentamer formation and transport to the cell surface, but it did not contribute to the ligand binding sites.
Photoaffinity labeling of Torpedo AChRs with radiolabeled d-tubocurarine (d-TC) further indicated that the β-subunit was not the subunit between the two α-subunits (206). d-TC was photo-incorporated into all four AChR subunits, but incorporation was inhibited by agonist only in the γ-and δ-subunits. Both the subunit omission and photolabeling studies showed that if the β-subunit was the subunit between the two α-subunits, the α-subunits would be predicted to position back-to-back within the pentameric ring so that each could form an interfacial binding site with the γ- and δ-subunits. However, rotational symmetry of the pentamer, a natural consequence of sequence homology among the subunits, predicted the subunits should position front-to-back. These experiments did not distinguish clockwise from anti-clockwise arrangements of the subunits; this final aspect of quaternary structure emerged with high-resolution structural data.
B. AChR Structure at the Atomic Scale
The era of atomic-scale resolution of the AChR began soon after the new millennium. Unexpectedly, the first atomistic insight emerged from discovery of a water-soluble homolog of the AChR ligand binding domain called AChBP. Produced in the nervous system of the freshwater snail Lymnaea stagnalis, AChBP was released by glial cells local to the synapse and bound nerve-released ACh, providing a novel means for attenuating the synaptic response (253). Cloning of AChBP immediately revealed that it was homologous to the superfamily of Cys-loop receptors, exhibiting 24% sequence identity to the major extracellular region of the neuronal α7 AChR. Because AChBP was water soluble, it was a good candidate for crystallization and structure determination by x-ray diffraction. Ultimately, well-diffracting crystals were formed and x-ray diffraction yielded an electron density map that was refined to a resolution of 2.7 Å (32). AChBP comprised a pentamer of identical subunits, each with an immunoglobulin-like assembly of β-strands that formed inner and outer β-sheets, and it contained many structural hallmarks of nicotinic receptors (FIGURE 4B): a size that coincided with that of the Torpedo AChR extracellular domain at 4.6 Å resolution, the eponymous Cys-loop, ligand binding sites composed of multiple loops at subunit interfaces, conserved aromatic residues at both faces of the ligand binding sites, vicinal cysteine residues at the ligand binding sites, and a main immunogenic region at the top of each subunit. The collective structural features exhibited excellent agreement with years of research on the AChR, including mutagenesis coupled with functional measurements, site-directed labeling, substituted cysteine accessibility studies, and spectroscopic determination of secondary structure (105, 135, 239).
Subsequently, AChBPs from other species of mollusks were discovered and their atomic structures determined (44, 110). The overall set of AChBPs allowed construction of homology models of the ligand binding domains of receptors from the Cys-loop superfamily (156, 252). The resulting models allowed far more precise investigations of structure-function relationships than were possible previously, and allowed application of computational methods such as molecular dynamics simulations to identify rigid and flexible regions, and in silico docking of candidate ligands for drug discovery.
Resolution of the cryo-EM images of the Torpedo AChR soon improved to 4 Å. This was achieved by averaging more images than were used to obtain the 4.6 Å structure, about one million receptors in all, and by implementing a series of image processing advances that corrected for spurious elements of the signal. Initially the method yielded the structure of the pore domain (179), which became visible as a bundle of α-helices, four from each subunit, in which the α-carbon atoms and large side chains could be discerned and identified by the known primary sequences of the subunits. Residues from the second of four hydrophobic regions, called TMD2, lined the pore with hydrophobic side chains and formed a constriction composed of a ring of leucine residues near the center of the membrane, presumed to form the barrier to ion flow. The TMD2s tilted radially, creating a wide opening at the extracellular entrance that narrowed toward the middle of the lipid bilayer and remained narrow at the intracellular end. The three other α-helices from each subunit, TMD1, TMD3, and TMD4, surrounded TMD2, but without forming extensive van der Waals contacts, creating a shield between it and the membrane lipids.
The improved cryo-EM method, along with further advances, resulted in a 4 Å resolution structure of the majority of the Torpedo AChR (271; FIGURE 4C). A major advance was fitting of candidate modules one at a time to the experimental densities. The extracellular module was modeled from the coordinates of the inner and outer β-sheet domains of AChBP, the transmembrane module from the previously determined 4 Å structure, and the intracellular module from tentative assignments in the 4.6 Å structure. The missing regions were then built in later stages of structural refinement. Additional refinement was achieved by cycles of molecular dynamics energy minimization, imposing backbone hydrogen bond restraints in regions of secondary structure, and manual rebuilding of loop regions. The final structure included ~80% of a total of 2,335 residues, but did not include about half of the intracellular loop, the β7-β8 loops from the extracellular domains of the non-α-subunits and the COOH termini of the γ- and δ-subunits.
The emerging 4 Å resolution structure allowed placement of nearly all previously recognized residues of interest in three dimensions. The large extracellular domain in each subunit was composed predominantly of β-strands, as in AChBP, and was joined with the pore domain composed of four α-helices. The pore domain then joined with a long α-helix that formed part of the intracellular domain. Perhaps the most significant structural insight emerged from the manner in which the ligand binding and pore domains joined. This inter-domain interface was previously modeled by connecting a homology model based on AChBP in register with the 4 Å structure of the Torpedo AChR pore domain (152). However, in the cryo-EM structure, the interdomain interface differed substantially from previous models, and for the first time the physical linkages between the two allosterically coupled domains were known. Three loops from the binding domain and the covalent link between the binding and pore domains articulated in a precise fashion with the loop spanning transmembrane domains TMD2 and TMD3 (FIGURE 5), provoking the immediate impression that this assembly of loops constituted an actuator that coupled agonist binding to channel opening. Key residues of this actuator, some of which were conserved, were ultimately tested for their ability to communicate structural changes following agonist binding to the channel.
FIGURE 5.

Interface dividing ligand binding and pore domains of the α-subunit from a homology model of the human AChR generated using the Torpdeo AChR as a template (276). Consecutive residues are highlighted in a single color and labeled. In the left panel, the pore runs vertically along the left side, and in the right panel, the pore is just beneath the β1-β2 loop coming out of the plane of the page.
Atomic resolution insight for Cys-loop receptors has lagged behind that for voltage-gated ion channels (77, 134). The chief reason was that bacterial genome sequences for voltage-gated channels were discovered first. Bacterial proteins could be expressed in their native host cells, allowing production of milligram quantities of protein for structural analyses. Bacterial homologs were also structurally simpler than their eukaryotic counterparts, having a smaller size and lacking glycosylation and disulfide bonds. Prior to the new millennium, however, bacterial homologs had not been identified for any member of the Cys-loop receptor superfamily. Then in 2005, a landmark bioinformatics study identified several classes of Cys-loop receptor homologs from bacterial genomic databases (262). One of these homologs from the cyanobacterium Gloeobacter violaceus, called GLIC, formed proton-activated ion-conductive channels (26), and soon the x-ray structure of another bacterial channel, called ELIC, was solved at a resolution of 3.3 Å (FIGURE 4D) (119). ELIC contains five identical subunits, each with a large extracellular domain of 10 β-strands and a pore domain of four transmembrane α-helices. The second transmembrane α-helix from each subunit, TMD2, formed a hydrophobic ion translocation pathway that was occluded by Phe residues stemming from its upper half, indicating the conformation in the crystal was closed to ions. Conspicuously, instead of the large cytoplasmic domain that linked TMD3 and TMD4 in eukaryotic AChRs, the bacterial channels contained only a short stretch of linkage residues.
After the ELIC structure was published, two high-resolution structures of GLIC were published (25, 118). Unlike the ELIC structure, the GLIC structures were determined at a low pH, a condition that maximally opened the channel. Differences between ELIC in the closed state and GLIC in the presumed open state suggested structural motions underlying channel opening. To open the channel, the large extracellular β-sandwich region appeared to rotate as a rigid body and produced small displacements of this structure at the interface between the extracellular and pore domains. In response to these displacements, the tops of the pore-lining α-helices tilted away from the central pore axis. The intracellular ends of the pore-lining α-helices remained close together, resulting in a funnel-shaped conduit through which ions presumably could flow.
IV. STRUCTURE-MECHANISM RELATIONSHIPS
Studies of structure-mechanism relationships of the AChR span the protein size spectrum from individual amino acid residues to the whole pentamer. In the following sections, functional contributions of individual or groups of residues will be considered together with descriptions of the atomic structures of the corresponding region. Then, structure-mechanism relationships will be considered for larger parts of the receptor.
Both direct and indirect approaches have been used to probe structure-mechanism relationships. Direct approaches include cryo-EM of the native AChR, x-ray crystallography, and NMR spectroscopy of AChBP, in some cases with and without bound agonist. However, most approaches have been indirect and used heterologous expression systems, such as Xenopus oocytes or HEK 293 cells, to produce wild-type and mutant AChRs for comparison of functional properties. Contributions of individual subunits to receptor function have been determined by deliberate omission of subunits (23, 240) or by substituting a subunit from a species of receptor in which the function of interest diverges (148, 222). Chimeric subunits, constructed from segments of subunits with different functional attributes, have been used to identify parts of the subunits responsible for those attributes (126, 238). Functional contributions of individual residues have been probed by substituting natural amino acids, or the essential chemical group within a residue has been probed by substituting unnatural amino acids (196). Covalent modification of substituted residues is a powerful approach and has been most successful with substituted cysteine. Cysteine substitution is often well tolerated, and a wealth of cysteine-reactive methane-thiosulfonate (MTS) reagents have been developed to probe solvent accessibility, serve as spectroscopic indicators of local environment, or directly perturb function.
To assess the consequences of a structural perturbation, combination with a functional measurement is essential. Concentrations of mutated proteins are typically small, from femtomolar to nanomolar, preventing direct determination of the structural change. Despite small quantities, however, alterations in function can be assessed from measurements of radiotracer binding to receptors from cells in suspension, or by electrophysiological or fluorescence measurements of receptors on individual cells. Among single-cell measurements, the most widespread are whole cell voltage-clamp recordings of macroscopic currents and patch-clamp recordings of single-channel currents. Macroscopic recording has the advantage that the signal is usually robust and can be analyzed in a short time, but the ability to decipher the underlying mechanism is limited. Single-channel recordings require large amounts of data collected over extended periods, but analyses of the recordings can give deep insight into mechanism; there are limitations, however, because mechanistic insights from single-channel kinetic analyses are only as reliable as the molecular mechanisms fitted to the data. For example, if an intermediate closed state between closed and open states is present (as in the Flip and Primed mechanisms) but not included in the mechanism fitted to the data (del Castillo and Katz mechanism), the resulting fitted parameters, such as the channel gating rate constants, will be apparent rate constants, encompassing multiple elementary steps.
An experimental approach called mutant cycle analysis (MCA) has been widely used to assess whether pairs of residues are interdependent in contributing to a particular function (122, 123). The first step is to measure a relevant functional parameter for both the mutant and the wild-type receptor, say the channel gating equilibrium constant θ2, and then convert that parameter to a free energy. A founding principle of MCA is that the free energy change due to mutation of a single residue (ΔG1) depends on other residues in the protein. If mutation of a second residue alters the free energy change produced by mutation of the first, yielding a free energy change (ΔG2), the two residues are deemed interdependent with a first-order coupling free energy (ΔΔG) which equals ΔG2 − ΔG1. On the other hand, the contributions of the two residues are independent if ΔG2 − ΔG1 = 0, even though mutations of the two residues may produce large free energy changes on their own. Because the free energy of inter-residue coupling is solely a thermodynamic parameter, coupling could arise through either a direct or an indirect interaction. However, if two residues are strongly coupled and contact each other in the structure, a likely interpretation is that in the context of the surrounding structure, the coupling arises through direct interaction between the two residues. In cases in which inter-residue energetic coupling was determined in parallel with structure determination of the mutants in the cycle, significant coupling between residues correlated with those within 7 Å of each other (228, 273).
In virtually all studies of structure-mechanism relationships, spatial and temporal resolution of the functional measurements is limited and impact interpretation. However, if atomic coordinates of the protein or a model of the protein are available, computational methods based on Newtonian physics can overcome these limitations. All-atom molecular dynamics simulations are considered the gold standard, although atomic polarizability is not included, force fields are approximate, and simulation time is limited to tens to hundreds of nanoseconds, briefer than most biological processes (138). To overcome the time scale limitation, course grained simulation methods have been developed, but with additional approximations (53). Continued increases in computer processor speed and advances in simulation methods and software give hope that the gap will ultimately be bridged between the time scales of practical simulations and those of biological processes.
A. The Ligand Binding Domain
The initial task of the ligand binding domain of the AChR is to bind nerve-released ACh. In the course of binding, however, ACh competes with other positively charged ions such as sodium, potassium, calcium, and its breakdown product choline, as well as polar water molecules. After ACh binds to the resting, inactive state of the AChR, transition to the active state increases affinity for ACh, stabilizing that state. Once the active state forms, conformational changes local to the binding site are relayed to the junction of the binding and pore domains, which in turn triggers opening of the ion channel. Reaction steps that increase ACh affinity and subsequently couple agonist occupancy to channel gating likely comprise multiple intermediate steps and are subjects of intense investigation.
As long ago as 1957, when the response to ACh was found to be positively cooperative (141), the endplate AChR was thought to contain more than one agonist binding site. Evidence for two binding sites emerged with the findings that two α-neurotoxins bound per receptor (191) and the subunit stoichiometry was α2βγδ (163, 220). Further support came from the observation of a 1:1 ratio of α-neurotoxin to ACh binding sites (191). Polypeptides derived from the α-subunit, but not from the non-α-subunits, bound α-neurotoxin (291), suggesting the binding sites were formed entirely by the α-subunits, an idea that prevailed for many years. However, this idea was overturned by the observations that competitive antagonists, such as d-TC, bound with different affinities to the two binding sites and that the different affinities arose from intrinsic structural differences rather than from negative cooperativity (191, 250). Structural differences between the two binding sites were further demonstrated using the monoclonal antibody mAb 383C that targets residues 187–199 of the α-subunit (85). Rather than binding to both α-subunits, the antibody bound only to the α-subunit that forms the site with high affinity for d-TC. The presence of two intrinsically different binding sites was most easily explained if the sites were located at interfaces between a principal α-subunit and a complementary non-α-subunit where the different non-α-subunits contributed different residues to each site. Further evidence for the interface concept came from mutagenesis experiments in which binding affinity for a d-TC analog could be transferred from the high-affinity γ-subunit to the low-affinity δ-subunit by swapping a few residues at positions of homology equivalence (238); these studies also showed that the subunits were built upon a common protein scaffold, an idea suggested by homology among the subunits. Photoaffinity labeling by radiolabeled d-TC, followed by microsequencing, gave further evidence for the subunit interface concept (206).
Long before atomic structures became available, studies of site-directed mutagenesis and affinity labeling defined the amino acid residues at the AChR ligand binding sites. The collective studies showed that the α-subunit formed the principal face of the binding site and contributed residues from three loops, named A, B, and C, each of which localized to separate sections of the primary sequence (239). Loop A contained a conserved Tyr at position 93, loop B a conserved Trp at position 149, and loop C conserved Cys at positions 192 and 193 and conserved Tyr at positions 190 and 198 (TABLE 1). The γ- and δ-subunits formed the complementary faces of the binding sites and contributed residues from four separate sections of the primary sequence, named loops D, E, F, and G. Loop D contained a conserved Trp at positions of homology equivalence γ55 and δ57; loop E a Tyr at γ117 and Thr at δ119; loop F a Phe at γ172 and Ile at δ178; and loop G a Lys at γ34 and Ser at δ36. Given the small sizes of the ligands used to probe the binding sites, the natural question arose of how seven distinct sections of the primary sequence could converge into such a small space. Subsequent atomic structural data confirmed that such a convergence was possible (FIGURE 6).
Table 1.
AChR ligand binding site multiloop structure
| Loop | Residue Span | Secondary Structure | Key Residues | Defining Ligands |
|---|---|---|---|---|
| A | α93-97 | β4-β5 linker | Tyr93, Ala96, Asp97 | Nicotine, ACh |
| B | α148-153 | β7-β8 linker | Trp149, Tyr151, Gly153 | Nicotine, ACh, d-tubocurarine |
| C | α189-200 | β9-β10 linker | Tyr190, Cys192, Cys193, Tyr198, Asp200 | Nicotine, ACh, bromo-ACh, lophotoxin |
| D | ε55-59, δ57-61 | β2-strand | εTrp55, εGly57, εAsp59, δTrp57, δGlu59, δGly61 | d-Tubocurarine, metocurine, nicotine, ACh, NmmI α-toxin, Waglerin I |
| E | ε109-119, δ111-121 | β5′-β6-strands | εLeu109, εTyr111, εVal116, εThr117, εLeu119, δLeu111, δTyr113, δVal118, δTyr119, δLeu121 | α-Conotoxin M1, d-tubocurarine, metocurine, α -bungarotoxin, NmmI α-toxin, Waglerin I |
| F | ε161-183, δ163-187 | β8-β9 linker | εAsp173, εAsp175, δIle178, δAsp180 | α-Conotoxin M1, metocurine, NmmI α-toxin, ACh, Waglerin I |
| G | ε29-45, δ31-47 | β1-strand | εLys34, δAla36 | α-Conotoxin M1, carbamylcholine |
Residue positions correspond to the human endplate AChR.
FIGURE 6.

Ligand binding domain of a homology model of the adult human AChR [276] highlighting the subunit interface and seven discontinuous loops A–C at the principal face and D–G at the complementary face. Each loop is highlighted in a single color.
The atomic structure of AChBP, together with the 4 Å resolution Torpedo structure, set the template for the ligand binding domain of the AChR. Within each subunit this domain contains 10 β-strands, the first 6 of which form an inner β-sheet core, while the next 4 β-strands form an outer β-sheet shell (FIGURE 7). The NH2 terminus begins with an α-helix at the top of the subunit, followed by a linker and strand β1 that spirals part way around and spans the length of the subunit, with successive residue side chains alternating between the protein surface and the hydrophobic core (252). The midpoint of strand β1 localizes to the complementary face of the ligand binding site and forms loop G from which δLys36 contributes to binding of carbamylcholine and α-conotoxin M1 (208, 243). Following the β1-β2 hairpin, strand β2 retraces strand β1 in an anti-parallel β-sheet, giving rise to binding site loop D at the complementary face, from which γTrp55, εIle58, and εAsp59 contribute to binding of d-TC (34, 54), and γTrp55 and its equivalent residue δTrp57 contribute to binding of nicotine (55) and Naja mossambica mossambica α-toxin (204). From the end of strand β2 at the top of the subunit, a short α-helix followed by an extended stretch of residues forms a structure homologous to the main immunogenic region (MIR) of the endplate AChR α-subunit (267). Following the MIR, four short β-strands, β3, β4, β5, and β5′, and intervening linkers encircle the subunit’s midsection facing the central lumen, with the β4-β5 linker forming binding site loop A at the principal face, from which αTyr93 stems to stabilize agonists (60). Next, strand β6 retraces strand β2 in an antiparallel β-sheet, giving rise to binding site loop E at the complementary face from which γTyr117 contributes to binding of d-TC (238), γLeu119 and δLeu 121 contribute to binding of α-bungarotoxin (237), and εTyr115 and δTyr117 contribute to binding of Waglerin-1, a peptide toxin from Wagler’s pit viper (180). Strand β6 then terminates near the bottom of the subunit with the beginning of the signature Cys-loop. Structural elements from strands β1 through β6 constitute the inner β-sheet domain of each subunit.
FIGURE 7.

Secondary structure of the ligand binding domain of the human AChR α-subunit. The two views differ by 90° rotation about an axis passing vertically through the domain.
The 15 residues spanning strands β6 and β7 comprise the Cys-loop, which extends across the bottom of the subunit at a shallow angle to the plane of the membrane. From the end of the Cys-loop, strand β7 extends toward the top of the subunit, giving rise to a short linker to strand β8 that forms a parallel β-sheet with strand β1; the β7-β8 linker forms binding site loop B at the principal face of the subunit from which αTrp149 stems to stabilize quaternary ammonium moieties of agonists and antagonists (45, 110, 298). The linker from strand β8 to β9 is long and extends from the top to the bottom of the subunit, giving rise to binding site loop F at the complementary face from which γSer161 contributes to binding of d-TC (238), δIle176 contributes to binding of α-conotoxin M1 (243), δAsp180 contributes to agonist and competitive antagonist binding (69, 167), εAsp173 contributes to binding of Waglerin-1 (181), and γAsp174 and γGlu176 contribute to binding of Naja mossambica mossambica α-toxin (203). Strand β9 then projects from the bottom to the top of the subunit, giving rise to a hairpin that forms binding site loop C at the principal face that contributes the signature binding site residues αCys192, αCys193, αTyr190 and αTyr198, which contribute to agonist and antagonist binding (202, 245, 264). Finally, strand β10 retraces strand β9 in an anti-parallel β-sheet as it courses to the bottom of the subunit where it concludes with the COOH terminus of the ligand binding domain. Structural elements from strands β7 through β10 constitute the outer β-sheet domain of each subunit.
The ligand binding pocket is lined primarily by aromatic side chains from which π electrons project to stabilize the cationic moiety of ACh (FIGURE 8). Key aromatic residues comprise αTrp149 from loop B, αTyr93 from loop A, αTyr190 and αTyr198 from loop C, and γTrp55 or δTrp57 from loop D. The crystal structure of AChBP with bound carbamylcholine showed that the five aromatic residues, at positions equivalent to those just listed, framed the quaternary ammonium group of the agonist, forming an aromatic cage (45). Of these residues, the residue equivalent to αTrp149 in the AChR appears central, as it presents the greatest contact area to the positive charge on the agonist, and studies of unnatural substitutions showed a linear relationship between EC50 for ACh and calculated π-cation interaction energies (298). Measurements of intrinsic Trp fluorescence of AChBP showed that agonists quench fluorescence by promoting interaction between the Trp equivalent to αTrp149 and that equivalent to εTrp55 and δTrp57 (93, 109). The findings concurred with parallel MD simulations showing that agonist binding reduced the mobility of the Trp pair, promoting an edge to face stacking of the indole rings that facilitated electron transfer and quenching of the fluorescence (93). The findings raised the possibility that agonist-mediated changes in aromatic-aromatic interactions could be important in molecular recognition and possibly downstream events that trigger channel gating. In support, ligand binding measurements and single channel kinetic analyses show that mutation of any of the five key aromatic residues not only affects rate constants for ACh association and dissociation, but also rate constants for channel gating (6, 8, 50).
FIGURE 8.

Aromatic residues (space-filling) that form the ligand binding pocket of AChBP (PDB code 1I9B) with acetylcholine (ball and stick) docked to the site (93).
Inspection of the AChBP crystal structure with different bound agonists showed that the backbone carbonyl group of the residue equivalent to αTrp149 in loop B stabilizes the electron-deficient carbon atom of the agonist vicinal to the quaternary nitrogen atom (28, 45, 110, 260). Furthermore, the residue equivalent to the conserved αAsp89 from loop A, situated behind αTrp149, is positioned to polarize the backbone carbonyl group of αTrp149, increasing the partial negative charge and further stabilize the agonist (45). Mutagenesis combined with single-channel kinetic analyses showed that main chain amide groups in loop B serve as hydrogen bond acceptors for the carboxyl group of αAsp89 in loop A (154), a stabilization that appears to optimize the position of the indole ring of αTrp149 for rapid association of ACh. Substitutions of unnatural amino acids for αAsp89 provided evidence that up to four hydrogen bond donors from loop B, including two main chain amide groups and hydroxyl side chains of the two residues flanking αTrp149, hydrogen bond to the carboxyl group of αAsp89 (42).
In the crystal structure of AChBP with bound carbamylcholine, the ester tail of the agonist projects away from the aromatic cage and approaches loop E at the complementary face of the binding site (45). Key residues from loop E include the AChR equivalent residues γTyr117 and γLeu119, both of which were identified as binding site determinants by mutagenesis combined with measurements of ligand binding (237, 238). Studies of photolabeling of Torpedo AChRs by a partial agonist and an antagonist, followed by micro-sequencing, confirmed that several residues along loop E, including γLeu109, γTyr111, and γTyr117, are physically close to the expected location of the ester tail of the agonist (192, 277).
The aromatic-rich agonist binding site clearly favors an organic cation like ACh (76). However, negatively charged residues are located at the periphery of the aromatic cage, suggesting that as ACh approaches the binding site it competes with inorganic cations in extracellular solution. Single-channel measurements of AChR activation kinetics showed that the rate of ACh association changed by up to threefold depending on the type and concentration of the major extracellular cation, with the rank order Na+ > K+ > Cs+ (7). This dependence on extracellular cations appeared electrostatic in origin, as it was mitigated by charge-neutralization of the conserved εGlu184 at the periphery of the binding site, and was eliminated by charge reversal (8).
In addition to forming the ligand binding site, the extracellular domain forms a central vestibule through which permeating ions pass and thus is a natural candidate for a first pass filter that selects cations over anions prior to translocation. Simulations of single cation translocation showed that translocating cations paused at a ring of negatively charged residues formed by αAsp97, βAsp97, γAsp97, and δAsp99 before continuing through the vestibule (276). Furthermore, charge reversal of three or more residues within this ring reduced the single-channel current amplitude (111). Prolonged pauses of the cation were also observed at two additional rings of charged or polar residues within the extracellular vestibule, corresponding to αAsn47 and αGlu83, suggesting these additional rings may also contribute to a first-pass cation filter.
In the related 5HT3A receptor, rings of Arg residues were identified along the portion of the cytoplasmic α-helix that precedes TMD4, and were found to confer the unusually low single-channel conductance of this receptor (142). Neutralization or charge reversal of the Arg residues increased unitary conductance from undetectable to readily detectable by patch-clamp recording; in the endplate AChR, residues at positions equivalent to the ring of Arg residues are negatively charged or neutral, and likely contribute to its larger unitary conductance. Both the extracellular and intracellular determinants of cation conductance seem to function analogously to the previously identified rings of charged residues that immediately flank TMD2 and contribute to the single-channel conductance (126). Thus ionic filters arranged in series along the ion translocation pathway contribute to cation conductance of the AChR (FIGURE 9).
FIGURE 9.

Rings of charged or polar residues along the ion permeation pathway through a homology model of the adult human AChR (276). The α-subunit is blue. Two subunits facing the viewer are removed for clarity.
All atom molecular dynamics simulations applied to AChBP and homology models of the AChR ligand binding domain revealed both rigid and flexible regions (93, 115, 116). The β-sheet regions, which comprise the majority of the extracellular domain, are very stable and exhibit relatively small fluctuations. Regions of largest flexibility include the hairpin spanning strands β9 and β10 known as the C-loop, the β6-β7 linker known as the Cys-loop, the β8-β9 linker known as loop F, the NH2-terminal α-helix, and the MIR. The mobility of the C-loop is of particular interest because it adopts two main conformations: an uncapped conformation that opens up the binding pocket and a capped conformation that encloses the bound agonist. The simulations also showed that when agonist was bound, fluctuations of the mobile regions were markedly reduced, and the subunits within the pentamer remained symmetrical, which contrasted with the unbound simulation in which the subunits became increasingly asymmetric (93).
B. The Pore Domain
The principal function of the pore domain is to enable rapid translocation of ions in response to agonist occupancy. Ion translocation is determined by the chemical environment of the pore-forming α-helices, and in the AChR this environment is hydrophobic. As a result, the water shell of an ion is not removed prior to translocation, and positively or negatively charged ions are not selected by the pore alone. Paradoxically, rapid ion translocation occurs despite the fact that the channel lining offers no stabilization for ions or water molecules. Studies of model hydrophobic nanopores indicate that small changes in pore diameter can produce large changes in the extent to which water fills the pore, a prerequisite for ion translocation (19, 124). Thus the adaptation to employ hydrophobic residues for rapid translocation offers the possibility that the pore could change from non-conducting to conducting with relatively small changes in its structure.
Once the AChR subunits were cloned, the physical nature of the pore came under intense investigation. The approaches included mutagenesis together with AChR expression in heterologous cells, photo-incorporation of radiolabeled noncompetitive inhibitors, and mutagenesis combined with electrophysiological measurements of pore function. Deletion of the amphipathic helix between M3 and M4 of the α-subunit did not abolish ion conduction (174), excluding this region as the principal component of the pore. On the other hand, deletion of any of the four hydrophobic segments of the α-subunit abolished AChR function. The second of the four hydrophobic segments, TMD2, emerged as a strong candidate for the pore because it was flanked by polar or negatively charged residues, mutations of which altered single-channel conductance (125). Ultimately, a residue numbering convention was established for TMD2 for comparison of sequence positions in different receptor types where −1′ designates the cytoplasmic end of TMD2 and 20′ the extracellular end. Radiolabeled chlorpromazine, a noncompetitive antagonist, covalently labeled a Ser residue at the 6′ position of the δ-subunit (97), and it labeled Ser, Thr, and Leu residues at positions 2′, 6′, and 9′ in the γ-subunit, a spacing suggesting an α-helix (219). The collective findings suggested TMD2 was an α-helix and lined the pore, but other hydrophobic probes could be photo-incorporated into TMD4 with spacing consistent with an α-helix (22).
The nature of the ion channel was also probed using local anesthetics combined with single-channel recording. Local anesthetic inhibition of the AChR was shown to result from physical block of the open channel, based on a decrease in mean channel open time as the local anesthetic concentration was increased, a concentration-dependent increase in the fraction of closed dwell times due to block and voltage dependence of the closed dwell times that indicated a binding site within the transmembrane electric field (190). To probe the pore, mutations were generated along TMD2, and single-channel recording was used to monitor the ability of local anesthetics to block the open channels. Substitution of Ala for a Ser residue at the 6′ position within TMD2 mitigated local anesthetic-induced channel block in a manner that depended on the number of subunits with Ala substitutions (157). Subsequent studies revealed contributions to local anesthetic-induced channel block by a pair of residues at positions 6′ and 10′ within TMD2, a spacing that again suggested an α-helix. Substitutions of the more extracellular 10′ residue showed that it was chemically compatible with the hydrophobic moiety of the local anesthetic, whereas substitutions of the more intracellular 6′ residue showed that it was compatible with the hydrophilic moiety (49). The collective evidence from covalent labeling, mutagenesis, and single-channel recording led to a consensus that TMD2 formed the pathway for ion translocation through the AChR channel.
Subsequent studies assessed the secondary structure of TMD2 through measurements of residue accessibility to small, water-soluble MTS reagents (5). The approach was to mutate one residue at a time to Cys, express the mutant AChRs in heterologous cells, and monitor agonist-mediated responses before and after exposure to the MTS reagent MTSEA. If MTSEA affected the response to applied ACh, the residue was deemed accessible to solvent, and the rate at which the response was altered was a measure of accessibility. In the α-subunit, accessible residues were detected at TMD2 positions −1′, 2′, 3′, 6′, 8′, 9′, 10′, 13′, 16′, and 20′, thus showing a periodicity consistent with an α-helix punctuated by an extended structure near the center (4).
Because the studies were conducted in the absence of agonist, it was surprising that residues along the entire length of TMD2 were accessible to MTSEA; the reagent was larger than a sodium ion, and accessibility should have been limited by the barrier to ion flow. Thus this finding served as motivation to determine the location and nature of the barrier. MTSEA is a primary amine and thus can cross the cell membrane and partition into the cytoplasm, although the partitioning rate was expected to be much lower than the rate of diffusion in extracellular solution. To prevent MTSEA from reacting with Cys residues accessible from the cytoplasm, cysteine was loaded into the whole cell recording pipette and allowed to diffuse into the interior of the cell. In this way, any partitioned MTSEA was quenched, and extracellular application of MTSEA should have reacted only with Cys residues accessible from the outside of the cell. The results revealed that in the resting state the barrier to ion flow was at the extreme cytoplasmic end of TMD2, between residues αT244 and αG240 (positions 2′ and −2′), and further, that the barrier was removed upon addition of agonist (290). The converse experiment applied MTSEA within the cytoplasm, again via the whole cell recording pipette, and measured reactivity of substituted Cys residues before and after extracellular application of ACh. The results confirmed that an agonist-removable gate localized to the cytoplasmic end of TMD2 between residues αG240 and αT244.
The emergence of the resting state structure of the Torpdeo AChR at a resolution of 4 Å allowed comparison with results from mutagenesis approaches to assess pore structure. All but one residue in TMD2 determined to be solvent accessible in the resting state were confirmed to line the ion translocation pathway in the structure. The exception, αLeu250 at position 8′, does not line the channel, but it may have been labeled because it projects into an intra-helical cavity created by the loose association of TMD2 with the surrounding α-helices. Another residue, αVal 259 at position 17′, showed no apparent reactivity to MTSEA, but it was solvent accessible in the structure; αVal 259 may have reacted with MTSEA without effecting receptor function. The emerging profile of the translocation pathway was funnel shaped, with the radius largest at the extracellular end and narrowing at the midsection through the intracellular end, in general agreement with the accessibility studies
Structural changes of the pore upon opening were initially probed by application of ACh to Torpedo AChRs followed by rapid freezing (269). To arrest the AChR channel in the open state it was essential to prevent desensitization. This was accomplished by dropping the AChR-containing membrane tubes into a pool of liquid ethane, but just before reaching the ethane, a mist of ACh was sprayed into the path of the falling tubes, allowing millisecond application times. With the inclusion of electron-dense ferritin particles in the ACh mist, exposure to agonist could be confirmed for each image included in the analysis. Resolution of the structure was 9 Å, sufficient to show that the TMD2 domains were shaped like rods with a bend near the center. Following ACh application, each rod appeared to rotate about an axis through its center and normal to the plane of the membrane, with the resulting lateral displacement of the bend widening the pore.
After the 4 Å resolution Torpedo AChR structure was obtained, structural changes of the pore upon opening were probed by substituting the basic residue lysine one position at a time along TMD2 of the δ-subunit, and then registering the extent of blockade of agonist-mediated single-channel currents (68). At neutral pH, the lysine side chain is protonated, and if it extended into the channel lumen, it would be expected to block current flow to an extent that depended on proximity to the central axis, giving rise to step changes in current amplitude. Because the step changes in current amplitude occurred only during channel openings, the measurements provided a direct indication of the relative proximity of the substituted residue to the central axis in the open state. After substituting lysine at each position of δ-TMD2, a plot of the extent of blockade against residue position exhibited a periodic spacing consistent with an α-helix. Moreover, the observed spacing mirrored the expectations from residue-axis distances along TMD2 in the 4 Å Torpedo AChR structure obtained in the absence of agonist. Analogous experiments, carried out on TMD1 and TMD3, gave similar results: periodic α-helical spacing was observed in which the extent of channel blockade was greatest for residues closest to the central axis in the closed structure (66). In contrast to the proposed rotation of the pore lining α-helices (269), the similarity between the inferred open state structure and the 4 Å resolution structure obtained without agonist indicated that the pore opens with minimal rotations of the α-helices, suggesting a simple dilation mechanism.
The structure of the AChR pore in the open state remains to be determined directly. However, comparison of high-resolution x-ray structures of ELIC in the closed state and GLIC in the presumed open state suggest that upon opening, the radius at the extracellular end of the TMD2 α-helices widens, while the radius at the intracellular end remains little changed (25, 118). Studies of water occupancy in hydrophobic nanotubes showed that small increases in pore diameter could produce large increases in water occupancy and thus the capacity for ion conduction (19, 124), suggesting that the small structural differences between the ELIC and GLIC pores might be sufficient to account for the all-or-none changes in conductance associated with gating of the channels.
Computational approaches have been applied to investigate dynamic changes in the pore during ion translocation. A homology model of the human muscle AChR was built, inserted in an explicit lipid bilayer, and a transmembrane potential imposed (276). All-atom MD simulations revealed translocation of several individual cations through the channel, and suggested three fundamental requirements for ion translocation. First, during translocation of each cation, the TMD2 α-helices moved in a peristaltic manner, tilting back and forth about an axis parallel to and through the center of the bilayer, consistent with a dilation mechanism inferred from studies of channel blockade following lysine substitution. Second, at a physiological membrane potential, cation translocation coincided with a spontaneous change from low to high water occupancy of the hydrophobic channel lumen. Third, simulations in which the protein atoms were held stationary, but ions and water were free to move, showed no ion translocation despite the pore being continuously filled with water and the diameter wide enough to pass a hydrated ion. Thus the following key requirements for ion translocation through the AChR channel emerged: tilting motions of the TMD2s, filling of the pore with water and dynamic fluctuations of the protein.
A paradoxical aspect of the simulations was that the template used to build the homology model of the human AChR was the Torpedo AChR at a resolution of 4 Å in the absence of agonist and with the channel presumably closed. However, in such a nominally closed state, it is possible that a low frequency of single ion translocation events occurs in real AChRs, but such single ion events would not be detectable in patch-clamp recordings owing to limited bandwidth.
C. Interface Between Binding and Pore Domains
The interface between the ligand binding and the pore domain is a structural transition zone where β-sheets from the binding domain merge with the tops of α-helices from the pore domain, and thus comprises loops that connect units of secondary structure (FIGURE 5). The binding domain contributes the β1-β2 loop, the Cys-loop (also called the β6-β7 loop), the β8-β9 loop, and the terminus of the β10 strand, while the pore domain contributes the pre-TMD1 linker and the TMD2-TMD3 loop. The β1-β2 and Cys-loops straddle opposite sides of the TMD2-TMD3 loop, while the β8-β9 loop approaches the β10 strand, the β1-β2 loop, and the principal side of the neighboring subunit. A chimeric receptor, constructed by joining AChBP with the pore domain of the 5-HT3A receptor, was not functional initially, but after substituting residues from the 5-HT3A receptor into the β1-β2, Cys- and β8-β9 loops of AChBP, agonist-mediated currents were obtained (31). The emerging concept was that communication between the binding and pore domains required structural compatibility among all the loops at the interface between the domains. Additional structural elements are present in this region and may also contribute to coupling, including the β10 strand that passes through the interface before joining TMD1 and the top of TMD4 proximal to the Cys-loop.
Although the overall structural fold of the interface is conserved from prokaryotes to eukaryotes, no single residue is absolutely conserved over this phylogenetic span. However, if only α-subunits from eukaryotes are considered, three of the five interface structures contain conserved residues: αArg209 in the β10 strand; αCys128, αCys142, αPro136, αAsp138, and αGln140 in the Cys-loop; and αPro272 in the TMD2-TMD3 loop. If only α-subunits from vertebrates are considered, all five interface structures contain conserved residues in addition to those just listed: αGlu45 and αGln48 in the β1-β2 loop, αPhe135 and αPhe137 in the β6-β7 loop, αGlu175 and αTrp176 in the β8-β9 loop, and αLeu273 in the TMD2-TMD3 loop.
Within the binding-pore interface of the α-subunit, two main inter-residue connectivity pathways contribute to coupling agonist binding to channel gating. The first, named the principal coupling pathway, is present in the cryo-EM Torpedo structure (271) and in crystal structures of the α-subunit ligand binding domain (73) and GLIC (25, 118). The core of the principal coupling pathway is a salt bridge formed by the conserved AChR residues αArg209 from strand β10 and αGlu45 from the β1-β2 loop, which are situated within the hydrophobic interior of the subunit. This inherently strong electrostatic interaction links strand β10 to the β1-β2 loop and positions that loop in register with the TMD2-TMD3 loop (FIGURE 10). Charge-reversal of either αArg209 or αGlu45 sharply attenuated agonist-mediated channel gating, but charge-reversal of both residues simultaneously restored channel gating to normal (155). Additionally, in the β1-β2 loop, αVal46 showed energetic coupling to αPro272 of the M2-M3 loop. Thus, within the principal coupling pathway, a series of physically contiguous residues show energetic coupling, thus linking the β10 strand, via the β1-β2 loop, to the M2-M3 loop at the top of the pore.
FIGURE 10.

Interface dividing ligand binding and pore domains in the α-subunit from the Torpedo AChR (PDB code 2BG9) showing energetically coupled residues from the principal (155) (top) and secondary (153) (bottom) coupling pathways.
Both the initial identification of the principal coupling pathway (155) and subsequent studies (209, 293) suggested that additional residues contribute to the pathway. Whereas all mutations of αArg209 markedly reduced ACh-mediated channel gating, certain mutations of its electrostatic partner, αGlu45, showed smaller reductions of gating. For example, whereas the mutation αE45K reduced θ2 to 0.2, compared with a wild-type value of 27, the mutant αE45A reduced θ2 to only 2.1 (155). The mutation αE45A removes a negative charge, but it is possible a nearby negatively charged residue compensated for its removal. Using macroscopic current recordings, a comprehensive study of charged residues at the binding-pore interface interface suggested that the overall pattern of charged residues, rather than their specific pairwise inter-residue interactions, affected coupling between the binding and pore domains (293). In particular, substitution of Lys for the universally conserved αAsp138 within the Cys-loop maintained receptor expression, as shown by retention of α-bungarotoxin binding to the cell surface, but agonist-mediated currents could not be detected. However, combining the mutation αD138K with either of the charge-reversal mutations, αK276D in the TMD2-TMD3 linker or αR429D at the end of TMD4, restored agonist-mediated currents. In the Torpedo AChR structure at 4 Å resolution, these compensating residues are located 10 and 14 Å away from αAsp138, preventing direct inter-residue interactions. The collective findings suggested that additional residues, beyond those initially identified, contribute to the principal coupling pathway.
Candidates for these additional coupling residues are suggested by the high-resolution GLIC and ELIC structures (25, 118, 119). In those structures, the residue at the position equivalent to αArg209 forms an electrostatic contact with the negatively charged residue at the position equivalent to αAsp138 from the β6-β7 loop, in addition to contacting either the residue equivalent to αGlu45 in the β1-β2 loop (in GLIC) or that equivalent to αGlu175 from the β8-β9 loop (in ELIC). These additional electrostatic contacts were not apparent in the 4 Å Torpedo structure, but were present in the 1.9 Å crystal structure of the α-subunit ligand binding domain bound to α-bungarotoxin (73). Thus negatively charged residues from up to three different loops in the AChR ligand binding domain may potentially converge upon αArg209 from the β10 strand, forming a tight assembly that articulates with the TMD2-TMD3 loop (FIGURE 11). Whether αAsp138 or αGlu175 contribute structurally or functionally to the principal coupling pathway remains to be investigated.
FIGURE 11.

Key residues from the ligand binding domain that juxtapose the pore domain: comparison of the AChR α1 crystal structure (PDB code 2QC1; Ref. 73), GLIC (PDB code 3EHZ; Ref. 118), and ELIC (PDB code 2VI0; Ref. 119).
In addition to the principal coupling pathway, a secondary pathway also contributes to coupling of agonist binding to channel gating. It comprises residues within the Cys-loop, the terminus of the β10 strand, and the TMD2-TMD3 linker (FIGURE 10). In the 4 Å Torpedo AChR structure, residues αVal132, αPhe135, αPro136, and αPhe137 in the Cys-loop contact residues αPro272 and αLeu273 in the TMD2-TMD3 loop and αLeu210 in the β10 strand. Studies of inter-residue energetic coupling among these residues, based on the channel gating equilibrium constant θ2 in the del Castillo and Katz mechanism, show that these residues form a network of strongly coupled residues that mirrors inter-residue contacts in the structure (153). Because the principal and secondary coupling pathways unite all the loops at the binding-pore interface, they together may constitute a grand coupling pathway that functionally couples the ligand binding to the pore domain.
D. Changes in Global Quaternary Structure
Allosteric interactions are central to activation of the end-plate AChR. Originally introduced in the context of oligomeric enzymes with multiple activator binding sites, the term allosteric originates from the Greek words allo, meaning other, and stereos, meaning space. Applied to the AChR, allosteric refers to the fact that the end point of biological activity, the ion channel, is spatially distinct from the agonist binding site. Allostery in AChR activation thus begins with small, localized perturbations at the subunit interfaces that form the agonist binding sites, and concludes with structural changes in the remote ion channel. These structural changes include changes in tertiary structures within the subunits as well as changes in quaternary structures among the subunits.
A central feature of endplate AChR quaternary structure is that the agonist binding sites form at only two of the five subunit interfaces. However, the founding ancestral AChR was homomeric and thus contained five identical subunit interfaces and five agonist binding sites. Natural questions arose of why the evolutionary winnowing of binding site interfaces stopped at two, and why nonconsecutive positioning of the sites was preferred over consecutive positioning. These questions were addressed using a homomeric neuronal α7 receptor chimera in which one to five agonist binding sites were disabled (218). In a method called “electrical finger printing,” a subunit bearing a mutation that disabled the binding site was tagged with a second mutation that altered single-channel conductance. After co-expressing mutant and nonmutant subunits, recordings of agonist-activated channel opening events revealed five different current amplitudes arising from receptors with one to five mutant subunits per pentamer. Moreover, the current amplitude of each channel opening event gave a simultaneous measure of the number of disabled binding sites, while the dwell time of the corresponding channel opening event indicated the stability of the open state. The findings showed that maximal open state stability was achieved with receptors in which three of five binding sites were intact, and one of the three sites was located at a nonconsecutive subunit interface. With two intact binding sites, the stability of the open state was reduced compared with three sites, and again the two intact sites had to be located at nonconsecutive subunit interfaces. However, with two consecutive or one intact binding site, the open state was markedly destabilized as indicated by very brief channel openings. Thus at the level of quaternary structure of the endplate AChR, formation of a stable open state requires at least two agonist binding sites and dispersal of the sites at nonconsecutive subunit interfaces.
Agonist-mediated changes in quaternary structure have been directly investigated by cryo-EM of Torpedo AChRs in native tubular membranes subjected to a brief (5 ms) pulse of ACh followed by rapid freezing (269, 272). The resulting structures, determined at a resolution of 4.6 Å, revealed changes in the major extracellular domain, both within and between the subunits. Without agonist, the overall extracellular domain was asymmetric, with the β-, γ-, and δ-subunits adopting similar compact structures, but the two α-subunits adopted structures in which the region encompassing the C-loop protruded peripherally from the binding sites. Following rapid agonist application, however, the α-subunits no longer showed protrusions and all five subunits adopted similar compact structures, giving an overall symmetrical structure. Comparison of the symmetrical AChBP structure to that of the AChR extracellular domain showed that the change in conformation of the α-subunits resulted from rotation of the inner core of β-sheets and contraction of the C-loop toward the center of the binding pocket.
Agonist-mediated changes in quaternary structure were also sought using all atom MD simulations of AChBP and of homology models of the AChR. Simulations of AChBP, approaching 50 ns, compared structural dynamics with and without bound ACh (93). For both simulation conditions, the starting structure was the crystal structure of AChBP obtained with HEPES buffer within the binding sites (32), but with the HEPES removed for simulation. The simulations began with AChBP in a symmetrical conformation in which the five subunits were approximately equivalent. In the simulation without ACh, the pentamer achieved an asymmetrical conformation in which the C-loops in two of the subunits were displaced peripherally, whereas in the simulation with ACh bound, the structure remained unchanged and symmetrical throughout. Analogous observations of asymmetrical agonist-free and symmetrical agonist-bound structures were obtained with MD simulations of models of the homomeric neuronal α7 ligand binding domain (115, 116) and of the whole α7 AChR (152). The overall findings from all-atom MD simulations paralleled the experimentally observed changes of the Torpedo AChR at low resolution (272), showing that binding of agonist induces a change in quaternary structure from asymmetric to symmetric.
Changes in AChR quaternary structure have been further addressed by normal mode analysis (NMA). NMA is a course-grained computational method in which an elastic system, subjected to excitation, exhibits characteristic vibrations, or normal modes (37, 158). Each normal mode frequency corresponds to motions of different-sized structures, with low-frequency modes corresponding to motions of the quaternary structure and high-frequency modes to motions of individual residues. To reduce computational demand, atomic coordinates of the protein are usually reduced to the α-carbon atoms, an approximation thought to have minor impact on protein elastic properties.
NMA was applied to homology models of the Torpedo and homomeric neuronal α AChRs with attention to large-scale changes in quaternary structure associated with opening and closing of the ion conductive pore. In all studies, the lowest frequency mode exhibited a twist to open motion in which the extracellular domain twisted clockwise, the pore domain twisted counterclockwise, and the diameter of the pore increased from ~7 to 9 Å (51, 225, 261), sufficient for passage of a hydrated sodium ion. In two studies, the increase in pore diameter occurred through a simple dilation of the TMD2 α-helices (225, 261), whereas in a third study the α-helices rotated about their axes (51). The overall quaternary twist was accompanied by rotation of the inner core of β-sheets of the extracellular domains, in qualitative accord with agonist-mediated rotations of the α-subunit extracellular regions in the Torpedo AChR (272). Comparison of the starting and ending structures showed that movement of the inner β-sheets of the extracellular domain correlated best with motions of TMD2, but not with motions of the other TMDs, suggesting that channel opening resulted from concerted motions of the two structures. Additional correlated motions were observed at the interface between ligand binding and pore domains, including correlations between the β1-β2, Cys- and TMD2-TMD3 loops, suggesting interactions among these structures also accompanied channel opening.
E. Changes Within Subunits
1. REFER analysis
A wide variety of single residue mutations of the AChR alter the terminal channel gating step, defined by the equilibrium constant θ2 within the extended del Castillo and Katz mechanism. A change in θ2 could occur through changes in either the rate constant for channel opening β2 or the rate constant for channel closing α2. In the hypothetical case where a mutation altered only β2, the difference in free energy between the resting closed state and the transition state is changed, whereas the difference in free energy between the transition state and the open state is not changed. The traditional interpretation is that at the site of the mutation, the structure is the same in the transition and the open state, and in the course of channel opening the structural change occurs solely between the closed and the transition state. Conversely, if only α2 changed, the structure at the site of the mutation is the same in the resting closed state and the transition state, and in the process of channel opening the structural change occurs between the transition and the open state. Furthermore, if a series of mutations of a given residue resulted in the same relative changes in β2 and θ2, a plot of logβ2 against logθ2 would yield a straight line with slope Φ, which is known as a linear rate-equilibrium free energy relationship (REFER; Refs. 88, 131). For the mutated region, the value of Φ is taken as a measure of the structural similarity of the transition state compared with the closed or open states.
In an extensive series of REFER analyses, Φ values were mapped upon the cryo-EM structure of the AChR (39, 46, 47, 67, 104, 133, 175, 176, 209, 210). The initial studies found that for mutations of residues near the agonist binding site, Φ was close to 1.0, but for mutations near the pore, Φ approached 0.2–0.3. Thus the studies concluded that at the binding site the structural change associated with channel opening was complete on attaining the transition state, whereas the structural change at the pore occurred primarily after exiting the transition state. Thus agonist-mediated channel opening was proposed to result from a wave of conformational change that propagated from the binding site to the pore (104). Further analyses revealed that residues with similar Φ values congregated in contiguous groups within the AChR structure (67, 210), suggesting that the postulated wave of conformational change resulted from segmental motions among the groups, starting with the group encompassing the C-loop at the agonist binding site and concluding with the group lining the pore.
An important consideration in interpreting REFER analyses is that most of this work was done before intermediate primed or flip states were discovered. Thus the fitted parameters β2, α2, and θ2, corresponding to the terminal step in the extended del Castillo and Katz model, are apparent rate or equilibrium constants with contributions from both the priming/flipping and channel gating steps. So the resulting Φ values correlate one or more of these microscopic steps with structural information.
F. Role of the C-Loop in Agonist Recognition and Initial Triggering
The role of the C-loop in AChR activation has been addressed using a variety of approaches, including all atom MD simulations, solution NMR, and x-ray structures of AChBP. The collective studies concluded that the C-loop was mobile and that in the absence of agonist it adopted an opened up, uncapped conformation, whereas with agonist bound, it adopted a closed in, capped conformation (45, 93, 95, 110) (FIGURE 12). When AChBP in solution was subjected to hydrogen-deuterium exchange, subsequent mass spectrometry showed increases in isotopic exchange within the C-loop compared with the NH2− and COOH-terminal regions of the subunit (234). Furthermore, exchange within the C-loop was reduced by AChR agonists and antagonists, supporting a change from an uncapped to a capped conformation. The x-ray structure of AChBP with bound α-cobratoxin showed that the toxin wrapped around the C-loop and maintained it in the uncapped conformation (29). An analogous interaction of α-bungarotoxin with the C-loop could potentially explain why binding of the toxin, that presumably arrested the C-loop in an uncapped conformation, suppressed spontaneous opening of the AChR channel (128).
FIGURE 12.

Comparison of AChBP ligand binding site without (yellow ribbons) and with (blue ribbons) bound ACh following prolonged all atom MD simulation (93). ACh is shown in space-filling representation.
By probing interactions between residues in the C-loop and the neighboring β7 strand, insights emerged into the earliest structural changes that trigger channel opening. Measurements of inter-residue energetic coupling, based on the measurements of the channel gating equilibrium constant for a series of mutant AChRs, revealed strong coupling between αTyr190 of the C-loop and αLys145 of strand β7. In addition, αLys145 coupled strongly to αAsp200 at the end of the C-loop (184). These inter-residue couplings mirrored inter-residue contacts in both the apo and agonist-bound structures of AChBP (32, 45). The collective studies concluded that change of the C-loop from an uncapped to a capped conformation is a key initiator of the chain of events that open the ion channel.
To further test whether capping of the C-loop initiates AChR channel opening, a free Cys was introduced at the tip of each C-loop and another Cys was introduced in each of the two non-α-subunits that form the complementary faces of the binding sites (185). In the absence of ACh, chemical oxidation of the Cys pair was expected to form a covalent bond, cap the C-loop, and open the channel. This expectation was not met when the Cys pair was introduced into the wild-type AChR, presumably because in the uncapped conformation the Cys-to-Cys spacing was too great for covalent reaction. However, the expectation was met in AChRs with Leu to Ser substitutions in the center of TMD2 that increased spontaneous opening of the channel; presumably the bidirectional nature of the link between the binding site and the channel increased spontaneous capping of the C-loop and thus promoted covalent reaction. The specificity of chemical oxidation on AChR function was demonstrated by reversal of channel opening with DTT and was further demonstrated by the requirement of the Cys pair at both agonist binding sites.
Steered MD (SMD) simulations further addressed the relationship between capping of the C-loop and channel opening (52, 280). SMD simulation begins with a protein of interest for which initial and targeted structures have been defined. During MD simulation, force is applied to specified atoms in the initial structure, while interatomic distances between the initial and targeted structures are monitored; atoms not subjected to force are allowed to undergo unbiased dynamic fluctuations. Once the specified atoms achieve the coordinates of the targeted structure, the simulation is complete. In an application of SMD to the AChR, the initial structure was a homology model of the human AChR based on the Torpedo AChR structure at a resolution of 4 Å, while in the target structure the extracellular domain was modeled using AChBP with bound carbamylcholine (45) as the template. Force was applied to the five aromatic side chains at each binding site, αTyr93, αTrp149, αTyr190, αTyr198, εTrp55, and δTrp57 (FIGURE 13). After the simulation was complete, the initial and final structures were compared for their ability to conduct sodium ions using a coarse-grained simulation method known as BioMOCA (71). The initial structure showed low conductance for sodium ions, as expected, but the final structure showed increased sodium ion conductance. Moreover, the difference in conductance between the two structures was close to the experimentally measured unitary conductance of the human AChR (280). Thus a variety of experimental approaches concluded that capping of the C-loop initiates the chain of events that open the AChR channel.
FIGURE 13.

Steered MD simulation of human AChR homology model beginning with the apo conformation (aromatic residues in red), using PDB code 2BG9 as a template, and ending with the agonist bound conformation (aromatic residues in yellow), using PDB code 1UV6 as a template. Arrows indicate direction of force application. Large inset shows TMD2s before (blue) and after (yellow) simulation. [From Wang et al. (280).]
Although individual steps within the binding-gating transduction process have been defined, the overall picture is only partially complete. The weight of evidence indicates that in the resting state the binding pockets maintain an open conformation with uncapped C-loops and the five aromatic residues in each binding pocket accessible to agonist. The uncapped conformation of the C-loops is stabilized by electrostatic interactions between the conserved residues αLys145 and αAsp200 from strands β7 and β10, respectively (184). When agonist enters the electron-rich pocket, the five conserved aromatic side chains coordinate about the charged nitrogen moiety of the agonist and the C-loop is drawn into the capped conformation (45). In the course of C-loop capping, the interaction between αLys145 and αAsp200 weakens, and a new electrostatic interaction is established between αTyr190 of the C-loop and αLys145 of strand β7 (45), resulting in positional shifts of strands β10 and β7. These shifts likely propagate to the ends of the β-strands at the interface separating the binding and pore domains. αArg209 that stems from strand β10 makes electrostatic contact with αGlu45 from the β1-β2 loop (155) and possibly αGlu175 from the β8-β9 loop and αAsp138 from the Cys-loop (β6-β7), stabilizing this assembly of loops that articulates with the TMD2-TMD3 loop at the top of the pore. Thus relayed through αArg209, structural changes at the binding site are communicated to the channel.
V. PHARMACOLOGY
Our understanding of how agonists and competitive antagonists interact with the AChR is approaching the atomic scale. Structures of the agonist binding sites in the Torpedo AChR have been determined at a resolution of 4 Å and in several species of AChBP at resolutions as low as 2 Å. A structurally diverse array of full and partial agonists is known, with affinities from hundreds of nanomolar to tens of micromolar, and for many of these, high-resolution crystalline and solution structures have been determined (FIGURE 14). Structures of competitive antagonists are more diverse and include the alkaloids curare and toxiferin, the synthetic compounds gallamine and pancuronium, the cone snail α-conotoxins, the snake α-toxins, the Waglerin snake toxins, and the cyclic diterpinoid from coral lophotoxin; ligand affinities range from subnanomolar to micromolar, with high-resolution structures available for many antagonists. Crystal structures of AChBP with bound agonists or antagonists have been solved, giving insights into primary stabilizing interactions. The action of agonists on the AChR has been interpreted according to the classical del Castillo and Katz mechanism, and more recently according to the Flip and Primed mechanisms, while the action of competitive antagonists can be interpreted in light of either mechanism.
FIGURE 14.

Structures of AChR agonists and antagonists.
A. Agonists
The structure of ACh has been determined in both crystalline and solution forms. The C-C bond within the N+-C-C-O- grouping can rotate freely, but in both the crystal and solution structures, the gauche conformation predominates over the trans conformation (40, 256). In the gauche conformation, the reduced distance of 3.3 Å between quaternary nitrogen and ether oxygen atoms increases intramolecular electrostatic stabilization, promoting that conformation. However, substituting sulfur or selenium for the ether oxygen atom results in absolute preference for the trans conformation (166, 231); steric crowding by the larger sulfur or selenium and changes in electron distribution likely offset electrostatic attraction to the quaternary nitrogen atom. Acetylthiocholine and acetylselenocholine are extremely weak agonists (229), suggesting the gauche conformation is essential for bioactivity. Solid-state NMR measurements showed that when bound to the Torpedo AChR, ACh adopted the same conformation as in the crystal (289), suggesting ACh rearranged minimally when bound to its target.
Both organic and inorganic cations bind to aromatic rings with high enthalpy (76). Because a permanently charged or chargeable nitrogen atom is present in every AChR agonist, the aromatic-rich binding pocket provides a natural source of stabilization through π-cation interactions. In the co-crystal structure of AChBP with carbamylcholine (45), the quaternary nitrogen atom lodges over Trp143 from the principal face of the binding site and is framed by Tyr89, Tyr185, and Tyr188 from the principal face and Trp53 from the complementary face. Similarly, in the structure of AChBP with bound nicotine, the tertiary nitrogen atom of the pyrrolidine ring lodges in a similar location within the aromatic pocket. Thus the center of the aromatic pocket, framed by five conserved aromatic residues, serves as the coordination site for the charged nitrogen atom of agonists (FIGURE 8).
The central element of an AChR agonist is a charged or chargeable nitrogen atom. The structurally simple agonist TMA is remarkably efficacious (150), which can be rationalized in terms of the cation coordination site within the aromatic binding pocket. However, to achieve greater efficacy, one of the four moieties on the quaternary nitrogen atom has additional requirements. Substitution of ethanol for one methyl group on TMA yields choline, which paradoxically is far less efficacious than TMA (151). A larger or chemically different substitution is apparently required, which in the case of ACh is ethyl acetate. When the crystal structure of ACh was computationally docked into the binding site of AChBP, the terminal ester moiety aligned between the tip of the C-loop at the principal face and the complementary face (93, 289). Residues stemming from the complementary face, however, are not conserved among AChRs; in AChBP the residue of closest approach is Leu112, whereas residues at equivalent positions in AChRs are Ser, Thr, or Tyr. Sequence variability at the complementary face may contribute to differences in ACh affinities among different species of AChR or between the two sites in a given species.
When a methyl group on TMA is replaced by a sufficiently large and polar group, agonist efficacy can increase beyond that of choline. For example, whereas the bis-quaternary ligand hexamethonium, with the structure (CH3)3N+-(CH2)n-N+(CH3)3 and n = 6, is an antagonist, decamethonium with n = 10 is a weak agonist. Similarly, the bis-choline ester succinylcholine, with the structure (CH3)3N+CH2CH2OCO-(CH2)n-COOCH2CH2N+(CH3)3 and n = 2, is a weak, low-affinity agonist (succinylcholine), but suberyldicholine with n = 6 is a strong, high-affinity agonist (41). In suberyldicholine, the inter-quaternary distance of 18 Å is long enough so that with one end lodged in the aromatic pocket at the principal face, the other end can extend to the complementary face.
In addition to linear chain agonists, a variety of heterocyclic alkaloids are partial agonists of the AChR. The prototypical heterocyclic alkaloid is the eponymous (−)-nicotine, which contains pyridine and pyrrolidine rings connected by a single carbon-carbon bond. The two rings in nicotine are free to rotate, but the conformation in the crystal structure shows a torsion angle of 119° about the inter-ring carbon-carbon bond (15). The nitrogen atoms in the two rings are separated by 4.8 Å, somewhat longer than the 3.3 Å separation distance between quaternary ammonium and carbonyl oxygen groups in the gauche conformation of ACh. In solution, (−)-nicotine adopts a similar conformation to that when bound to crystals of AChBP (45).
Heterocyclic alkaloid agonists from other natural sources are structurally similar to nicotine, including cytisine from the seeds of Cytisus laburnum, epibatidine from the Ecuadoran frog Epipedobates tricolor, and anatoxin from cyanobacteria. Structures of all three alkaloids have been determined, and common elements of their structures can be superimposed upon the nicotine scaffold (16). In the crystal structure of AChBP with bound epibatidine, the ring with the bridging nitrogen atom in epibatidine is framed by aromatic residues from the principal face of the binding site (FIGURE 15), while the pyridine ring contacts the complementary face via a bridging water molecule (110). In both the nicotine and epibatidine complexes, the chargeable nitrogen atom is stabilized by both π-electrons of Trp143 and its main chain carbonyl oxygen atom. For heterocyclic alkaloids, the emerging pharmacophore comprises a pair of ring systems that are not coplanar and an inter-nitrogen distance of ~4.5 Å (233).
FIGURE 15.

Orientation of epibatidine bound to Aplysia AChBP (PDB code 2BYQ; Ref. 110). Left: principal and complementary faces are shown as ribbons, while epibatidine is shown as space-filling. Right: residues of closest approach in AChBP are shown as space-filling (Trp143, Tyr185, Tyr192, Trp53, Met114) and epibatidine as ball and stick.
Mechanistic descriptions of activation by full and partial agonists are currently in flux. When interpreted according to the extended del Castillo and Katz mechanism, full agonists promote a large extent of channel opening, defined by the equilibrium constant θ2, whereas partial agonists promote a smaller extent of channel opening with a smaller θ2. However, with the recent identification of a closed state intermediate, called Flip or Primed, between resting closed and open channel states (150, 185), it is clear that when the extended del Castillo and Katz mechanism is fitted to sequences of single-channel currents, the result is an apparent value for θ2 rather than the true value. However, according to either the Primed or Flip mechanisms, the apparent channel gating equilibrium constant θ2apparent equals θ2P/(1 + P), where θ2 is the true channel gating equilibrium constant and P is the priming equilibrium constant. The flip equilibrium constant F, analogous to P, has been determined for ACh, TMA, and choline and was found to decrease as agonist efficacy decreased (150, 151). This correlation between F and agonist efficacy suggested that the classical del Castillo and Katz interpretation of partial agonism is over-simplified and that the Flip and Primed models are more realistic; however, further evaluation of these more recent mechanisms is required using the full spectrum of AChR agonists.
B. Antagonists
Competitive antagonists lie at the opposite end of the efficacy spectrum. Although competitive antagonists fully occupy the AChR binding sites, they are poor at promoting the channel gating reaction. According to the del Castillo and Katz mechanism, a competitive antagonist has a vanishingly small channel gating equilibrium constant θ2, whereas according to the Primed or Flip mechanisms, the equilibrium constants P or F are small. Regardless of the mechanism, however, substantial evidence shows that occupancy of one site by agonist opens the channel with low probability, whereas occupancy of two sites opens the channel with highest probability (250). From the perspective of a macroscopic response, occupancy of both binding sites by an agonist is required to elicit a full response, while occupancy of only one site by an antagonist is sufficient to block the response to agonist. If the two binding sites have equal affinity for the antagonist, the measured EC50 for functional antagonism is lower than the intrinsic dissociation constant for antagonist binding, whereas if the sites have very different affinity, the EC50 for functional antagonism approaches the intrinsic dissociation constant of the binding site with highest affinity. Here functional antagonism corresponds to steady-state exposure of the antagonist followed by application of agonist for a time sufficient to occupy the sites not occupied by antagonist, but not long enough for antagonist and agonist to reach steady state. Because the two binding sites on the AChR are formed at α-δ and either α-ε or α-γ subunit interfaces, a given antagonist may or may not distinguish between the two binding sites, and the measured EC50 for functional antagonism can differ from the antagonist concentration that occupies half of the binding sites. In fact, studies of competitive antagonists were among the first to show that the two AChR binding sites were not equivalent owing to their formation at subunit interfaces (191, 250).
d-TC is the prototypical competitive antagonist of the end-plate AChR and is produced by the vines of Chondrodenron tomentosum found the rain forest canopy in regions of South America. At the level of single-channel currents, d-TC is a very weak agonist, opening single AChR channels with <1% the efficacy of ACh (183, 258), but at the level of macroscopic currents, it is a nondepolarizing, competitive antagonist (90, 132). The d-TC scaffold is rigid, containing four interconnected six-member rings and a quaternary and a tertiary nitrogen atom separated by 11 Å; in the synthetic derivative metocurine, both nitrogen atoms are quaternary (59, 254). One quaternary nitrogen atom lodges within the aromatic-rich region near the principal face of the binding site, while the other nitrogen extends toward the complementary face (FIGURE 16). Owing to structural differences in the δ-, γ- and ε-subunits, both d-TC and metocurine distinguish between the two AChR binding sites, binding with affinities that differ by nearly 100-fold. In the fetal AChR, site-selectivity of metocurine originates from different contributions of the γ- and δ-subunits (238) owing to different residues at equivalent positions of sequence homology. In the adult AChR, site-selectivity also arises from different contributions of the ε- and δ-subunits, but the residues that determine selectivity localize to different microregions of the complementary face of the binding site (34).
FIGURE 16.

Orientation of d-tubocurarine bound to AChBP (92). Left: principal subunit is yellow and the complementary subunit blue, while d-tubocurarine is shown as space-filling. Right: key contact residues are shown as space-filling (Y192, Y89, W143, M114, L112), while d-tubocurarine is shown as ball and stick.
The distinction between d-TC and metocurine is important regarding binding to a given AChR binding site. Methylation of d-TC to give metocurine produces relatively small changes in the structure, but for both AChBP and the AChR, the two congeners bind in distinctly different orientations (92, 278). The initial evidence for different orientations emerged from molecular docking simulations, which predicted distinct contact residues for each congener as a consequence of their different orientations; these predictions were confirmed by mutation of the specified contact residues followed by measurements of binding affinity for each congener. Given the lack of conserved residues at the complementary face of the binding site, the findings further suggested that a given antagonist may bind in different orientations to AChRs from different species or to different subtypes of AChRs from the same species. Divergence in the complementary face of the binding site may explain the observation that one group of residues at the complemantary face confers metocurine selectivity for the fetal receptor, whereas a different group of complementary face residues confers selectivity for the adult receptor (34, 238).
In the muscle AChR, the α-ε and α-γ sites bind d-TC with high affinity, whereas the α-δ site binds with up to 100-fold lower affinity (23, 191, 240, 250). Competitive antagonism is thus explained by a one-site occupancy model in which the measured EC50 for functional antagonism approaches the intrinsic Kd for binding to the high-affinity site. Measurements of the kinetics of onset of functional antagonism by d-TC reveal an association rate constant of 1.2 × 108 M−1·s−1 (288), lower than the diffusion limit, but similar to that of ACh. Measurements of the kinetics of recovery from functional antagonism reveal a slow rate constant for dissociation of 6 s−1, indicating d-TC forms a stable complex with the AChR that far outlasts the endplate potential.
α-Conotoxins are peptide neurotoxins from the venoms of carnivorous marine snails and bind to and competitively antagonize AChRs. α-Conotoxins GI, MI, and SI are the prototypical toxins that target endplate AChRs; they contain two disulfide bonded loops, the first of which brackets three non-cysteine residues while the second brackets five noncysteine residues (99, 100, 295); as a result, they are called 3/5 α-conotoxins. Analogously to d-TC, the α-conotoxins bind with different affinities to the two sites on the endplate AChR, with the degree of site-selectivity depending on the type of α-conotoxin and subtype and species of AChR. For example, α-conotoxin M1 distinguishes between the two sites on the fetal mouse AChR with affinities that differ by almost 10,000-fold (145), with the α-δ site exhibiting high affinity and the α-γ site low affinity. The complex between α-conotoxin M1 and the α-δ site is unusually stable, showing negligible dissociation over the course of 7 h (207). By further analogy to d-TC, site-selectivity of α-conotoxin M1 can be transferred from the α-γ to the α-δ site by swapping just a few residues at regions of homology equivalence between the γ- and δ-subunits (243). d-TC is an alkaloid and α-conotoxin MI is a peptide, so it is not surprising that d-TC prefers the α-γ site, whereas α-conotoxin MI prefers the α-δ site. Accordingly, residues that confer site-selectivity for d-TC and α-conotoxin MI localize to different microregions at the complementary faces of the binding sites. In fact, one of the three selectivity determinants for α-conotoxin MI provided the first evidence for a seventh binding site loop, called loop G, within strand β1 at the complementary face of the AChR binding site (243).
α-Conotoxin MI folds into a compact structure in which three lobes required for bioactivity localize to one contiguous region (98). These lobes contain the hydrophobic residues Ala, Pro, and Tyr, while the remaining surface of the toxin is hydrophilic. Double-mutant cycle analyses, based on mutations in both the toxin and the AChR α-δ binding site, revealed strong inter-residue coupling between the Ala of the toxin and residues at the principal face of the binding site, and between the Tyr and Pro of the toxin and residues at the complementary face (33). The large coupling energy between the Tyr of the toxin and the binding site was noteworthy because the coupling involved all three determinants in the δ-subunit that together conferred selectivity of α-conotoxin MI for the α-δ binding site. In contrast, the hydrophilic residues contribute only modestly to α-conotoxin MI affinity, with mutations of individual residues reducing affinity by 10-fold or less; these residues may increase solubility or promote transit through the hydrophilic periphery of the binding site. The overall studies showed that α-conotoxin MI antagonizes AChR function through hydrophobic contacts established at the α-δ binding site interface.
The orientation of α-conotoxins at the ligand binding site was initially addressed using double-mutant cycle analyses and subsequently by crystal structures of AChBP in complex with the same α-conotoxins. These studies represent model studies for the endplate AChR because homomeric rather than heteromeric ligand binding sites were studied, and residues at the binding sites, although homologous, differed in many cases. For α-conotoxin ImI, a 4/3 α-conotoxin, strong inter-residue coupling was observed between the toxin residue Arg7 and the residue in the homomeric α7 AChR equivalent to αTyr198 from the C-loop at the principal face of the binding site. On the other hand, toxin residue Trp10 extends in a direction opposite to Arg7 and couples to residues from strands β3 and β6 from the complementary face of the binding site (213). For α-conotoxin PnIB, a 4/7 α-conotoxin, strong pairwise coupling was observed between toxin residue Leu10 and residues in the α7 AChR equivalent to αTrp149 and αTyr93 from loops B and A, respectively, at the principal face of the binding site (214). The overall findings predicted toxin orientations and inter-residue stabilizing interactions within the ligand binding site. These predicted orientations were consistent with orientations in crystal structures of complexes between AChBP and each α-conotoxin (FIGURE 17). Furthermore, the cocrystal structures showed that the α-conotoxins displaced the C-loop outward relative to the center of the binding cavity (43, 110, 268).
FIGURE 17.

Orientation of α-conotoxin ImI bound to Aplysia AChBP (PDB code 2BYP; Ref. 110). Left: principal and complementary faces are shown as ribbons, while α-conotoxin ImI is shown as space-filling. Right: key AChBP determinants of affinity, based on mutagenesis analyses of α7 AChRs (213, 215), are shown as space-filling (Tyr91, Tyr186,Tyr193, Asp195, Met114, Arg77), while key determinants of α-conotoxin ImI affinity (216) are shown as ball and stick (Asp5, Pro6, Arg7, Trp10).
α-Neurotoxins are peptide toxins from the venoms of snakes of the family Elapidae and bind to and competitively antagonize the endplate AChR (48). They are basic peptides of 60–70 residues that, through multiple intramolecular disulfide bonds, form three characteristic fingers that extend from a common globular base (20). For some species of AChR, α-neurotoxin binding is nearly irreversible (rat, mouse, Torpedo; Ref. 198), for others it is slowly reversible (humans, hedgehog; Ref. 14), while for select species binding is negligible (snakes, mongoose; Refs. 13, 145). α-Neurotoxins associate with forward rate constants some three to four orders of magnitude slower than the diffusion limit (281, 284), suggesting multiple points of attachment, conformational changes at the binding site, conformational changes in the toxin, or a combination of these. Binding of AChR agonists, competitive antagonists, and α-neurotoxins is mutually exclusive (191), indicating these three classes of ligands bind to a common, overlapping site.
α-Neurotoxins bind to α-subunits isolated from the intact endplate AChR (106), monomeric α-subunits (24), proteolytic fragments from the α-subunit (266), and short peptides derived from the C-loop of the α-subunit (291). Both NMR and crystallographic structures showed that fingers I and II of α-neurotoxin wrapped around a peptide derived from the C-loop (1, 18, 112). The cocrystal structure of α-bungarotoxin and the ligand binding domain from the muscle α1 subunit confirmed that fingers I and II wrapped around the C-loop, but the resolution of 1.9 Å disclosed additional intermolecular contacts (73). These included extensive hydrogen bonds and van der Waals contacts, as well as cation-π interactions between the conserved Phe32 and Arg36 residues from finger II of the toxin and the conserved triad of Tyr residues from the α-subunit (αTyr93, αTyr190, αTyr198); the guanidinium group of Arg36 assumed the same position as the quaternary ammonium group of agonists bound to AChBP (45). Also, hydrogen bonds between an N-linked sugar in the α1 subunit and main chain atoms in finger I of the toxin revealed additional stabilization. In an additional key contact, αPhe189 of the C-loop lodged within a hydrophobic crevice between fingers I and II of the toxin. Comparison of α-subunit sequences from different species indicates that receptors with highest affinity for α-neurotoxins contain Phe or Tyr at position 189 (rat, mouse, Torpedo), whereas those with reduced affinity contain Thr (human, hedgehog, mongoose) (1, 198). Receptors that do not bind α-neurotoxins contain Lys (α2-α4 neuronal AChRs) at position 189 or N-linked glycosylation sites along strand β9 [αAsn187 in mongoose (13), αAsn189 in cobra (145)].
In the cocrystal structure of AChBP with α-cobratoxin at 4.2 Å resolution (29), finger II penetrates some 10 Å into the subunit interface where its conserved Arg33 assumes a position equivalent to that of the carbamyl moiety of carbamylcholine bound to AChBP (45); the position of Arg33 in the complex is proximal to the complementary face of the binding site, unlike the equivalent Arg36 of α-bungarotoxin bound to the α1 ligand binding domain (73), which lodges proximal to the principal face (FIGURE 18). The different bound orientations may owe to the presence of both principal and complementary subunits in the complex of α-co-bratoxin with AChBP, or to contacts with sugars in the complex between α-bungarotoxin and the α1 ligand binding domain. In the complex between AChBP and α-cobratoxin, the binding site adopts an expanded, uncapped conformation, with the C-loop at the principal face and the F loop at the complementary face opening outward. Finally, in both the AChBP-α-cobratoxin and α1-subunit-α-bungarotoxin complexes, the bound conformation of the toxin differs from that in solution (226, 296). Thus conformational changes in both the binding site and the α-neurotoxin, together with multiple points of attachment, contribute to the slow association of α-neurotoxins with the AChR.
FIGURE 18.

Orientation of cobra α-neurotoxin bound to Lymnaea AChBP (PDB code 1YI5; Ref. 29). Left: principal (yellow) and complementary (blue) faces of AChBP and cobra α-neurotoxin (green) are shown as ribbons. Right: close-up view of the complex, with residues in finger II of α-neurotoxin shown as ball and stick (Trp25, Asp27, Phe29, Arg33, Arg36) and residues of the aromatic binding pocket of AChBP (Trp143, Tyr185, Tyr192, Trp53) shown as space-filling.
The overall structural studies suggested that binding of α-neurotoxins to the AChR depends little on the complementary face of the binding site. This idea was further supported by kinetic studies showing only single association and dissociation rate constants for α-cobratoxin or α-bungarotoxin to the nonequivalent binding sites on the Torpedo AChR (281, 284). However, α-bungarotoxin was photo-incorporated into the α-, γ-, and δ-subunits of the Torpedo AChR (205), suggesting the toxin closely approached the complementary face of the binding site. Furthermore, when cysteine was substituted for γLeu119, δLeu121, or εLeu119 at the complementary face of the AChR, treatment with MTSET prevented subsequent binding of α-bungarotoxin only to the binding site that contained the substituted cysteine (237). In the complex between AChBP and α-cobratoxin, Arg33 at the tip of finger II of the toxin approaches Met114 of strand β6 of the complementary face, which is equivalent to γLeu119, δLeu121, and εLeu119 in the AChR; the quaternary ammonium moiety of MTSET, when covalently bound to substituted Cys at these positions, likely repeled Arg33 of the toxin, preventing binding.
Studies of Naja mossambica mossambica α-toxin showed that although it bound with high affinity to the α-γ and α-δ sites from the fetal AChR, it bound with low affinity to the α-ε site from the adult AChR (203). The origin of the low affinity was traced to residue differences in the linker that joins the β8 and β9 strands at the complementary face of the binding site. After generating mutations in both the α-toxin and the binding site, double mutant cycle analyses showed that Arg33 at the tip of finger II interacted with γLeu119 on strand β6, while Lys27 at the midpoint of finger II interacted with γGlu174 on the β9-β9 linker (204). Thus, in binding to the AChR, α-neurotoxins not only wrap around the C-loop of the principal face, but they also closely approach the complementary face.
Waglerin peptides are found in the venom of Wagler’s pit viper Tropidolaemus wagleri and competitively antagonize muscle AChRs (227, 286). Waglerins contain 22–24 residues, an unusually high proline content, and a single disulfide bond that forms a central loop with only three intervening residues. In solution, Waglerin-1 assumes the shape of the letter Y in which the two branches are flexible and their conformations depend on the polarity of the environment, potentially enabling the peptide to conform to the binding site (56, 230). Toxicity of Waglerin-1 depends on species, with mice very sensitive and rats insensitive (159). The origin of this species selectivity was traced to differences in residues at equivalent positions of sequence homology in the region of the ε-subunit that forms the complementary face of the AChR binding site. Studies of site-directed mutations showed that high affinity for Waglerin-1 originated from interactions between the toxin and charged and hydrophobic residues in strands β1, β2, β6, and the β8-β9 loop from the complementary face of the binding site, which accounted for species-dependent toxicity (180, 181).
Lophotoxin is a cyclic diterpenoid antagonist of the AChR found in gorgonian soft corals plentiful in the Pacific Ocean from Panama to California (12, 87). The diterpenoid ring contains furanoaldehyde and epoxy-lactone functional groups, and lophotoxin is unique among nicotinic AChR ligands because it contains only carbon, hydrogen, and oxygen atoms and thus lacks a nitrogen atom. Lophotoxin reacts covalently with the AChR via αTyr190, which forms a nucleophilic tyrosinate anion intermediate that attacks one of the electron-deficient epoxide carbon atoms in the toxin (2). Both lophotoxin and an analog generated by chemical reduction bind with different affinities to the two AChR ligand binding sites (65, 240), showing highest affinity for the α-δ site, like α-conotoxin M1, but opposite to d-TC.
VI. AChR DISEASE MECHANISMS
The clinical disorder myasthenia is derived from the Greek words myo, meaning muscle, and asthenia, meaning weakness, and arises from failure of the muscle endplate potential (EPP) to reach the AP threshold, which compromises the safety margin of neuromuscular transmission. Myasthenia gravis is the most common type of myasthenia and is characterized by serum auto-antibodies directed against the MIR (main immunogenic region) of the AChR α1-subunit, progressive degeneration of the postjunctional folds, and widening of the synaptic cleft (82). Congenital myasthenic syndromes (CMS) are less common but more diverse and are caused by genetic defects in presynaptic, synaptic, or postsynaptic proteins (84). Defects in postsynaptic proteins account for >75% of CMS, and most frequently arise from mutations of the AChR subunits and the receptor-associated protein rapsyn (242).
The molecular bases of congenital myasthenia were first recognized in 1995 (199). The general approach was to combine clinical, electrophysiological, and ultrastructural studies with candidate gene sequencing and in vitro genetic reconstitution and functional analyses of proteins engineered with the identified mutations. In cases involving mutations of endplate AChR subunits, functional analyses included measurements of radiolabeled α-bungarotoxin binding, an indicator of AChR expression on the cell surface, and steady-state binding of ACh and patch-clamp recordings of single-channel currents, indicators of augmented or impaired AChR function (201). Kinetic analyses of single-channel currents were especially powerful because they could pinpoint the elementary reaction step altered by the mutation, giving insight into structure-function relationships of the AChR as well as guiding rational therapy.
Pathogenic mutations in the endplate AChR have been identified in each of its subunits and in major domains within the subunits, and divide into two main classes: those that reduce or eliminate expression of AChR on the cell surface and those that alter the kinetics of AChR activation. Mutations that alter the kinetics of activation divide further into slow channel mutations that prolong the ACh-mediated postsynaptic response, and fast channel mutations that curtail the response (FIGURE 19). The slow and fast channel mutations have opposite effects on the time course and amplitude of the postsynaptic response and on elementary reaction steps in AChR activation; treatment of slow and fast channel CMS is thus achieved by drugs with opposing mechanisms of action.
FIGURE 19.

Overview of congenital myasthenic syndromes due to mutations in AChR transmembrane domains. Top panel compares time courses of miniature endplate currents from control (black), fast channel (green), and slow channel (red) endplates [From Engel et al. (84)]. Bottom panel shows a homology model of the AChR α-subunit (based on PDB code 2BG9) as space-filling, with TMD2 highlighted in blue, TMD3 in magenta, and TMD1 in orange. Identified CMS mutations are indicated and shown with side chains white.
A majority of postsynaptic CMS arise from homozygous or heterozygous recessive mutations of AChR subunit genes that reduce or prevent formation of AChRs on the cell surface. Such null mutations occur predominantly in the ε-subunit, likely because the fetal γ-subunit rescues enough AChRs to enable a postsynaptic response above the AP threshold, whereas a null mutation of a non-ε-subunit would not allow formation of the AChR, preventing survival (200). Identified null mutations include those that alter the subunit signal peptide, cause premature chain termination, or prevent N-linked glycosylation, disulfide bond formation, protein folding, subunit assembly, or trafficking to the cell surface (84).
A. Slow Channel CMS
Slow channel CMS are characterized by dominant inheritance, a repetitive rather than a single compound muscle AP elicited by single nerve stimulation, prolonged biexponential EPPs (endplate potentials), spontaneous as well as prolonged AChR channel openings, degenerating postjunctional folds, and widening of the synaptic cleft (83). The compound muscle AP becomes repetitive because the prolonged EPP persists beyond the refractory period of the AP, enabling a second AP. Furthermore, at physiological rates of nerve stimulation, the prolonged EPPs sum in a staircase fashion, depolarizing the muscle and blocking synaptic transmission. For some slow channel AChRs, desensitization is accelerated so that at physiological rates of stimulation, the postsynaptic response decreases within seconds, further impairing neuromuscular transmission (79). Degeneration of the postjunctional folds and loss of AChR are secondary to the activation of proteases and lipases and stimulation of nuclear apoptosis caused by intracellular calcium accumulation, a result of both spontaneous and prolonged AChR channel openings. The severity of slow channel CMS is variable, likely depending on the extent of calcium overloading, the expression efficiency of the mutant relative to the normal AChR, and the degree of increase in spontaneous and ACh-induced channel openings. Both the immediate and long-term consequences of slow channel mutations compromise the safety margin of neuromuscular transmission. Thus early diagnosis and drugs that accelerate the EPC decay, such as long-lived channel blockers, currently offer the best means of treatment (81).
The first slow channel CMS described at the molecular level arose from a Thr to Pro mutation in TMD2 of the ε-subunit (199; FIGURE 20). When placed in an α-helix, proline distorts the normal helical geometry and removes an intrahelical hydrogen bond. Substitution of proline within the TMD2 α-helix was therefore expected to alter AChR channel function. This expectation was tested by engineering the Pro substitution in the cloned ε-subunit, coexpressing it with normal α-, β-, and δ-subunits in heterologous cells and recording single-channel currents. The recordings revealed a striking increase in spontaneous single-channel openings of the AChR and prolonged durations of ACh-induced channel openings that mirrored those recorded from the patient endplates. The overall findings showed that a single mutation of the AChR accounted for both the abnormal synaptic response and cation overloading, and explained the degeneration and simplification of the postsynaptic membrane.
FIGURE 20.

First identified slow channel CMS. Left: Subunit TMDs are shown with the location of the mutant residue εT264 indicated by arrow. Right: compares single-channel currents for wild-type and mutant AChRs (channel openings are upward deflections). [From Ohno et al. (199).]
A majority of slow channel CMS arise from mutations in TMD2 that increase spontaneous and prolong ACh-induced channel openings (84). Within the mechanistic framework of the two-site MWC model, an increase in spontaneous channel opening indicates an increase in the unliganded channel gating equilibrium constant θ0. Because the ligand binding sites and ion channel are separated by several tens of angstroms, changes in the intrinsic dissociation constants for ACh binding to closed and open states, K and K*, are not expected. Thus given the relationship θ2 = θ0(KR/KO)2 based on the MWC model, an increase of θ0 predicts an increase of θ2, suggesting prolonged ACh-induced channel openings; θ2 could increase through either an increase of the channel opening rate constant β or a decrease in the channel closing rate constant α. Single-channel currents activated by low concentrations of ACh showed an increase of the apparent open time, suggesting α increased and the open state was stabilized.
On closer inspection, however, the ACh-induced channel openings appeared as long bursts of successive channel openings flanked by brief closings (113, 172, 244, 275). Because the durations of brief closings approached the time resolution limit of the patch clamp, burst rather than open duration was often measured. Thus the prolonged ACh-induced bursts may have arisen from changes in elementary reaction steps that determine whether a channel that just closed reopens and consequently continues the burst. In the extended del Castillo and Katz mechanism, the number of brief closings per burst is given by the ratio β2/k−2, indicating burst duration could increase owing to an increase in β2 or a decrease in k−2. Alternatively, in the Flip or Primed mechanisms, the number of brief closings per burst obtained by analogy to Equation 3.9 in Reference 62 yields (β2/p− + β2p+/k−2p−); thus burst duration could increase owing to increases in β2 or p+, decreases in k−2 or p− or a combination of these. For slow channel CMS arising from mutations in TMD2, fitting kinetic schemes to single-channel data has not been accomplished so far, so the changes in elementary reaction steps remain to be determined. A major barrier to kinetic fitting is that in these mutants the exponential components of closed times exceeds the number of closed states within tractable kinetic mechanisms (172).
On the other hand, a slow channel CMS due to a mutation near the ligand binding site, αG153S, yielded sequences of single-channel dwell times that could be described by a scheme with relatively few stable states (244). When dwell times from αG153S receptors were analyzed according to the extended del Castillo and Katz mechanism, the observed increase in burst duration arose from a slowing of the rate constant for ACh dissociation k−2 without a change in the channel opening rate constant β2, predicting an increase in the number of openings per burst. The fitting also revealed a modest slowing of the rate constant for channel closing α2 that further increased burst duration. The Flip or Primed mechanisms were not envisioned at the time this mutation was discovered, but either model is physically plausible given that the mutation is close to the ligand binding site where priming is initiated (185). In these alternative mechanisms, p+ or β2 may have increased or p− or k−2 decreased.
Slow channel mutations have also been identified in TMD1 (αN217K, εL221F) and analyzed by fitting kinetic schemes to ACh-induced single-channel dwell times. When receptors bearing the mutation αN217K were analyzed according to the extended del Castillo and Katz mechanism, the rate constant for ACh dissociation from closed, di-liganded receptors decreased 10-fold, again predicting increased reopening of the channel before the agonist could dissociate, while the channel gating rate constants α2 and β2 showed minor changes (275). Substitutions of Lys at equivalent positions of the non-α-subunits were without effect, as were substitutions of Gln or Glu at position 217 of the α-subunit. Thus the functional consequences were specific to substitutions in the α-subunit and Lys as the substituting residue. When receptors bearing the mutation εL221F were analyzed according to an extended del Castillo and Katz mechanism that allowed independent rather than sequential binding of ACh, the rate constant for dissociation of ACh from closed, di-liganded receptors decreased, while the channel opening rate constant β2 increased and the closing rate constant α2 decreased, all of which predicted prolonged bursts of ACh-induced channel openings (113). In either of these mutant AChRs, slowing of ACh dissociation from the di-liganded closed state may have arisen from propagation of the mutational effects in TMD1 to the binding site via the bidirectional binding-gating transduction pathway. So, although a point mutation implies a localized structural perturbation, it may alter more than one elementary reaction step in AChR activation. Alternatively, limitations in time resolution or choice of activation mechanism for kinetic fitting may have eluded identification of the precise state transitions affected by the mutations. Continued progress toward understanding the activation mechanism is thus essential to defining disease mechanisms.
B. Fast Channel CMS
Fast channel CMS are characterized by recessive inheritance, a single compound muscle AP elicited by nerve stimulation, small and rapidly decaying EPPs, absence of spontaneous AChR channel openings, brief ACh-induced channel openings, and nearly normal endplate morphology (83). Fast channel CMS can arise from either homozygous or heterozygous inheritance, but if the inheritance is heterozygous, the second allele is either a null mutation or a second fast channel mutation. Vesicular release of ACh and its hydrolysis are normal, but the safety margin of neuromuscular transmission is compromised because the EPP is too small to reach the muscle AP threshold. As a consequence, drugs that increase synaptic ACh, such as 3,4-di-aminopyridine, with or without an AChE inhibitor, currently offer the best means of treatment (81).
The first fast channel CMS described at the molecular level arose from a Pro to Leu mutation (εP121L) in strand β6 of the ε-subunit (201). Strand β6 contributes to the complementary face of the α-ε binding site, where εPro121 is the second of two consecutive Pro residues. Strand β6 and the flanking strands β2 and β5′ form an anti-parallel β-sheet, so in the wild-type AChR, the consecutive Pro residues remove intersheet hydrogen bonds, potentially increasing flexibility of the local β-sheet structure. Also a Pro residue can increase the cis- relative to the trans-conformation of the peptide bond, further affecting the conformation of strand β6. Substitution of Leu for εPro121 is expected to introduce an interstrand hydrogen bond and promote the trans-conformation, thus altering strand β6 at the complementary face of the α-ε site, in turn affecting ACh binding.
Functional consequences of εP121L were tested by engineering the Leu for Pro substitution in the ε-subunit, coexpressing it with normal α-, β-, and δ-subunits in heterologous cells and recording ACh-induced single-channel currents (201). The recordings showed that successive channel openings were flanked by prolonged channel closings, even at a saturating concentration of ACh. Fitting the extended del Castillo and Katz mechanism to the sequences of dwell times revealed little change in affinity of ACh for the resting, closed state, but revealed reduced ACh affinity for the open channel state and a marked slowing of the rate constant for channel opening. Channel opening was slowed apparently because the energetic driving force for opening was attenuated through reduced affinity of ACh for the open relative to the closed channel state. The slow rate of channel opening increased the probability that a closed channel would lose bound agonist rather than open, and decreased the probability that a newly closed channel would reopen; these twin consequences accounted for the reduced size and rapid decay of the EPP that compromised the safety margin for neuromuscular transmission.
Two other fast channel mutations were identified at the complementary face of the α-ε binding site, εD175N and εN182Y (246). Located within loop F at the periphery of the binding site, both mutations were identified in the same patient, but on different ε-subunit alleles. The two mutations exerted opposite effects on resting state affinity of ACh for the α-ε site, while they did not affect affinity for the α-δ site. εD175N slowed ACh association and increased dissociation, likely through reduced electrostatic attraction of ACh toward the binding site (FIGURE 21). εN187Y enhanced association and slowed dissociation, perhaps through increased π-cation stabilization of ACh. However, independent of their effects on ACh binding, both mutations slowed the rate and reduced the extent of channel opening. These findings echoed an emerging theme that key residues at the ACh binding site contribute not only locally to stabilize bound ACh, but also globally to stabilize the closed relative to the open state of the channel.
FIGURE 21.

Fast channel CMS due to mutation at the entrance to the α-ε binding site (246). Left: extracellular domains of the α- and ε-subunits are shown with the mutant residue εAsp175 in standard atom colors. Right: compares single-channel currents for wild-type and mutant AChRs (channel openings are upward deflections). [From Sine et al. (246).]
A severe fast channel CMS caused by the mutation δL42P highlighted the importance of the subunit interface in conferring rapid and efficient channel opening of the endplate AChR (232). δLeu42 localizes to the lower end of the extracellular region within strand β2, which forms an anti-parallel β-sheet with strand β1, but shows no obvious contribution to the α-δ ligand binding site. Instead, δLeu42 neighbors δAsn41, which forms an intersubunit linkage with αTyr127 required for rapid channel opening (186). A functionally analogous Asn-Tyr linkage is also present at the α-ε subunit interface. Substitution of Pro for δLeu42 removes a hydrogen bond between the antiparallel β-sheets and would be expected to locally weaken the β-sheet, potentially disrupting the intersubunit linkage to αTyr127. Evidence that δL42P disrupted the intersubunit linkage was obtained by double-mutant cycle analysis that disclosed a large coupling free energy for the mutant pair αY127T and δL42P, based on the channel gating equilibrium constant in the extended del Castillo and Katz mechanism; coupling free energy approached that observed for the primary inter-subunit linkage between δAsn41 and αTyr127 that approached 4 kcal/mol. Although δLeu42 is conserved among AChR subunits, substitution of Pro for Leu in the β-subunit did not affect channel gating, but substitution in the α-subunit enhanced channel gating. Thus, at four of the five AChR subunit interfaces, an intersubunit linkage in this extracellular region contributes to the efficiency of channel gating.
A novel category of fast channel CMS resulted from mutations in the long linker between TMD3 and TMD4 and forms a V-shaped structure that extends into the cytoplasm. Only the second half of this linker was resolved in the Torpedo structure at a resolution of 4 Å, and it comprised a long α-helix that extended from the base of TMD4 to the apex of the V (271). The TMD3-TMD4 linker was first recognized to contribute to the kinetics of AChR activation when it was found to confer the majority of the fetal to adult decrease in channel open time that occurs when the γ-subunit is replaced by the ε-subunit (30). The functionally relevant region was a 30-residue stretch at the COOH-terminal end of the TMD3-TMD4 linker, the same region resolved in the Torpedo structure.
The first fast channel CMS of this kind arose from duplication of six residues, STRDQE, in the center of the α-helical region of the TMD3-TMD4 linker of the ε-subunit, while the second allele harbored a null mutation (173). Patch-clamp recording, in the presence of a high concentration of ACh, revealed that the six residue duplication caused abrupt mode switches from normal to intermediate or low channel open probability. Furthermore, each kinetic mode exhibited a second population of very brief channel openings not observed in normal receptors; these brief openings were suggested to represent a transient intermediate in the normal activation pathway that was unmasked by the mutation. Together, the mode switches to low channel open probability and the increase of brief channel openings accounted for the compromised safety margin of neuromuscular transmission.
A second fast channel CMS caused by mode switching kinetics arose from substitution of Pro for Ala at position 411 of the ε-subunit (εA411P) located near the apex of the V-shaped cytoplasmic domain (279; FIGURE 22). However, the kinetic signature of εA411P was novel: whereas individual episodes of channel openings were kinetically homogeneous, each episode exhibited a different open probability, ranging from very low to high open probability. To identify the elementary kinetic steps that were altered within individual channel opening episodes, an analysis method called Hidden Markov modeling (274) was used to determine channel opening and closing rate constants within each episode. For the overall set of channel opening episodes, the channel gating rate constants distributed across an unusually wide range, while the ACh association and dissociation rate constants spanned normal ranges. Substituting Pro at positions flanking Ala 411 in the ε-subunit also increased the range of channel gating rate constants similar to εA411P, whereas substituting Pro at equivalent positions of the non-ε-subunits were without effect. Thus, in the ε-subunit, the TMD3-TMD4 linker is crucial for maintaining stable channel gating kinetics.
FIGURE 22.

Fast channel CMS due to a mode-switching mutation in the intracellular domain of the ε-subunit (279). Left: a homology model of the AChR (based on PDB code 2BG9) in ribbon representation with regions in which CMS localize highlighted in white space-filling representation. Right: compares individual episodes of single-channel currents for wild-type and ε411P AChRs. [From Wang et al. (279).]
Because the kinetics of channel gating were homogeneous within individual channel opening episodes, the mode switches occurred during the long closed periods between episodes. Thus switches from one kinetic mode to the next were suggested to occur by thermal navigation over a large energy barrier. However, close inspection revealed that even normal receptors exhibited occasional abnormal kinetic modes, suggesting the CMS mutation amplified a rare but normal process. The overall findings could be explained by postulating a corrugated structure of the energy landscape that governs channel gating modes (91). In the normal receptor, the corrugations overlay a steeply pitched, funnel-shaped foundation that minimizes mode switching, but in the mutant receptor, the corrugations overlay a relatively flat foundation, enabling thermal navigation about the landscape and frequent mode switching.
VII. PROSPECTUS
A. Activation Mechanism
Mechanistic understanding of AChR activation has advanced considerably since the time of del Castillo and Katz, yet there is a pressing need for further advances. The briefest dwell time detectable by single-channel recording is ~10 μs, which translates to a state with an energy basin 10 kcal/mol deep. For each kcal/mol decrease in the depth of the energy basin, roughly an order of magnitude increase in temporal resolution is required to detect state transitions out of that basin. Time resolution of the patch clamp improved rapidly following its introduction in the 1980s, but resolution has remained essentially unchanged since the new millennium. Thus qualitative improvements in patch-clamp circuitry and experimental configuration are sorely needed (283). Data analysis is also a significant limitation, in particular, the labor-intensive process of operator-assisted detection and idealization of single channel currents required to generate sequences of open and closed dwell times; automation of this step will thus facilitate further mechanistic advances. Software for single-channel kinetic analysis is freely available, allowing fitting of schemes with relatively small numbers of states to sequences of single-channel dwell times (211). Although current computer processor speeds are adequate for most fitting problems, continued increases in processing speed and development of next generation analysis approaches will enable fitting of schemes with increasing complexity. Thus prospects are good for achieving the goal of delineating the energy landscapes that dictate state transitions of single AChR channels.
B. AChR Structure
A major short-term goal is a high-resolution structure of the ligand binding domain from a eukaryotic source. In the most promising approach so far, the ligand binding domain of the homomeric neuronal α7 AChR has become an attractive target because it circumvents the problem of heteromeric subunit assembly (287). Initial attempts yielded milligram quantities of the ligand binding domain that bound α-neurotoxin and appeared as pentameric rosettes by electron microscopy (11, 300). Because the binding and pore domains are allosterically coupled, it will be important to arrest the binding domains in a single functional state. Once this is achieved and a high-resolution structure is determined, in silico studies of ligand docking will provide a powerful means to design drugs with target specificity.
A significant but elusive goal is a high-resolution structure of a full-length nicotinic receptor. Functional state will again be of particular importance, as the ultimate goal is a set of structures in the resting, active, and desensitized states. By combining these structures with single molecule functional measurements, investigators can begin to decipher the symphony of molecular motions that underpin conversion among functional states. We will then be equipped to explain how inherited mutations cause malfunction of the receptor, and to design drugs with the specificity to target particular receptor subtypes.
Acknowledgments
I gratefully acknowledge my senior colleagues for guidance, lessons, and contributions over many years: Drs. Andrew Engel, Palmer Taylor, Joe Henry Steinbach, and Fred Sigworth. I am also endebted to my junior colleagues for their dedication, hard work, and insights: Drs. Cecilia Bouzat, Hai-Long Wang, Richard Prince, Won Yong Lee, Nuriya Mukhtasimova, Da-Xiong Fu, Fan Gao and Polly Quiram.
GRANTS
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS-31744.
Footnotes
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author.
References
- 1.Abraham SO, Scherf T, Eisenstein M, Chill JH, Anglister J. The mechanism for acetylcholine receptor inhibition by α-neurotoxins and species-specific resistance to α-bungarotoxin revealed by NMR. Neuron. 2002;35:319–332. doi: 10.1016/s0896-6273(02)00773-0. [DOI] [PubMed] [Google Scholar]
- 2.Abramson SN, Li Y, Culver P, Taylor P. An analog of lophotoxin reacts covalently with Tyr190 in the alpha-subunit of the nicotinic acetylcholine receptor. J Biol Chem. 1989;264:12666–12672. [PubMed] [Google Scholar]
- 3.Adams PR. An analysis of the dose-response curve at voltage-clamped frog-endplates. Pflügers Arch. 1975;360:145–153. doi: 10.1007/BF00580537. [DOI] [PubMed] [Google Scholar]
- 4.Akabas MH, Kaufmann C, Archdeacon P, Karlin A. Identification of acetylcholine receptor channel-lining residues in the entire M2 segment of the alpha subunit. Neuron. 1994;13:919–927. doi: 10.1016/0896-6273(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 5.Akabas MH, Stauffer DA, Xu M, Karlin A. Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science. 1992;258:307–310. doi: 10.1126/science.1384130. [DOI] [PubMed] [Google Scholar]
- 6.Akk G. Aromatics at the murine nicotinic receptor agonist binding site: mutational analysis of the alphaY93 and alphaW149 residues. J Physiol. 2001;535:729–740. doi: 10.1111/j.1469-7793.2001.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akk G, Auerbach A. Inorganic, monovalent cations compete with agonists for the transmitter binding site of nicotinic acetylcholine receptors. Biophys J. 1996;70:2652–2658. doi: 10.1016/S0006-3495(96)79834-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akk G, Zhou M, Auerbach A. A mutational analysis of the acetylcholine receptor channel transmitter binding site. Biophys J. 1999;76:207–218. doi: 10.1016/S0006-3495(99)77190-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson CR, Stevens CF. Voltage clamp analysis of acetylcholine produced end-plate current fluctuations at frog neuromuscular junction. J Physiol. 1973;235:655–691. doi: 10.1113/jphysiol.1973.sp010410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Auerbach A, Lingle CJ. Activation of the primary kinetic modes of large- and small-conductance cholinergic ion channels in Xenopus myocytes. J Physiol. 1987;393:437–466. doi: 10.1113/jphysiol.1987.sp016832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Avramopoulou V, Mamalaki A, Tzartos SJ. Soluble, oligomeric, ligand-binding extracellular domain of the human alpha7 acetylcholine receptor expressed in yeast: replacement of the hydrophobic cysteine loop by the hydrophilic loop of the ACh-binding protein enhances protein solubility. J Biol Chem. 2004;279:38287–38293. doi: 10.1074/jbc.M402533200. [DOI] [PubMed] [Google Scholar]
- 12.Bakus GJ. Chemical defense mechanisms on the Great Barrier Reef, Australia. Science. 1981;211:497–499. doi: 10.1126/science.7455691. [DOI] [PubMed] [Google Scholar]
- 13.Barchan D, Kachalsky S, Neumann D, Vogel Z, Ovadia M, Kochva E, Fuchs S. How the mongoose can fight the snake: the binding site of the mongoose acetylcholine receptor. Proc Natl Acad Sci USA USA. 1992;89:7717–7721. doi: 10.1073/pnas.89.16.7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barchan D, Ovadia M, Kochva E, Fuchs S. The binding site of the nicotinic acetylcholine receptor in animal species resistant to alpha-bungarotoxin. Biochemistry. 1995;34:9172–9176. doi: 10.1021/bi00028a029. [DOI] [PubMed] [Google Scholar]
- 15.Barlow RB, Howard JAK, Johnson O. Structures of nicotine monomethyl iodide and nicotine monohydrogen iodide. Acta Cryst. 1986;C42:853–856. [Google Scholar]
- 16.Barlow RB, Johnson O. Relations between structure and nicotine-like activity: X-ray crystal structure analysis of (−)-cytisine and (−)-lobeline hydrochloride and a comparison with (−)-nicotine and other nicotine-like compounds. Br J Pharmacol. 1989;98:799–808. doi: 10.1111/j.1476-5381.1989.tb14608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartol TM, Jr, Land BR, Salpeter EE, Salpeter MM. Monte Carlo simulation of miniature endplate current generation in the vertebrate neuromuscular junction. Biophys J. 1991;59:1290–1307. doi: 10.1016/S0006-3495(91)82344-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basus VJ, Song G, Hawrot E. NMR solution structure of an alpha-bungarotoxin/nicotinic receptor peptide complex. Biochemistry. 32:12290–12298. doi: 10.1021/bi00097a004. [DOI] [PubMed] [Google Scholar]
- 19.Beckstein O, Sansom MS. The influence of geometry, surface character, and flexibility on the permeation of ions and water through biological pores. Phys Biol. 2004;1:42–52. doi: 10.1088/1478-3967/1/1/005. [DOI] [PubMed] [Google Scholar]
- 20.Betzel C, Lange G, Pal GP, Wilson KS, Maelicke A, Saenger W. The refined crystal structure of alpha-cobratoxin from Naja naja siamensis at 2.4-A resolution. J Biol Chem. 1991;266:21530–21536. doi: 10.2210/pdb2ctx/pdb. [DOI] [PubMed] [Google Scholar]
- 21.Biesecker G. Molecular properties of the cholinergic receptor purified from Electrophorus electricus. Biochemistry. 1973;12:4403–4409. doi: 10.1021/bi00746a017. [DOI] [PubMed] [Google Scholar]
- 22.Blanton MP, Cohen JB. Mapping the lipid-exposed regions in the Torpedo californica nicotinic acetylcholine receptor. Biochemistry. 1992;31:3738–3750. doi: 10.1021/bi00130a003. [DOI] [PubMed] [Google Scholar]
- 23.Blount P, Merlie JP. Molecular basis of the two nonequivalent ligand binding sites of the muscle nicotinic acetylcholine receptor. Neuron. 1989;3:349–357. doi: 10.1016/0896-6273(89)90259-6. [DOI] [PubMed] [Google Scholar]
- 24.Blount P, Merlie JP. Native folding of an acetylcholine receptor alpha subunit expressed in the absence of other receptor subunits. J Biol Chem. 1988;263:1072–1080. [PubMed] [Google Scholar]
- 25.Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- 26.Bocquet N, Prado de Carvalho L, Cartaud J, Neyton J, Le Poupon C, Taly A, Grutter T, Changeux JP, Corringer PJ. A prokaryotic proton-gated ion channel from the nicotinic acetylcholine receptor family. Nature. 2007;445:116–119. doi: 10.1038/nature05371. [DOI] [PubMed] [Google Scholar]
- 27.Bon F, Lebrun E, Gomel J, Van Rapenbusch R, Cartaud J, Popot JL, Changeux JP. Image analysis of the heavy form of the acetylcholine receptor from Torpedo marmorata. J Mol Biol. 1984;176:205–237. doi: 10.1016/0022-2836(84)90421-2. [DOI] [PubMed] [Google Scholar]
- 28.Bourne Y, Radic Z, Aráoz R, Talley TT, Benoit E, Servent D, Taylor P, Molgó J, Marchot P. Structural determinants in phycotoxins and AChBP conferring high affinity binding and nicotinic AChR antagonism. Proc Natl Acad Sci USA USA. 2010;107:6076–6081. doi: 10.1073/pnas.0912372107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bourne Y, Talley TT, Hansen SB, Taylor P, Marchot P. Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake alpha-neurotoxins and nicotinic receptors. EMBO J. 2005;24:1512–1522. doi: 10.1038/sj.emboj.7600620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bouzat C, Bren N, Sine SM. Structural basis of the different gating kinetics of fetal and adult acetylcholine receptors. Neuron. 1994;13:1395–1402. doi: 10.1016/0896-6273(94)90424-3. [DOI] [PubMed] [Google Scholar]
- 31.Bouzat C, Gumilar F, Spitzmaul G, Wang HL, Rayes D, Hansen SB, Taylor P, Sine SM. Coupling of agonist binding to channel gating in an ACh-binding protein linked to an ion channel. Nature. 2004;430:896–900. doi: 10.1038/nature02753. [DOI] [PubMed] [Google Scholar]
- 32.Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 33.Bren N, Sine SM. Hydrophobic pairwise interactions stabilize a-conotoxin MI in the muscle acetylcholine receptor binding site. J Biol Chem. 2000;275:12692–12700. doi: 10.1074/jbc.275.17.12692. [DOI] [PubMed] [Google Scholar]
- 34.Bren N, Sine SM. Identification of residues in the adult nicotinic acetylcholine receptor that confer selectivity for curariform antagonists. J Biol Chem. 1997;272:30793–30798. doi: 10.1074/jbc.272.49.30793. [DOI] [PubMed] [Google Scholar]
- 35.Brett RS, Dilger JP, Adams PR, Lancaster B. A method for the rapid exchange of solutions bathing excised membrane patches. Biophys J. 1986;50:987–992. doi: 10.1016/S0006-3495(86)83539-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brisson A, Unwin PNT. Tubular crystals of acetylcholine receptor. J Cell Biol. 1984;99:1202–1211. doi: 10.1083/jcb.99.4.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brooks B, Karplus M. Normal modes for specific motions of macromolecules: application to the hinge-bending mode of lysozyme. Proc Natl Acad Sci USA USA. 1985;82:4995–4999. doi: 10.1073/pnas.82.15.4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burzomato V, Beato M, Groot-Kormelink PJ, Colquhoun D, Sivilotti LG. Single-channel behavior of heteromeric alpha1beta glycine receptors: an attempt to detect a conformational change before the channel opens. J Neurosci. 2004;24:10924–10940. doi: 10.1523/JNEUROSCI.3424-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cadugan DJ, Auerbach A. Conformational dynamics of the alphaM3 transmembrane helix during acetylcholine receptor channel gating. Biophys J. 2007;93:859–865. doi: 10.1529/biophysj.107.105171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Canepa FG, Pauling P, Sörum H. Structure of acetylcholine and other substrates of cholinergic systems. Nature. 1966;210:907–909. doi: 10.1038/210907a0. [DOI] [PubMed] [Google Scholar]
- 41.Carter CR, Cao L, Kawai H, Smith PA, Dryden WF, Raftery MA, Dunn SM. Chain length dependence of the interactions of bisquaternary ligands with the Torpedo nicotinic acetylcholine receptor. Biochem Pharmacol. 2007;73:417–426. doi: 10.1016/j.bcp.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 42.Cashin AL, Torrice MM, McMenimen KA, Lester HA, Dougherty DA. Chemical-scale studies on the role of a conserved aspartate in preorganizing the agonist binding site of the nicotinic acetylcholine receptor. Biochemistry. 2007;46:630–639. doi: 10.1021/bi061638b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Celie PH, Kasheverov IE, Mordvintsev DY, Hogg RC, van Nierop P, van Elk R, van Rossum-Fikkert SE, Zhmak MN, Bertrand D, Tsetlin V, Sixma TK, Smit AB. Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an alpha-conotoxin PnIA variant. Nat Struct Mol Biol. 2005;12:582–588. doi: 10.1038/nsmb951. [DOI] [PubMed] [Google Scholar]
- 44.Celie PH, Klaassen RV, van Rossum-Fikkert SE, van Elk R, van Nierop P, Smit AB, Sixma TK. Crystal structure of acetylcholine-binding protein from Bulinus truncatus reveals the conserved structural scaffold and sites of variation in nicotinic acetylcholine receptors. J Biol Chem. 2005;280:26457–26466. doi: 10.1074/jbc.M414476200. [DOI] [PubMed] [Google Scholar]
- 45.Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 2004;41:907–914. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 46.Chakrapani S, Bailey TD, Auerbach A. Gating dynamics of the acetylcholine receptor extracellular domain. J Gen Physiol. 2004;123:341–356. doi: 10.1085/jgp.200309004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chakrapani S, Bailey TD, Auerbach A. The role of loop 5 in acetylcholine receptor channel gating. J Gen Physiol. 2003;122:521–539. doi: 10.1085/jgp.200308885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang CC, Lee CY. Isolation of neurotoxins from the venom of Bungarus multicinctus and their modes of neuromuscular blocking action. Arch Int Pharmacodyn Ther. 1963;144:241–57. [PubMed] [Google Scholar]
- 49.Charnet P, Labarca C, Leonard RJ, Vogelaar NJ, Czyzyk L, Gouin A, Davidson N, Lester HA. An open-channel blocker interacts with adjacent turns of alpha-helices in the nicotinic acetylcholine receptor. Neuron. 1990;4:87–95. doi: 10.1016/0896-6273(90)90445-l. [DOI] [PubMed] [Google Scholar]
- 50.Chen J, Zhang Y, Akk G, Sine SM, Auerbach A. Activation kinetics of recombinant mouse nicotinic acetylcholine receptors with mutations at α subunit residue tyrosine 190. Biophys J. 1995;69:849–859. doi: 10.1016/S0006-3495(95)79959-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng X, Lu B, Grant B, Law RJ, McCammon JA. Channel opening motion of alpha7 nicotinic acetylcholine receptor as suggested by normal mode analysis. J Mol Biol. 2006;355:310–324. doi: 10.1016/j.jmb.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 52.Cheng X, Wang HL, Grant B, Sine SM, McCammon JA. Targeted molecular dynamics study of C-loop closure and channel gating in nicotinic receptors. PLos Comp Biol. 2006;2:1173–1184. doi: 10.1371/journal.pcbi.0020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chennubhotla C, Rader AJ, Yang LW, Bahar I. Elastic network models for understanding biomolecular machinery: from enzymes to supramolecular assemblies. Phys Biol. 2005;2:S173–180. doi: 10.1088/1478-3975/2/4/S12. [DOI] [PubMed] [Google Scholar]
- 54.Chiara DC, Cohen JB. Identification of amino acids contributing to high and low affinity d-tubocurarine sites in the Torpedo nicotinic acetylcholine receptor. J Biol Chem. 1997;272:32940–32950. doi: 10.1074/jbc.272.52.32940. [DOI] [PubMed] [Google Scholar]
- 55.Chiara DC, Middleton RE, Cohen JB. Identification of tryptophan 55 as the primary site of [3H]nicotine photoincorporation in the gamma-subunit of the Torpedo nicotinic acetylcholine receptor. FEBS Lett. 1998;423:223–226. doi: 10.1016/s0014-5793(98)00093-3. [DOI] [PubMed] [Google Scholar]
- 56.Chuang LC, Yu HM, Chen C, Huang TH, Wu SH, Wang KT. Determination of three-dimensional solution structure of waglerin I, a toxin from Trimeresurus wagleri, using 2D-NMR and molecular dynamics simulation. Biochim Biophys Acta. 1996;1292:145–155. doi: 10.1016/0167-4838(95)00181-6. [DOI] [PubMed] [Google Scholar]
- 57.Claudio T, Ballivet M, Patrick J, Heinemann S. Nucleotide and deduced amino acid sequences of Torpedo californica acetylcholine receptor gamma subunit. Proc Natl Acad Sci USA. 1983;80:1111–1115. doi: 10.1073/pnas.80.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cockcroft VB, Osguthorpe DJ, Barnard EA, Lunt GG. Modeling of agonist binding to the ligand-gated ion channel superfamily of receptors. Proteins. 1990;8:386–397. doi: 10.1002/prot.340080412. [DOI] [PubMed] [Google Scholar]
- 59.Codding PW, James MNG. The crystal and molecular structure of a potent neuromuscular blocking agent: d-tubocurarine dichloride pentahydrate. Acta Cryst. 1973;29:935–942. [Google Scholar]
- 60.Cohen JB, Sharp SD, Liu WS. Structure of the agonist-binding site of the nicotinic acetylcholine receptor. [3H]acetylcholine mustard identifies residues in the cation-binding subsite. J Biol Chem. 1991;266:23354–23364. [PubMed] [Google Scholar]
- 61.Colquhoun D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Colquhoun D, Hawkes AG. On the stochastic properties of single ion channels. Proc R Soc Lond B Biol Sci. 1981;211:205–235. doi: 10.1098/rspb.1981.0003. [DOI] [PubMed] [Google Scholar]
- 63.Colquhoun D, Sakmann B. Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol. 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Colquhoun D, Sakmann B. Fluctuations in the microsecond time range of the current through single acetylcholine receptor ion channels. Nature. 1981;294:464–466. doi: 10.1038/294464a0. [DOI] [PubMed] [Google Scholar]
- 65.Culver P, Fenical W, Taylor P. Lophotoxin irreversibly inactivates the nicotinic acetylcholine receptor by preferential association at one of the two primary agonist sites. J Biol Chem. 1984;259:3763–3770. [PubMed] [Google Scholar]
- 66.Cymes GD, Grosman C. Pore-opening mechanism of the nicotinic acetylcholine receptor evinced by proton transfer. Nat Struct Mol Biol. 2008;15:389–396. doi: 10.1038/nsmb.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cymes GD, Grosman C, Auerbach A. Structure of the transition state of gating in the acetylcholine receptor channel pore: a phi-value analysis. Biochemistry. 2002;41:5548–5555. doi: 10.1021/bi011864f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cymes GD, Ni Y, Grosman C. Probing ion-channel pores one proton at a time. Nature. 2005;438:975–980. doi: 10.1038/nature04293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Czajkowski C, Kaufmann C, Karlin A. Negatively charged amino acid residues in the nicotinic receptor delta subunit that contribute to the binding of acetylcholine. Proc Natl Acad Sci USA. 1993;90:6285–6289. doi: 10.1073/pnas.90.13.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Damle VN, Karlin A. Affinity labeling of one of two alpha-neurotoxin binding sites in acetylcholine receptor from Torpedo californica. Biochemistry. 1978;17:2039–2045. doi: 10.1021/bi00604a002. [DOI] [PubMed] [Google Scholar]
- 71.Dan Der Straaten TA, Kathawala G, Trellakis A, Eisenberg RS, Ravaioli U. BioMO-CHA–a Boltzmann transport Monte Carlo model for ion channel simulation. Mol Simul. 2005;31:151–171. [Google Scholar]
- 72.Del Castillo J, Katz B. Interaction at end-plate receptors of different choline derivatives. Proc R Soc Lond B Biol Sci. 1957;146:369–381. doi: 10.1098/rspb.1957.0018. [DOI] [PubMed] [Google Scholar]
- 73.Dellisanti CD, Yao Y, Stroud JC, Wang ZZ, Chen L. Crystal structure of the extracellular domain of nAChR alpha1 bound to alpha-bungarotoxin at 1.94 Å resolution. Nat Neurosci. 2007;10:953–962. doi: 10.1038/nn1942. [DOI] [PubMed] [Google Scholar]
- 74.Dionne VE, Steinbach JH, Stevens CF. An analysis of the dose-response relationship at voltage-clamped frog neuromuscular junctions. J Physiol. 1978;281:421–444. doi: 10.1113/jphysiol.1978.sp012431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Donizelli M, Djite MA, Le Novère N. LGICdb: a manually curated sequence database after the genomes. Nucleic Acids Res. 2006;34:D267–269. doi: 10.1093/nar/gkj104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dougherty DA. Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 77.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 78.Dreyer F, Peper K, Sterz R. Determination of dose-response curves by quantitative ionophoresis at the frog neuromuscular junction. J Physiol. 1978;281:395–419. doi: 10.1113/jphysiol.1978.sp012430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Elenes S, Decker M, Cymes GD, Grosman C. Decremental response to high-frequency trains of acetylcholine pulses but unaltered fractional Ca2+currents in a panel of “slow-channel syndrome” nicotinic receptor mutants. J Gen Physiol. 2009;133:151–169. doi: 10.1085/jgp.200810089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Elliott J, Blanchard SG, Wu W, Miller J, Strader CD, Hartig P, Moore HP, Racs J, Raftery MA. Purification of Torpedo californica post-synaptic membranes and fractionation of their constituent proteins. Biochem J. 1980;185:667–677. doi: 10.1042/bj1850667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Engel AG. The therapy of congenital myasthenic syndromes. Neurotherapeutics. 2007;4:252–257. doi: 10.1016/j.nurt.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Engel AG, Hohlfeild R. Disturbances of neuromuscular transmission: acquired auto-immune gyasthenia gravis. In: Engel AG, Francini-Arm-strong C, editors. Myology. 3. New York: McGraw-Hill; 2004. pp. 1755–1790. [Google Scholar]
- 83.Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes. In: Engel AG, Francini-Armstrong C, editors. Myology. 3. New York: McGraw-Hill; 2004. pp. 1801–1844. [Google Scholar]
- 84.Engel AG, Ohno K, Sine SM. Sleuthing molecular targets for neurological diseases: the neuromuscular junction as a paradigm. Nature Rev Neurosci. 2003;4:339–352. doi: 10.1038/nrn1101. [DOI] [PubMed] [Google Scholar]
- 85.Fairclough RH, Twaddle GM, Gudipati E, Lin MY, Richman DP. Differential surface accessibility of alpha(187–199) in the Torpedo acetylcholine receptor alpha subunits. J Mol Biol. 1998;282:317–330. doi: 10.1006/jmbi.1998.2001. [DOI] [PubMed] [Google Scholar]
- 86.Fatt P, Katz B. An analysis of the end-plate potential recorded with an intracellular electrode. J Physiol. 1951;115:320–370. doi: 10.1113/jphysiol.1951.sp004675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fenical W, Okuda RK, Bandurraga MM, Culver P, Jacobs RS. Lophotoxin: a novel neuromuscular toxin from Pacific sea whips of the genus Lophogorgia. Science. 1981;212:1512–1514. doi: 10.1126/science.6112796. [DOI] [PubMed] [Google Scholar]
- 88.Fersht AR, Leatherbarrow RJ, Wells TNC. Quantitative analysis of structure-activity relationships in engineered proteins by linear free-energy relationships. Nature. 1986;322:284–286. doi: 10.1021/bi00393a013. [DOI] [PubMed] [Google Scholar]
- 89.Fertuck HC, Salpeter MM. Quantitation of junctional and extrajunctional acetylcholine receptors by electron microscope autoradiography after 125I-alpha-bungarotoxin binding at mouse neuromuscular junctions. J Cell Biol. 1976;69:144–158. doi: 10.1083/jcb.69.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fletcher GH, Steinbach JH. Ability of nondepolarizing neuromuscular blocking drugs to act as partial agonists at fetal and adult mouse muscle nicotinic receptors. Mol Pharmacol. 1996;49:938–947. [PubMed] [Google Scholar]
- 91.Frauenfelder H, Leeson DT. The energy landscape in non-biological and biological molecules. Nat Struct Biol. 1998;5:757–759. doi: 10.1038/1784. [DOI] [PubMed] [Google Scholar]
- 92.Gao F, Bren N, Little A, Wang HL, Hansen S, Talley T, Taylor P, Sine SM. Curariform antagonists bind in different orientations to acetylcholine binding protein. J Biol Chem. 2003;278:23020–23026. doi: 10.1074/jbc.M301151200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gao F, Burghardt T, Bren N, Hansen SB, Henchman R, Taylor P, McCammon JA, Sine SM. Acetylcholine-mediated conformational changes in acetylcholine-binding protein revealed by simulation and intrinsic tryptophan fluorescence. J Biol Chem. 2005;280:8443–8451. doi: 10.1074/jbc.M412389200. [DOI] [PubMed] [Google Scholar]
- 94.Galzi JL, Revah F, Black D, Goeldner M, Hirth C, Changeux JP. Identification of a novel amino acid alpha-tyrosine 93 within the cholinergic ligands-binding sites of the acetylcholine receptor by photoaffinity labeling. Additional evidence for a three-loop model of the cholinergic ligands-binding sites. J Biol Chem. 1990;265:10430–1047. [PubMed] [Google Scholar]
- 95.Gao F, Mer G, Toneli M, Hansen SB, Taylor P, Sine SM. Solution NMR of acetylcholine binding protein reveals agonist-mediated conformational change of the C-loop. Mol Pharmacol. 2006;70:1230–1235. doi: 10.1124/mol.106.027185. [DOI] [PubMed] [Google Scholar]
- 96.Ginsborg BL, McLachlan EM, Martin AR, Searl JW. The computation of simulated endplate potentials. Proc R Soc Lond B Biol Sci. 1981;213:233–242. doi: 10.1098/rspb.1981.0064. [DOI] [PubMed] [Google Scholar]
- 97.Giraudat J, Dennis M, Heidmann T, Chang JY, Changeux JP. Structure of the high-affinity binding site for noncompetitive blockers of the acetylcholine receptor: serine-262 of the delta subunit is labeled by [3H]chlorpromazine. Proc Natl Acad Sci USA. 1986;83:2719–2723. doi: 10.1073/pnas.83.8.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gouda H, Yamazaki K, Hasegawa J, Kobayashi Y, Nishiuchi Y, Sakakibara S, Hirono S. Solution structure of alpha-conotoxin MI determined by 1H-NMR spectroscopy and molecular dynamics simulation with the explicit solvent water. Biochim Biophys Acta. 1997;1343:327–334. doi: 10.1016/s0167-4838(97)00140-4. [DOI] [PubMed] [Google Scholar]
- 99.Gray WR, Luque A, Olivera BM, Barrett J, Cruz LJ. Peptide toxins from Conus geographus venom. J Biol Chem. 1981;256:4734–4740. [PubMed] [Google Scholar]
- 100.Gray WR, Rivier JE, Galyean R, Cruz LJ, Olivera BM. Conotoxin MI disulfide bonding and conformational states. J Biol Chem. 1983;258:12247–12251. [PubMed] [Google Scholar]
- 101.Green WN, Claudio T. Acetylcholine receptor assembly: subunit folding and oligomerization occur sequentially. Cell. 1993;74:57–69. doi: 10.1016/0092-8674(93)90294-z. [DOI] [PubMed] [Google Scholar]
- 102.Grosman C. Free-energy landscapes of ion-channel gating are malleable: changes in the number of bound ligands are accompanied by changes in the location of the transition state in acetylcholine-receptor channels. Biochemistry. 2003;42:14977–14987. doi: 10.1021/bi0354334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Grosman C, Auerbach A. Kinetic, mechanistic, and structural aspects of unliganded gating of acetylcholine receptor channels: a single channel study of second transmembrane segment 12′ mutants. J Gen Physiol. 2000;115:621–635. doi: 10.1085/jgp.115.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Grosman C, Zhou M, Auerbach A. Mapping the conformational wave of acetylcholine receptor channel gating. Nature. 2000;403:773–776. doi: 10.1038/35001586. [DOI] [PubMed] [Google Scholar]
- 105.Grutter T, Changeux JP. Nicotinic receptors in wonderland. Trends Biochem Sci. 2001;26:459–463. doi: 10.1016/s0968-0004(01)01921-1. [DOI] [PubMed] [Google Scholar]
- 106.Haggerty JG, Froehner SC. Restoration of 125I-alpha-bungarotoxin binding activity to the alpha subunit of Torpedo acetylcholine receptor isolated by gel electrophoresis in sodium dodecyl sulfate. J Biol Chem. 1981;256:8294–8297. [PubMed] [Google Scholar]
- 107.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 108.Hamilton SL, McLaughlin M, Karlin A. Formation of disulfide-linked oligomers of acetylcholine receptor in membrane from Torpedo electric tissue. Biochemistry. 1979;18:155–163. doi: 10.1021/bi00568a024. [DOI] [PubMed] [Google Scholar]
- 109.Hansen SB, Radic’ Z, Talley TT, Molles BE, Deerinck T, Tsigelny I, Taylor P. Tryptophan fluorescence reveals conformational changes in the acetylcholine binding protein. J Biol Chem. 2002;277:41299–41302. doi: 10.1074/jbc.C200462200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P, Bourne Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005;24:3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hansen S, Wang HL, Taylor P, Sine SM. On the origin of ion conductance in pentameric Cys-loop receptors. J Biol Chem. 2008;283:36066–36070. doi: 10.1074/jbc.C800194200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Harel M, Kasher R, Nicolas A, Guss JM, Balass M, Fridkin M, Smit AB, Brejc K, Sixma TK, Katchalski-Katzir E, Sussman JL, Fuchs S. The binding site of acetylcholine receptor as visualized in the X-ray structure of a complex between alpha-bungarotoxin and a mimotope peptide. Neuron. 2001;32:265–275. doi: 10.1016/s0896-6273(01)00461-5. [DOI] [PubMed] [Google Scholar]
- 113.Hatton CJ, Shelley C, Brydson M, Beeson D, Colquhoun D. Properties of the human muscle nicotinic receptor, of the slow-channel myasthenic syndrome mutant epsilonL221F, inferred from maximum likelihood fits. J Physiol. 2003;547:729–760. doi: 10.1113/jphysiol.2002.034173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Heidmann T, Bernhardt J, Neumann E, Changeux JP. Rapid kinetics of agonist binding and permeability response analyzed in parallel on acetylcholine receptor rich membranes from Torpedo marmorata. Biochemistry. 1983;22:5452–5459. doi: 10.1021/bi00292a029. [DOI] [PubMed] [Google Scholar]
- 115.Henchman RH, Wang HL, Sine SM, Taylor P, McCammon JA. Asymmetric structural motions of the homomeric α7 nicotinic receptor ligand binding domain revealed by molecular dynamics simulation. Biophys J. 2003;85:3007–3018. doi: 10.1016/S0006-3495(03)74720-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Henchman RH, Wang HL, Sine SM, Taylor P, McCammon JA. Ligand-induced conformational changes in the α7 nicotinic receptor ligand binding domain. Biophys J. 2005;88:2564–2576. doi: 10.1529/biophysj.104.053934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Heuser JE, Salpeter SR. Organization of acetylcholine receptors in quick-frozen, deep-etched, rotary-replicated Torpedo postsynaptic membrane. J Cell Biol. 1979;82:150–173. doi: 10.1083/jcb.82.1.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hilf RJ, Dutzler R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature. 2009;457:115–118. doi: 10.1038/nature07461. [DOI] [PubMed] [Google Scholar]
- 119.Hilf RJ, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- 120.Hodgkin AL, Huxley AF. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J Physiol. 1952;116:449–472. doi: 10.1113/jphysiol.1952.sp004717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hodgkin AL, Huxley AF, Katz B. Measurement of current-voltage relations in the membrane of the giant axon of Loligo. J Physiol. 1952;116:424–448. doi: 10.1113/jphysiol.1952.sp004716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Horovitz A, Fersht AR. Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J Mol Biol. 1990;214:613–617. doi: 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- 123.Horovitz A, Serrano L, Avron B, bycroft M, Fersht AR. Strength and co-operativity of contributions of surface salt bridges to protein stability. J Mol Biol. 1990;216:1031–1044. doi: 10.1016/S0022-2836(99)80018-7. [DOI] [PubMed] [Google Scholar]
- 124.Hummer G, Rasaiah JC, Noworyta JP. Water conduction through the hydrophobic channel of a carbon nanotube. Nature. 2001;414:188–190. doi: 10.1038/35102535. [DOI] [PubMed] [Google Scholar]
- 125.Imoto K, Busch C, Sakmann B, Mishina M, Konno T, Nakai J, Bujo H, Mori Y, Fukuda K, Numa S. Rings of negatively charged amino acids determine the acetylcholine receptor channel conductance. Nature. 1988;335:645–648. doi: 10.1038/335645a0. [DOI] [PubMed] [Google Scholar]
- 126.Imoto K, Methfessel C, Sakmann B, Mishina M, Mori Y, Konno T, Fukuda K, Kurasaki M, Bujo H, Fujita Y. Location of a delta-subunit region determining ion transport through the acetylcholine receptor channel. Nature. 1986;324:670–674. doi: 10.1038/324670a0. [DOI] [PubMed] [Google Scholar]
- 127.Jackson MB. Dependence of acetylcholine receptor channel kinetics on agonist concentration in cultured mouse muscle fibres. J Physiol. 1988;397:555–583. doi: 10.1113/jphysiol.1988.sp017019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jackson MB. Kinetics of unliganded acetylcholine receptor channel gating. Biophys J. 1986;49:663–672. doi: 10.1016/S0006-3495(86)83693-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jackson MB. Perfection of a synaptic receptor: kinetics and energetics of the acetylcholine receptor. Proc Natl Acad Sci USA. 1989;86:2199–2203. doi: 10.1073/pnas.86.7.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jackson MB. Spontaneous openings of the acetylcholine receptor channel. Proc Natl Acad Sci USA. 1984;81:3901–3904. doi: 10.1073/pnas.81.12.3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jencks WP. A primer for the Bema Hapothle. An empirical approach to the characterization of changing transition state structures. Chem Rev. 1985;85:511–527. [Google Scholar]
- 132.Jenkinson DH. The antagonism of tubocurarine and substances which depolarize the motor end-plate. J Physiol. 1960;152:309–324. doi: 10.1113/jphysiol.1960.sp006489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jha A, Cadugan DJ, Purohit P, Auerbach A. Acetylcholine receptor gating at extracellular transmembrane domain interface: the cys-loop and M2–M3 linker. J Gen Physiol. 2007;130:547–558. doi: 10.1085/jgp.200709856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 135.Karlin A. Emerging structure of the nicotinic acetylcholine receptors. Nat Rev Neurosci. 2002;3:102–114. doi: 10.1038/nrn731. [DOI] [PubMed] [Google Scholar]
- 136.Karlin A, Holtzman E, Yodh N, Lobel P, Wall J, Hainfeld J. The arrangement of the subunits of the acetylcholine receptor of Torpedo californica. J Biol Chem. 1983;258:6678–6681. [PubMed] [Google Scholar]
- 137.Karlin A, Winnik M. Reduction and specific alkylation of the receptor for acetylcholine. Proc Natl Acad Sci USA. 1968;60:668–674. doi: 10.1073/pnas.60.2.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Struct Biol. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 139.Katz B, Miledi R. The measurement of synaptic delay and the time course of acetylcholine release at the neuromuscular junction. Proc R Soc Lond B Biol Sci. 1965;161:483–495. doi: 10.1098/rspb.1965.0016. [DOI] [PubMed] [Google Scholar]
- 140.Katz B, Miledi R. The statistical nature of the acetycholine potential and its molecular components. J Physiol. 1972;224:665–699. doi: 10.1113/jphysiol.1972.sp009918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Katz B, Thesleff S. A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol. 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kelley SP, Dunlop JI, Kirkness EF, Lambert JJ, Peters JA. A cytoplasmic region determines single-channel conductance in 5-HT3 receptors. Nature. 2003;424:321–324. doi: 10.1038/nature01788. [DOI] [PubMed] [Google Scholar]
- 143.Kistler J, Stroud RM. Crystalline arrays of membrane-bound acetylcholine receptor. Proc Natl Acad Sci USA. 1981;78:3678–36782. doi: 10.1073/pnas.78.6.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Korn SJ, Horn R. Statistical discrimination of fractal and Markov models of single-channel gating. Biophys J. 1988;54:871–877. doi: 10.1016/S0006-3495(88)83023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Krienkamp H, Sine SM, Maeda R, Taylor P. Glycosylation sites selectively interfere with α-toxin binding to the nicotinic acetylcholine receptor. J Biol Chem. 1994;269:8108–8114. [PubMed] [Google Scholar]
- 146.Kubalek E, Ralston S, Lindstrom J, Unwin N. Location of subunits within the acetylcholine receptor by electron image analysis of tubular crystals from Torpedo marmorata. J Cell Biol. 1987;105:9–18. doi: 10.1083/jcb.105.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kuffler SW, Yoshikami D. The number of transmitter molecules in a quantum: an estimate from iontophoretic application of acetylcholine at the neuromuscular synapse. J Physiol. 1975;251:465–482. doi: 10.1113/jphysiol.1975.sp011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kurosaki T, Fukuda K, Konno T, Mori Y, Tanaka K, Mishina M, Numa S. Functional properties of nicotinic acetylcholine receptor subunits expressed in various combinations. FEBS Lett. 1987;214:253–258. doi: 10.1016/0014-5793(87)80065-0. [DOI] [PubMed] [Google Scholar]
- 149.Land BR, Salpeter EE, Salpeter MM. Acetylcholine receptor site density affects the rising phase of miniature endplate currents. Proc Natl Acad Sci USA. 1980;77:3736–3740. doi: 10.1073/pnas.77.6.3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lape R, Colquhoun D, Sivilotti LG. On the nature of partial agonism in the nicotinic receptor superfamily. Nature. 2008;454:722–727. doi: 10.1038/nature07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lape R, Krashia P, Colquhoun D, Sivilotti LG. Agonist and blocking actions of choline and tetramethylammonium on human muscle acetylcholine receptors. J Physiol. 2009;587:5045–5072. doi: 10.1113/jphysiol.2009.176305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Law RJ, Henchman RH, McCammon JA. A gating mechanism proposed from a simulation of a human alpha7 nicotinic acetylcholine receptor. Proc Natl Acad Sci USA. 2005;102:6813–6818. doi: 10.1073/pnas.0407739102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lee WY, Free CR, Sine SM. Binding to gating transduction in nicotinic receptors: Cys-loop energetically couples to pre-M1 and M2–M3 regions. J Neurosci. 2009;29:3189–3199. doi: 10.1523/JNEUROSCI.6185-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lee WY, Sine SM. Invariant aspartic acid in muscle nicotinic receptor contributes selectively to the kinetics of agonist binding. J Gen Physiol. 2004;124:555–567. doi: 10.1085/jgp.200409077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lee WY, Sine SM. Principal pathway coupling agonist binding to channel gating in the nicotinic receptor. Nature. 2005;438:243–247. doi: 10.1038/nature04156. [DOI] [PubMed] [Google Scholar]
- 156.Le Novere N, Grutter T, Changeux JP. Models of the extracellular domain of the nicotinic receptors and of agonist and Ca2+ binding sites. Proc Natl Acad Sci USA. 2002;99:3210–3215. doi: 10.1073/pnas.042699699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Leonard RJ, Labarca CG, Charnet P, Davidson N, Lester HA. Evidence that the M2 membrane-spanning region lines the ion channel pore of the nicotinic receptor. Science. 1988;242:1578–1581. doi: 10.1126/science.2462281. [DOI] [PubMed] [Google Scholar]
- 158.Levitt M, Sander C, Stern PS. Protein normal-mode dynamics: trypsin inhibitor, crambin, ribonuclease and lysozyme. J Mol Biol. 1985;181:423–447. doi: 10.1016/0022-2836(85)90230-x. [DOI] [PubMed] [Google Scholar]
- 159.Lin WW, Smith LA, Lee CY. A study on the cause of death due to waglerin-I, a toxin from Trimeresurus wagleri. Toxicon. 1995;33:111–114. doi: 10.1016/0041-0101(94)00134-t. [DOI] [PubMed] [Google Scholar]
- 160.Linder TM, Quastel DM. A voltage-clamp study of the permeability change induced by quanta of transmitter at the mouse end-plate. J Physiol. 1978;281:535–558. doi: 10.1113/jphysiol.1978.sp012438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lindstrom J, Anholt R, Einarson B, Engel A, Osame M, Montal M. Purification of acetylcholine receptors, reconstitution into lipid vesicles, and study of agonist-induced cation channel regulation. J Biol Chem. 1980;255:8340–8350. [PubMed] [Google Scholar]
- 162.Lindstrom J, Einarson B, Merlie J. Immunization of rats with polypeptide chains from Torpedo acetylcholine receptor causes an autoimmune response to receptors in rat muscle. Proc Natl Acad Sci USA. 1978;75:769–773. doi: 10.1073/pnas.75.2.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lindstrom J, Merlie J, Yogeeswaran G. Biochemical properties of acetylcholine receptor subunits from Torpedo californica. Biochemistry. 1979;18:4465–4470. doi: 10.1021/bi00588a003. [DOI] [PubMed] [Google Scholar]
- 164.Magleby KL, Stevens CF. A quantitative description of end-plate currents. J Physiol. 1972;223:173–197. doi: 10.1113/jphysiol.1972.sp009840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Maconochie DJ, Steinbach JH. Block by acetylcholine of mouse muscle nicotinic receptors, stably expressed in fibroblasts. J Gen Physiol. 1995;106:113–147. doi: 10.1085/jgp.106.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Makriyannis A, Sullivan RF, Mautner HG. Conformational relationships between analogs of acetylcholine and those of local anesthetics in solution. Proc Natl Acad Sci USA. 1972;69:3416–3419. doi: 10.1073/pnas.69.11.3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Martin M, Czajkowski C, Karlin A. The contributions of aspartyl residues in the acetylcholine receptor gamma and delta subunits to the binding of agonists and competitive antagonists. J Biol Chem. 1996;271:13497–134503. doi: 10.1074/jbc.271.23.13497. [DOI] [PubMed] [Google Scholar]
- 168.Mayer ML, Armstrong N. Structure and function of glutamate receptor ion channels. Annu Rev Physiol. 2004;66:161–181. doi: 10.1146/annurev.physiol.66.050802.084104. [DOI] [PubMed] [Google Scholar]
- 169.McManus OB, Weiss DS, Spivak CE, Blatz AL, Magleby KL. Fractal models are inadequate for the kinetics of four different ion channels. Biophys J. 1988;54:859–870. doi: 10.1016/S0006-3495(88)83022-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Meunier JC, Sealock R, Olsen R, Changeux JP. Purification and properties of the cholinergic receptor protein from Electrophorus electricus electric tissue. Eur J Biochem. 1974;45:371–394. doi: 10.1111/j.1432-1033.1974.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 171.Miledi R, Molinoff P, Potter LT. Isolation of the cholinergic receptor protein of Torpedo electric tissue. Nature. 1971;229:554–557. doi: 10.1038/229554a0. [DOI] [PubMed] [Google Scholar]
- 172.Milone M, Wang HL, Ohno K, Fukudome T, Pruitt JN, Bren N, Sine SM, Engel AG. Slow-channel myasthenic syndrome caused by enhanced activation, desensitization, and agonist binding affinity attributable to mutation in the M2 domain of the acetylcholine receptor alpha subunit. J Neurosci. 1997;17:5651–5665. doi: 10.1523/JNEUROSCI.17-15-05651.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Milone M, Wang HL, Ohno K, Prince R, Fukudome T, Shen XM, Brengman J, Griggs R, Sine SM, Engel AG. Mode switching kinetics by a naturally occurring mutation in the cytoplasmic loop of the human acetylcholine receptor ε subunit. Neuron. 1998;20:575–588. doi: 10.1016/s0896-6273(00)80996-4. [DOI] [PubMed] [Google Scholar]
- 174.Mishina M, Tobimatsu T, Imoto K, Tanaka K, Fujita Y, Fukuda K, Kurasaki M, Takahashi H, Morimoto Y, Hirose T. Location of functional regions of acetylcholine receptor alpha-subunit by site-directed mutagenesis. Nature. 1985;313:364–369. doi: 10.1038/313364a0. [DOI] [PubMed] [Google Scholar]
- 175.Mitra A, Bailey TD, Auerbach AL. Structural dynamics of the M4 transmembrane segment during acetylcholine receptor gating. Structure. 2004;12:1909–1918. doi: 10.1016/j.str.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 176.Mitra A, Cymes GD, Auerbach A. Dynamics of the acetylcholine receptor pore at the gating transition state. Proc Natl Acad Sci USA. 2005;102:15069–15074. doi: 10.1073/pnas.0505090102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mitra AK, McCarthy MP, Stroud RM. Three-dimensional structure of the nicotinic acetylcholine receptor and location of the major associated 43-kD cytoskeletal protein, determined at 22 Å by low dose electron microscopy and x-ray diffraction to 12.5 Å. J Cell Biol. 1989;109:755–774. doi: 10.1083/jcb.109.2.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Miyazawa A, Fujiyoshi Y, Stowell M, Unwin N. Nicotinic receptor at 4.6 Å resolution: transverse tunnels in the channel wall. J Mol Biol. 1999;288:765–786. doi: 10.1006/jmbi.1999.2721. [DOI] [PubMed] [Google Scholar]
- 179.Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- 180.Molles BE, Rezai P, Kline EF, McArdle JJ, Sine SM, Taylor P. Identification of residues at the alpha and epsilon subunit interfaces mediating species selectivity of Waglerin-1 for nicotinic acetylcholine receptors. J Biol Chem. 2002;277:5433–5440. doi: 10.1074/jbc.M109232200. [DOI] [PubMed] [Google Scholar]
- 181.Molles BE, Tsigelny I, Nguyen PD, Gao SX, Sine SM, Taylor P. Residues in the epsilon subunit of the nicotinic acetylcholine receptor interact to confer selectivity of waglerin-1 for the alpha-epsilon subunit interface site. Biochemistry. 2002;41:7895–7906. doi: 10.1021/bi025732d. [DOI] [PubMed] [Google Scholar]
- 182.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 183.Morris CE, Wong BS, Jackson MB, Lecar H. Single-channel currents activated by curare in cultured embryonic rat muscle. J Neurosci. 1983;3:2525–2531. doi: 10.1523/JNEUROSCI.03-12-02525.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mukhtasimova N, Free C, Sine SM. Initial coupling of binding to gating mediated by conserved residues in muscle nicotinic receptor. J Gen Physiol. 2005;126:23–39. doi: 10.1085/jgp.200509283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mukhtasimova N, Lee WY, Wang HL, Sine SM. Detection and trapping of intermediate states priming nicotinic receptor channel opening. Nature. 2009;459:451–454. doi: 10.1038/nature07923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mukhtasimova N, Sine SM. An intersubunit trigger of channel gating in the muscle nicotinic receptor. J Neurosci. 2007;27:4110–4119. doi: 10.1523/JNEUROSCI.0025-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nastuk WL. Membrane potential changes at a single muscle end-plate produced by transitory application of acetylcholine with an electrically controlled microjet. Federation Proc. 1953;12:102. [Google Scholar]
- 188.Nastuk WL, Hodgkin AL. The electrical activity of single muscle fibers. J Cell Comp Physiol. 1950;35:39. [Google Scholar]
- 189.Neher E, Sakmann B, Steinbach JH. The extracellular patch clamp: a method for resolving currents through individual open channels in biological membranes. Pflügers Arch. 1978;375:219–228. doi: 10.1007/BF00584247. [DOI] [PubMed] [Google Scholar]
- 190.Neher E, Steinbach JH. Local anaesthetics transiently block currents through single acetylcholine-receptor channels. J Physiol. 1978;277:153–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Neubig RR, Cohen JB. Equilibrium binding of [3H]tubocurarine and [3H]acetylcholine by Torpedo postsynaptic membranes: stoichiometry and ligand interactions. Biochemistry. 1979;18:5464–5475. doi: 10.1021/bi00591a032. [DOI] [PubMed] [Google Scholar]
- 192.Nirthanan S, Ziebell MR, Chiara DC, Hong F, Cohen JB. Photolabeling the Torpedo nicotinic acetylcholine receptor with 4-azido-2,3,5,6-tetrafluorobenzoylcholine, a partial agonist. Biochemistry. 2005;44:13447–13456. doi: 10.1021/bi051209y. [DOI] [PubMed] [Google Scholar]
- 193.Noda M, Takahashi H, Tanabe T, Toyosato M, Furutani Y, Hirose T, Asai M, Inayama S, Miyata T, Numa S. Primary structure of alpha-subunit precursor of Torpedo californica acetylcholine receptor deduced from cDNA sequence. Nature. 1982;299:793–797. doi: 10.1038/299793a0. [DOI] [PubMed] [Google Scholar]
- 194.Noda M, Takahashi H, Tanabe T, Toyosato M, Kikyotani S, Furutani Y, Hirose T, Takashima H, Inayama S, Miyata T, Numa S. Structural homology of Torpedo californica acetylcholine receptor subunits. Nature. 1983;302:528–532. doi: 10.1038/302528a0. [DOI] [PubMed] [Google Scholar]
- 195.Noda M, Takahashi H, Tanabe T, Toyosato M, Kikyotani S, Hirose T, Asai M, Takashima H, Inayama S, Miyata T, Numa S. Primary structures of beta- and delta-subunit precursors of Torpedo californica acetylcholine receptor deduced from cDNA sequences. Nature. 1983;301:251–255. doi: 10.1038/301251a0. [DOI] [PubMed] [Google Scholar]
- 196.Nowak MW, Gallivan JP, Silverman SK, Labarca CG, Dougherty DA, Lester HA. In vivo incorporation of unnatural amino acids into ion channels in Xenopus oocyte expression system. Methods Enzymol. 1998;293:504–529. doi: 10.1016/s0076-6879(98)93031-2. [DOI] [PubMed] [Google Scholar]
- 197.Ogden DC, Colquhoun D. Ion channel block by acetylcholine, carbachol and suberyl-dicholine at the frog neuromuscular junction. Proc R Soc Lond B Biol Sci. 1985;225:329–355. doi: 10.1098/rspb.1985.0065. [DOI] [PubMed] [Google Scholar]
- 198.Ohana B, Gershoni JM. Comparison of the toxin binding sites of the nicotinic acetylcholine receptor from Drosophila to human. Biochemistry. 1990;29:6409–6415. doi: 10.1021/bi00479a011. [DOI] [PubMed] [Google Scholar]
- 199.Ohno K, Hutchinson DO, Milone M, Brengman JM, Bouzat C, Sine SM, Engel AG. Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the epsilon subunit. Proc Natl Acad Sci USA. 1995;92:758–762. doi: 10.1073/pnas.92.3.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ohno K, Quiram P, Milone M, Wang HL, Harper M, Pruitt JN, Brengman JM, Pao L, Fischbeck K, Crawford T, Sine SM, Engel AG. Congenital myasthenic syndromes due to heteroallelic nonsense/missense mutations in the acetylcholine receptor ε subunit gene: identification and functional characterization of six new mutations. Hum Mol Genet. 1997;6:753–766. doi: 10.1093/hmg/6.5.753. [DOI] [PubMed] [Google Scholar]
- 201.Ohno K, Wang HL, Milone M, Bren N, Brengman JM, Nakano S, Quiram P, Pruitt JN, Sine SM, Engel AG. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit. Neuron. 1996;17:157–170. doi: 10.1016/s0896-6273(00)80289-5. [DOI] [PubMed] [Google Scholar]
- 202.O’Leary ME, White MM. Mutational analysis of ligand-induced activation of the Torpedo acetylcholine receptor. J Biol Chem. 1992;267:8360–8365. [PubMed] [Google Scholar]
- 203.Osaka H, Malany S, Kanter JR, Sine SM, Taylor P. Subunit interface selectivity of the alpha-neurotoxins for the nicotinic acetylcholine receptor. J Biol Chem. 1999;274:9581–9586. doi: 10.1074/jbc.274.14.9581. [DOI] [PubMed] [Google Scholar]
- 204.Osaka H, Malany S, Molles BE, Sine SM, Taylor P. Pairwise electrostatic interactions between α-neurotoxins and γ, δ and ε subunits of the nicotinic acetylcholine receptor. J Biol Chem. 2000;275:5478–5484. doi: 10.1074/jbc.275.8.5478. [DOI] [PubMed] [Google Scholar]
- 205.Oswald RE, Changeux JP. Crosslinking of alpha-bungarotoxin to the acetylcholine receptor from Torpedo marmorata by ultraviolet light irradiation. FEBS Lett. 1982;139:225–229. doi: 10.1016/0014-5793(82)80857-0. [DOI] [PubMed] [Google Scholar]
- 206.Pedersen SE, Cohen JB. d-Tubocurarine binding sites are located at alpha-gamma and alpha-delta subunit interfaces of the nicotinic acetylcholine receptor. Proc Natl Acad Sci USA. 87:2785–2789. doi: 10.1073/pnas.87.7.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Prince RJ, Sine SM. Acetylcholine and epibatidine binding to muscle acetylcholine receptors distinguish between concerted and uncoupled models. J Biol Chem. 1999;274:19623–19629. doi: 10.1074/jbc.274.28.19623. [DOI] [PubMed] [Google Scholar]
- 208.Prince RJ, Sine SM. Molecular dissection of subunit interfaces in the acetylcholine receptor: identification of residues that determine agonist selectivity. J Biol Chem. 1996;271:25770–25777. doi: 10.1074/jbc.271.42.25770. [DOI] [PubMed] [Google Scholar]
- 209.Purohit P, Auerbach A. Acetylcholine receptor gating at extracellular transmembrane domain interface: the “pre-M1” linker. J Gen Physiol. 2007;130:559–568. doi: 10.1085/jgp.200709857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Purohit P, Auerbach A. Acetylcholine receptor gating: movement in the alpha-subunit extracellular domain. J Gen Physiol. 2007;130:569–79. doi: 10.1085/jgp.200709858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Qin F, Auerbach A, Sachs F. Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys J. 1996;70:264–280. doi: 10.1016/S0006-3495(96)79568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Quast U, Schimerlik M, Lee T, Witzemann TL, Blanchard S, Raftery MA. Ligand-induced conformation changes in Torpedo californica membrane-bound acetylcholine receptor. Biochemistry. 1978;17:2405–2014. doi: 10.1021/bi00605a024. [DOI] [PubMed] [Google Scholar]
- 213.Quiram P, Jones J, Sine SM. Pairwise interactions between neuronal α7 receptors and α-conotoxin ImI. J Biol Chem. 1999;274:19517–19525. doi: 10.1074/jbc.274.28.19517. [DOI] [PubMed] [Google Scholar]
- 214.Quiram P, McIntosh M, Sine SM. Pairwise interactions between neuronal α7 receptors and α-conotoxin PnIB. J Biol Chem. 2000;275:4889–4996. doi: 10.1074/jbc.275.7.4889. [DOI] [PubMed] [Google Scholar]
- 215.Quiram P, Sine SM. Identification of residues in the neuronal α7 receptor that confer selectivity for conotoxin ImI. J Biol Chem. 1998;273:11001–11006. doi: 10.1074/jbc.273.18.11001. [DOI] [PubMed] [Google Scholar]
- 216.Quiram P, Sine SM. Structural elements in α-conotoxin ImI essential for binding to neuronal α7 receptors. J Biol Chem. 1998;273:11007–11011. doi: 10.1074/jbc.273.18.11007. [DOI] [PubMed] [Google Scholar]
- 217.Raftery MA, Hunkapiller MW, Strader CD, Hood LE. Acetylcholine receptor: complex of homologous subunits. Science. 1980;208:1454–1456. doi: 10.1126/science.7384786. [DOI] [PubMed] [Google Scholar]
- 218.Rayes D, De Rosa MJ, Sine SM, Bouzat C. Number and locations of agonist binding sites required to activate homomeric Cys-loop receptors. J Neurosci. 2009;29:6022–6032. doi: 10.1523/JNEUROSCI.0627-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Revah F, Galzi JL, Giraudat J, Haumont PY, Lederer F, Changeux JP. The noncompetitive blocker [3H]chlorpromazine labels three amino acids of the acetylcholine receptor gamma subunit: implications for the alpha-helical organization of regions MII and for the structure of the ion channel. Proc Natl Acad Sci USA. 1990;87:4675–4679. doi: 10.1073/pnas.87.12.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Reynolds JA, Karlin A. Molecular weight in detergent solution of acetylcholine receptor from Torpedo californica. Biochemistry. 1978;17:2035–2038. doi: 10.1021/bi00604a001. [DOI] [PubMed] [Google Scholar]
- 221.Rosenberry TL. Quantitative simulation of endplate currents at neuromuscular junctions based on the reaction of acetylcholine with acetylcholine receptor and acetylcholinesterase. Biophys J. 1979;26:263–289. doi: 10.1016/S0006-3495(79)85249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Sakmann B, Methfessel C, Mishina M, Takahashi T, Takai T, Kurasaki M, Fukuda K, Numa S. Role of acetylcholine receptor subunits in gating of the channel. Nature. 1985;318:538–543. doi: 10.1038/318538a0. [DOI] [PubMed] [Google Scholar]
- 223.Sakmann B, Patlak J, Neher E. Single acetylcholine-activated channels show burst-kinetics in presence of desensitizing concentrations of agonist. Nature. 1980;286:71–73. doi: 10.1038/286071a0. [DOI] [PubMed] [Google Scholar]
- 224.Salpeter MM. The Vertebrate Neuromuscular Junction. New York: Liss; 1987. Vertebrate neuromuscular junctions: general morphology, molecular organization and functional consequences; pp. 1–54. [Google Scholar]
- 225.Samson AO, Levitt M. Inhibition mechanism of the acetylcholine receptor by alpha-neurotoxins as revealed by normal-mode dynamics. Biochemistry. 2008;47:4065–4070. doi: 10.1021/bi702272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Scarselli M, Spiga O, Ciutti A, Bernini A, Bracci L, Leilli B, Lozzi L, Calamandrei D, Di Maro D, Klein S, Niccolai N. NMR structure of alpha-bungarotoxin free and bound to a mimotope of the nicotinic acetylcholine receptor. Biochem. 2002;41:1457–1463. doi: 10.1021/bi011012f. [DOI] [PubMed] [Google Scholar]
- 227.Schmidt JJ, Weinstein SA, Smith LA. Molecular properties and structure-function relationships of lethal peptides from venom of Wagler’s pit viper, Trimeresurus wagleri. Toxicon. 1992;30:1027–1036. doi: 10.1016/0041-0101(92)90047-9. [DOI] [PubMed] [Google Scholar]
- 228.Schreiber G, Fersht AR. Energetics of protein-protein interactions: analysis of the barnase-barstar interface by single mutations and double mutant cycles. J Mol Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- 229.Scott KA, Mautner HG. Sulfur and selenium isologs related to acetylcholine and choline. IX. Further comparative studies of the pharmacological effects of acetylcholine and its thio and seleno analogs and their hydrolysis products. Biochem Pharmacol. 1967;16:1903–1918. doi: 10.1016/0006-2952(67)90301-2. [DOI] [PubMed] [Google Scholar]
- 230.Sellin LC, Mattila K, Annila A, Schmidt JJ, McArdle JJ, Hyvönen M, Rantala TT, Kivistö T. Conformational analysis of a toxic peptide from Trimeresurus wagleri which blocks the nicotinic acetylcholine receptor. Biophys J. 1996;70:3–13. doi: 10.1016/S0006-3495(96)79559-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Shefter E, Mautner HG. Acetylcholine and its thiolester and selenolester analogs: conformation, electron distribution, and biological activity. Proc Natl Acad Sci USA. 1969;63:1253–1260. doi: 10.1073/pnas.63.4.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Shen XM, Fukuda T, Ohno K, Sine SM, Engel AG. Novel AChR δ subunit mutation causes myasthenia by interfering with inter-subunit communication essential for channel gating. J Clin Invest. 2008;180:1867–1876. doi: 10.1172/JCI34527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Sheridan RP, Nilakantan R, Dixon JS, Venkataraghavan R. The ensemble approach to distance geometry: application to the nicotinic pharmacophore. J Med Chem. 1986;29:899–906. doi: 10.1021/jm00156a005. [DOI] [PubMed] [Google Scholar]
- 234.Shi J, Koeppe JR, Komives EA, Taylor P. Ligand-induced conformational changes in the acetylcholine-binding protein analyzed by hydrogen-deuterium exchange mass spectrometry. J Biol Chem. 2006;281:12170–71217. doi: 10.1074/jbc.M600154200. [DOI] [PubMed] [Google Scholar]
- 235.Sigworth FJ. An example of analysis. In: Sakmann B, Neher E, editors. Single-Channel Recording. 1. New York: Plenum; 1983. [Google Scholar]
- 236.Sigworth FJ. Design of the EPC-9, a computer-controlled patch-clamp amplifier. 1. Hardware. J Neurosci Methods. 1995;56 ES/*ME2. [PubMed] [Google Scholar]
- 237.Sine SM. Identification of equivalent residues in the γ, ε, and δ subunits of the nicotinic receptor that contribute to α-bungarotoxin binding. J Biol Chem. 1997;272:23521–23527. doi: 10.1074/jbc.272.38.23521. [DOI] [PubMed] [Google Scholar]
- 238.Sine SM. Molecular dissection of subunit interfaces in the acetylcholine receptor: identification of residues that determine curare selectivity. Proc Natl Acad Sci USA. 1993;90:9436–9440. doi: 10.1073/pnas.90.20.9436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Sine SM. The nicotinic receptor ligand binding domain. J Neurobiol. 2002;53:431–46. doi: 10.1002/neu.10139. [DOI] [PubMed] [Google Scholar]
- 240.Sine SM, Claudio T. Gamma- and delta-subunits regulate the affinity and the cooperativity of ligand binding to the acetylcholine receptor. J Biol Chem. 1991;266:19369–19377. [PubMed] [Google Scholar]
- 241.Sine SM, Claudio T, Sigworth FJ. Activation of Torpedo acetylcholine receptors expressed in mouse fibroblasts. Single channel current kinetics reveal distinct agonist binding affinities. J Gen Physiol. 1990;96:395–437. doi: 10.1085/jgp.96.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Sine SM, Engel AG. Recent advances in Cys-loop structure and function. Nature. 2006;440:448–455. doi: 10.1038/nature04708. [DOI] [PubMed] [Google Scholar]
- 243.Sine SM, Kreienkamp HJ, Bren N, Maeda R, Taylor P. Molecular dissection of subunit interfaces in the acetylcholine receptor: identification of determinants of α-Conotoxin M1 selectivity. Neuron. 1995;15:205–211. doi: 10.1016/0896-6273(95)90077-2. [DOI] [PubMed] [Google Scholar]
- 244.Sine SM, Ohno K, Bouzat C, Auerbach A, Milone M, Pruitt AG, Engel JN. Mutation of the acetylcholine receptor α-subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron. 1995;15:229–239. doi: 10.1016/0896-6273(95)90080-2. [DOI] [PubMed] [Google Scholar]
- 245.Sine SM, Quiram P, Papanikolaou F, Kreienkamp HJ, Taylor P. Conserved tyrosines in the α subunit of the nicotinic acetylcholine receptor stabilize quaternary ammonium groups of agonists and curariform antagonists. J Biol Chem. 1994;269:8808–8816. [PubMed] [Google Scholar]
- 246.Sine SM, Shen XM, Wang HL, Brengmann JM, Lee WY, Bren N, Engel AG. Naturally-occurring mutations at the acetylcholine receptor binding site independently alter ACh binding and channel gating. J Gen Physiol. 2002;120:483–496. doi: 10.1085/jgp.20028568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Sine SM, Steinbach JH. Acetylcholine receptor activation by a site-selective ligand: nature of brief open and closed states in BC3H-1 cells. J Physiol. 1986;370:357–379. doi: 10.1113/jphysiol.1986.sp015939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Sine SM, Steinbach JH. Activation of acetylcholine receptors on clonal mammalian BC3H-1 cells by low concentrations of agonist. J Physiol. 1986;373:129–162. doi: 10.1113/jphysiol.1986.sp016039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Sine SM, Steinbach JH. Agonists block currents through acetylcholine receptor channels. Biophys J. 1984;46:277–284. doi: 10.1016/S0006-3495(84)84022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Sine SM, Taylor P. Relationships between reversible antagonist occupancy and the functional capacity of the acetylcholine receptor. J Biol Chem. 1981;256:6692–6699. [PubMed] [Google Scholar]
- 251.Sine SM, Taylor P. The relationship between agonist occupation and the permeability response of the cholinergic receptor revealed by bound cobra alpha-toxin. J Biol Chem. 1980;255:10144–10156. [PubMed] [Google Scholar]
- 252.Sine SM, Wang HL, Bren N. Lysine scanning mutagenesis delineates structural model of the nicotinic receptor ligand binding domain. J Biol Chem. 2002;277:28210–29223. doi: 10.1074/jbc.M203396200. [DOI] [PubMed] [Google Scholar]
- 253.Smit AB, Syed NI, Schaap D, van Minnen J, Klumperman J, Kits KS, Lodder H, van der Schors RC, van Elk R, Sorgedrager B, Brejc K, Sixma TK, Geraerts WP. A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature. 2001;411:261–268. doi: 10.1038/35077000. [DOI] [PubMed] [Google Scholar]
- 254.Sobell HM, Sakore TD, Tavale SS, Canepa FG, Pauling P, Petcher TJ. Stereochemistry of a curare alkaloid: O,O′,N-trimethyl-d-tubocurarine. Proc Natl Acad Sci USA. 1972;69:2212–225. doi: 10.1073/pnas.69.8.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Suchyna TM, Markin VS, Sachs F. Biophysics and structure of the patch and the gigaseal. Biophys J. 2009;97:738–747. doi: 10.1016/j.bpj.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Svinning T, Sörum H. A re-investigation of the crystal structure of acetylcholine bromide. Acta Cryst. 1975;B31:1581–1586. [Google Scholar]
- 257.Takai T, Noda M, Mishina M, Shimizu S, Furutani Y, Kayano T, Ikeda T, Kubo T, Takahashi H, Takahashi T. Cloning, sequencing and expression of cDNA for a novel subunit of acetylcholine receptor from calf muscle. Nature. 1985;315:761–764. doi: 10.1038/315761a0. [DOI] [PubMed] [Google Scholar]
- 258.Takeda K, Trautmann A. A patch-clamp study of the partial agonist actions of tubocurarine on rat myotubes. J Physiol. 1984;349:353–374. doi: 10.1113/jphysiol.1984.sp015160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Takeuchi A, Takeuchi N. On the permeability of end-plate membrane during the action of transmitter. J Physiol. 1960;154:52–67. doi: 10.1113/jphysiol.1960.sp006564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Talley TT, Harel M, Hibbs RE, Radic Z, Tomizawa M, Casida JE, Taylor P. Atomic interactions of neonicotinoid agonists with AChBP: molecular recognition of the distinctive electronegative pharmacophore. Proc Natl Acad Sci USA. 2008;105:7606–7611. doi: 10.1073/pnas.0802197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Taly A, Delarue M, Grutter T, Nilges M, Le Novère N, Corringer PJ, Changeux JP. Normal mode analysis suggests a quaternary twist model for the nicotinic receptor gating mechanism. Biophys J. 2005;88:3954–3965. doi: 10.1529/biophysj.104.050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Tasneem A, Iyer LM, Jakobsson E, Aravind L. Identification of the prokaryotic ligand-gated ion channels and their implications for the mechanisms and origins of animal Cys-loop ion channels. Genome Biol. 2005;6:R4. doi: 10.1186/gb-2004-6-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Tierney ML, Osborn KE, Milburn PJ, Stowell MH, Howitt SM. Phylogenetic conservation of disulfide-linked, dimeric acetylcholine receptor pentamers in southern ocean electric rays. J Exp Biol. 2004;207:3581–3590. doi: 10.1242/jeb.01204. [DOI] [PubMed] [Google Scholar]
- 264.Tomaselli GF, McLaughlin JT, Jurman ME, Hawrot E, Yellen G. Mutations affecting agonist sensitivity of the nicotinic acetylcholine receptor. Biophys J. 1991;60:721–727. doi: 10.1016/S0006-3495(91)82102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Toyoshima C, Unwin N. Ion channel of acetylcholine receptor reconstructed from images of postsynaptic membranes. Nature. 1988;336:247–250. doi: 10.1038/336247a0. [DOI] [PubMed] [Google Scholar]
- 266.Tzartos SJ, Changeux JP. High affinity binding of alpha-bungarotoxin to the purified alpha-subunit and to its 27,000-dalton proteolytic peptide from Torpedo marmorata acetylcholine receptor. Requirement for sodium dodecyl sulfate. EMBO J. 1983;2:381–387. doi: 10.1002/j.1460-2075.1983.tb01434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Tzartos SJ, Lindstrom JM. Monoclonal antibodies used to probe acetylcholine receptor structure: localization of the main immunogenic region and detection of similarities between subunits. Proc Natl Acad Sci USA. 1980;77:755–759. doi: 10.1073/pnas.77.2.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Ulens C, Hogg RC, Celie PH, Bertrand D, Tsetlin V, Smit AB, Sixma TK. Structural determinants of selective alpha-conotoxin binding to a nicotinic acetylcholine receptor homolog AChBP. Proc Natl Acad Sci USA. 2006;103:3615–3620. doi: 10.1073/pnas.0507889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- 270.Unwin N. Nicotinic acetylcholine receptor at 9 A resolution. J Mol Biol. 1993;229:1101–1124. doi: 10.1006/jmbi.1993.1107. [DOI] [PubMed] [Google Scholar]
- 271.Unwin N. Refined structure of the nicotinic receptor at 4 Å resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 272.Unwin N, Miyazawa A, Li J, Fujiyoshi Y. Activation of the nicotinic acetylcholine receptor involves a switch in conformation of the alpha subunits. J Mol Biol. 2002;319:1165–1176. doi: 10.1016/S0022-2836(02)00381-9. [DOI] [PubMed] [Google Scholar]
- 273.Vaughan CK, Harryson P, Buckle AM, Fersht A. A structural double-mutant cycle: estimating the strength of a buried salt bridge in barnase. Acta Cryst D Biol Cryst. 2002;58:591–600. doi: 10.1107/s0907444902001567. [DOI] [PubMed] [Google Scholar]
- 274.Venkataramanan L, Sigworth FJ. Applying hidden Markov models to the analysis of single ion channel activity. Biophys J. 2002;82:1930–1942. doi: 10.1016/S0006-3495(02)75542-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Wang HL, Auerbach A, Bren N, Ohno K, Engel AG, Sine SM. Mutation in the M1 domain of the acetylcholine receptor alpha subunit decreases the rate of agonist dissociation. J Gen Physiol. 1997;109:757–766. doi: 10.1085/jgp.109.6.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Wang HL, Cheng X, Taylor P, McCammon JA, Sine SM. Control of cation permeation through the nicotinic receptor. PLos Comp Biol. 2008;4:1–9. doi: 10.1371/journal.pcbi.0040041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Wang D, Chiara DC, Xie Y, Cohen JB. Probing the structure of the nicotinic acetylcholine receptor with 4-benzoylbenzoylcholine, a novel photoaffinity competitive antagonist. J Biol Chem. 2000;275:28666–28674. doi: 10.1074/jbc.M005441200. [DOI] [PubMed] [Google Scholar]
- 278.Wang HL, Gao F, Bren N, Sine SM. Curariform antagonists bind in different orientations to the nicotinic receptor ligand binding domain. J Biol Chem. 2003;278:32284–32291. doi: 10.1074/jbc.M304366200. [DOI] [PubMed] [Google Scholar]
- 279.Wang HL, Ohno K, Milone M, Brengman J, Evoli A, Batocchi AP, Middleton L, Christodoulou K, Engel AG, Sine SM. Fundamental gating mechanism of nicotinic receptor channel gating revealed by mutation causing a congenital myasthenic syndrome. J Gen Physiol. 2000;116:449–460. doi: 10.1085/jgp.116.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Wang HL, Toghraee R, Papke D, Cheng XL, McCammon JA, Ravaioli U, Sine SM. Single channel current through nicotinic receptor produced by closure of binding site C-loop. Biophys J. 2009;96:3582–3590. doi: 10.1016/j.bpj.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Weber M, Changeux JP. Binding of Naja nigricollis [3H]alpha-toxin to membrane fragments from Electrophorus and Torpedo electric organs. I. Binding of the tritiated alpha-neurotoxin in the absence of effector. Mol Pharmacol. 1974;10:1–14. [PubMed] [Google Scholar]
- 282.Weber M, David-Pfeuty T, Changeux JP. Regulation of binding properties of the nicotinic receptor protein by cholinergic ligands in membrane fragments from Torpedo marmorata. Proc Natl Acad Sci USA. 1975;72:3443–3447. doi: 10.1073/pnas.72.9.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Weerakoon P, Culurciello E, Yang Y, Santos-Sacchi J, Kindlmann PJ, Sigworth FJ. Patch clamp amplifiers on a chip. J Neurosci Methods. 2010;192:187–192. doi: 10.1016/j.jneumeth.2010.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Weiland G, Georgia B, Lappi S, Chignell CF, Taylor P. Kinetics of agonist-mediated transitions in state of the cholinergic receptor. J Biol Chem. 1977;252:7648–7656. [PubMed] [Google Scholar]
- 285.Weiland G, Georgia B, Wee VT, Chignell CF, Taylor P. Ligand interactions with cholinergic receptor-enriched membranes from Torpedo: influence of agonist exposure on receptor properties. Mol Pharmacol. 1976;12:1091–1105. [PubMed] [Google Scholar]
- 286.Weinstein SA, Schmidt JJ, Bernheimer AW, Smith LA. Characterization and amino acid sequences of two lethal peptides isolated from venom of Wagler’s pit viper, Trimeresurus wagleri. Toxicon. 1991;29:227–236. doi: 10.1016/0041-0101(91)90107-3. [DOI] [PubMed] [Google Scholar]
- 287.Wells GB, Anand R, Wang F, Lindstrom J. Water-soluble nicotinic acetylcholine receptor formed by alpha7 subunit extracellular domains. J Biol Chem. 1998;273:964–973. doi: 10.1074/jbc.273.2.964. [DOI] [PubMed] [Google Scholar]
- 288.Wenningmann I, Dilger JP. The kinetics of inhibition of nicotinic acetylcholine receptors by (+)-tubocurarine and pancuronium. Mol Pharmacol. 2001;60:790–796. [PubMed] [Google Scholar]
- 289.Williamson PT, Verhoeven A, Miller KW, Meier BH, Watts A. The conformation of acetylcholine at its target site in the membrane-embedded nicotinic acetylcholine receptor. Proc Natl Acad Sci USA. 2007;104:18031–18036. doi: 10.1073/pnas.0704785104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Wilson GG, Karlin A. The location of the gate in the acetylcholine receptor channel. Neuron. 1998;20:1269–1281. doi: 10.1016/s0896-6273(00)80506-1. [DOI] [PubMed] [Google Scholar]
- 291.Wilson PT, Lentz TL, Hawrot E. Determination of the primary amino acid sequence specifying the alpha-bungarotoxin binding site on the alpha subunit of the acetylcholine receptor from Torpedo californica. Proc Natl Acad Sci USA. 1985;82:8790–8794. doi: 10.1073/pnas.82.24.8790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Wise DS, Schoenborn BP, Karlin A. Structure of acetylcholine receptor dimer determined by neutron scattering and electron microscopy. J Biol Chem. 1981;256:4124–4126. [PubMed] [Google Scholar]
- 293.Xiu X, Hanek AP, Wang J, Lester HA, Dougherty DA. A unified view of the role of electrostatic interactions in modulating the gating of Cys loop receptors. J Biol Chem. 2005;280:41655–41666. doi: 10.1074/jbc.M508635200. [DOI] [PubMed] [Google Scholar]
- 294.Young EF, Ralston E, Blake J, Ramachandran J, Hall ZW, Stroud RM. Topological mapping of acetylcholine receptor: evidence for a model with five transmembrane segments and a cytoplasmic COOH-terminal peptide. Proc Natl Acad Sci USA. 1985;82:626–630. doi: 10.1073/pnas.82.2.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Zafaralla GC, Ramilo C, Gray WR, Karlstrom R, Olivera BM, Cruz LJ. Phylogenetic specificity of cholinergic ligands: alpha-conotoxin SI. Biochemistry. 1988;27:7102–7105. doi: 10.1021/bi00418a065. [DOI] [PubMed] [Google Scholar]
- 296.Zeng H, Moise L, Grant MA, Hawrot E. The solution structure of the complex formed between alpha-bungarotoxin and an 18-mer cognate peptide derived from the alpha 1 subunit of the nicotinic acetylcholine receptor from Torpedo californica. J Biol Chem. 2001;276:22930–22940. doi: 10.1074/jbc.M102300200. [DOI] [PubMed] [Google Scholar]
- 297.Zhang Y, Chen J, Auerbach A. Activation of recombinant mouse acetylcholine receptors by acetylcholine, carbamylcholine and tetramethylammonium. J Physiol. 1995;486:189–206. doi: 10.1113/jphysiol.1995.sp020802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Zhong W, Gallivan JP, Zhang Y, Li L, Lester HA, Dougherty DA. From ab initio quantum mechanics to molecular neurobiology: a cation-pi binding site in the nicotinic receptor. Proc Natl Acad Sci USA. 1998;95:12088–12093. doi: 10.1073/pnas.95.21.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Zingsheim HP, Barrantes FJ, Frank J, Hänicke W, Neugebauer DC. Direct structural localization of two toxin-recognition sites on an ACh receptor protein. Nature. 1982;299:81–84. doi: 10.1038/299081a0. [DOI] [PubMed] [Google Scholar]
- 300.Zouridakis M, Zisimopoulou P, Eliopoulos E, Poulas K, Tzartos SJ. Design and expression of human alpha7 nicotinic acetylcholine receptor extracellular domain mutants with enhanced solubility and ligand-binding properties. Biochim Biophys Acta. 2009;1794:355–66. doi: 10.1016/j.bbapap.2008.11.002. [DOI] [PubMed] [Google Scholar]